Novel triphenylamine-modified ruthenium(II) terpyridine complexes for nickel oxide-based cathodic dye-sensitized solar cells†
Christopher J. Wooda,
Kiyoshi C. D. Robsonb,
Paul I. P. Elliottc,
Curtis P. Berlinguette‡
b and
Elizabeth A. Gibson*a
aSchool of Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: elizabeth.gibson@nottingham.ac.uk
bDepartment of Chemistry, University of Calgary, 2500 University Drive Northwest, Calgary, Alberta T2N 1N4, Canada
cDepartment of Chemistry, University of Huddersfield, Queensgate, Huddersfield, HD1 3DH, UK
First published on 18th December 2013
Abstract
A pair of ruthenium based donor-π-chromophore sensitizers (K1 and K2) have been synthesized for use in NiO based p-type dye sensitized solar cells (p-DSCs). The optical and electrochemical properties of these dyes were determined experimentally and interpreted by DFT modelling. NiO p-DSC devices incorporating these dyes gave photocurrents of 2.9 for K1 and 2.0 mA cm−2 for K2 (IPCE of 14% and 9%); this is a vast improvement on the photocurrents of p-DSC devices incorporating ‘traditional’ ruthenium sensitizers.
Introduction
The dye-sensitized solar cell (DSC) is a low cost alternative to crystalline silicon photovoltaic devices that converts sunlight into electricity using a dye adsorbed on a transparent, nanostructured semiconductor electrode, surrounded by a redox electrolyte.1–4 Almost all the current research in photocatalysis and dye-sensitized solar cells is focused on n-type semiconductors typically based on TiO2. Unlike the standard DSC, which has a passive counter electrode, tandem devices use two photoelectrodes, simultaneously, to harvest more light more efficiently.5 Incorporation of a photocathode in tandem with a TiO2-based n-type photoanode in a single device should give rise to a substantial increase in voltage and efficiency.6–8 By choosing sensitizers which absorb the high energy photons on one electrode and low energy photons on the other, more of the solar spectrum can be harvested. Yet tandem DSCs have not yet beaten the best n-type DSCs because the poor performance of dye-sensitized photocathodes limits the overall efficiency.9One of the limitations to NiO-based photocathodes is the fast recombination between the photoreduced dye and holes in the NiO. Previously, it has been shown that significant improvements to NiO-based p-DSCs can be made by designing organic dyes which promote charge separation and limit the recombination between the sensitizer and/or electrolyte and the semiconductor.8,10 Currents obtained in these devices have reached over 5.5 mA cm−2 at 1 sun illumination but this is considerably less than that achieved with n-DSCs. Typically, when organic photosensitizers are used, the charge-separated state is short-lived (τ ≈ 20–200 ps) compared to the regeneration reaction, which must compete in order to collect the charges and produce a photocurrent.10–13 Organic dyes perform much better in p-DSCs than ruthenium-based sensitizers.14 This is possibly a result of the film thickness of NiO films typically used (ca. 1–3 μm) being much less than that typically used for TiO2 in n-DSCs (ca. 10 μm) so dyes with higher extinction coefficients maximise the light harvesting efficiency. In addition the optimum geometry for the anchoring group for NiO sensitizers may not have been found. Many organic dyes use one or two benzoic acid groups to couple the dye to the NiO.7,15,16 Although ruthenium bipyridyl complexes with various anchoring motifs have been applied, the essential requirements of good charge injection and stability have not been met.17,18 We postulated that, if a more stable and conjugated anchoring motif was chosen, we could develop a better ruthenium sensitizer that would make use of the favourable optoelectronic properties of ruthenium sensitizers, in particular the long-lived charge separated state.
Others have since tried to develop ruthenium-based sensitizers using a similar strategy of increasing the spatial separation between the electron acceptor region of the dye and the anchoring group/NiO surface to substantially increase the charge-separated state lifetime.19,20 However, whilst incorporating charge-transfer character in the dye design appears to improve the charge-separated state lifetime, the photocurrents generated have still remained low (below 1 mA cm−2 in ref. 19 and below 2 mA cm−2 in ref. 20). Previous success with ruthenium-based push–pull photosensitizers for TiO2 prompted us to modify our dye-design to suit photocathode sensitization.21,22 Here, we report the first of our photosensitizers which are equipped with stable, bidentate anchoring groups, an electron donating amine–thiophene linker with bis-terpyridyl (K1) and cyclometalated (K2) ruthenium chromophores, depicted in Fig. 1.
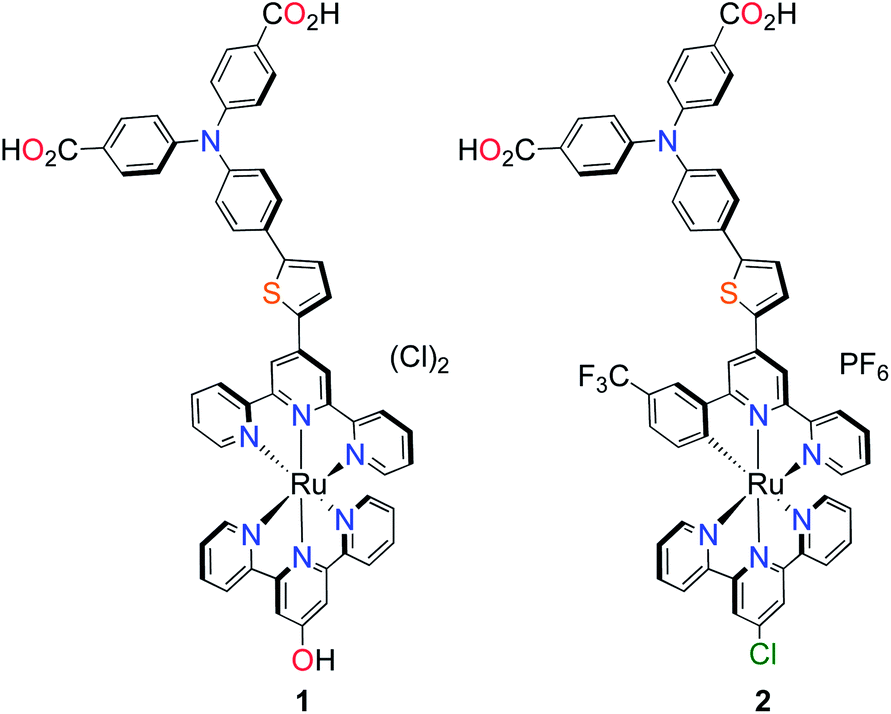 | ||
| Fig. 1 Molecular structures of p-type polypyridyl (K1) and cyclometalated (K2) bichromic sensitizers. | ||
Experimental section
Preparation of compounds
All reactions and manipulations were performed using solvents passed through an MBraun solvent purification system prior to use. All reagents were purchased from Aldrich. Purification by column chromatography was carried out using silica (Silicycle: Ultrapure Flash Silica). Analytical thin-layer chromatography (TLC) was performed on aluminum-backed sheets pre-coated with silica 60 F254 adsorbent (0.25 mm thick; Merck, Germany) and visualized under UV light. Routine 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz, respectively, on a Bruker AV 400 instrument at ambient temperature. Chemical shifts (δ) are reported in parts per million (ppm) from low- to high-field and referenced to residual non-deuterated solvent. Standard abbreviations indicating multiplicity are used as follows: s = singlet; d = doublet; t = triplet; m = multiplet. The compounds of 4,4′-(4-bromophenylazanediyl)dibenzaldehyde,23,24 4,4′-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenylazanediyl)dibenzoic acid,23,24 4′-(5-bromo-2-thienyl)-2,2′:6′,2′-terpyridine (P1c),25,26 4′-chloro-2,2′:6′,2′-terpyridine (L2),27 4-(5-bromothiophen-2-yl)-6-(3-(trifluoromethyl)phenyl)-2,2′-bipyridine (P2a),28 Ru(L2)Cl3 (ref. 29) were synthesized as previously reported.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), KMnO4 (2.08 g, 13.2 mmol) was added portion wise at 60 °C. The reaction brought to refluxing temperature for 5 h. The solvent was removed in vacuo and basic water (2.0 M KOH, 50 mL) was added. The suspension was filtered through celite to remove the black precipitate. The filtrate was acidified with conc. HCl to give a white precipitate that was filtered and washed with water and dried under an air stream. The solid was dried further by azeotrope with toluene. The white solid was dissolved in MeOH (100 mL) and conc. HCl (1 mL) was added. The mixture was refluxed overnight. After cooling to room temperature, the solvent was removed in vacuo. The product was purified by recrystallization from hot MeOH to yield 0.977 g (84.4%) of the product as a white solid. 1H NMR (400 MHz, CDCl3): δ = 7.90 (d, 4H, 3J = 8.9 Hz Hk), 7.42 (d, 2H, 3J = 8.8 Hz, Hh), 7.05 (d, 4H, 3J = 8.8 Hz, Hi), 7.00 (d, 2H, 3J = 8.8 Hz, Hi), 3.88 (s, 6H, Hl); 13C NMR (100 MHz, CDCl3): δ = 166.7, 150.8, 145.5, 133.2, 131.4, 127.9, 124.9, 122.9, 118.4, 55.2; HRMS (ESI): m/z = 440.0484 [(M + H)+] (calcd for C22H19BrNO4+: m/z = 440.0492).
:1, v/v), KMnO4 (2.08 g, 13.2 mmol) was added portion wise at 60 °C. The reaction brought to refluxing temperature for 5 h. The solvent was removed in vacuo and basic water (2.0 M KOH, 50 mL) was added. The suspension was filtered through celite to remove the black precipitate. The filtrate was acidified with conc. HCl to give a white precipitate that was filtered and washed with water and dried under an air stream. The solid was dried further by azeotrope with toluene. The white solid was dissolved in MeOH (100 mL) and conc. HCl (1 mL) was added. The mixture was refluxed overnight. After cooling to room temperature, the solvent was removed in vacuo. The product was purified by recrystallization from hot MeOH to yield 0.977 g (84.4%) of the product as a white solid. 1H NMR (400 MHz, CDCl3): δ = 7.90 (d, 4H, 3J = 8.9 Hz Hk), 7.42 (d, 2H, 3J = 8.8 Hz, Hh), 7.05 (d, 4H, 3J = 8.8 Hz, Hi), 7.00 (d, 2H, 3J = 8.8 Hz, Hi), 3.88 (s, 6H, Hl); 13C NMR (100 MHz, CDCl3): δ = 166.7, 150.8, 145.5, 133.2, 131.4, 127.9, 124.9, 122.9, 118.4, 55.2; HRMS (ESI): m/z = 440.0484 [(M + H)+] (calcd for C22H19BrNO4+: m/z = 440.0492).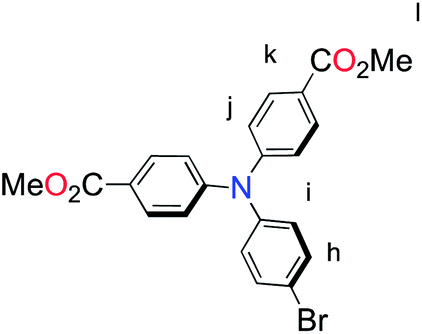
 :1, v/v; 50 mL) solution containing P1c (0.327 g, 0.83 mmol), P1b (0.570 g, 1.00 mmol), K2CO3 (0.881 g, 8.31 mmol) and Pd(PPh3)4 (96 mg, 0.08 mmol) was refluxed for 20 h under N2. The mixture was cooled to room temperature and the solvent removed in vacuo. The residue was purified by column chromatography [SiO2: CH2Cl2–EtOH, 9.5:0.5 v/v Rf = 0.31] to yield 0.196 g (74.3%) of the product as a yellow oil that solidified upon standing. 1H NMR (400 MHz, CDCl3): δ = 8.74 (ddd, 2H, 3J = 4.7, 4J = 2.5, 5J = 0.8 Hz, Ha), 8.69 (s, 2H, He), 8.64 (d, 2H, 3J = 7.9 Hz, Hd), 7.93 (d, 4H, 3J = 6.9 Hz, Hk), 7.86 (ddd, 2H, 3J = 7.7, 7.7, 4J = 1.7 Hz, Hc), 7.75 (d, 1H, 3J = 3.8 Hz, Hf), 7.63 (d, 2H, 3J = 8.7 Hz, Hh), 7.37–7.34 (m, 3H, Hb, Hg), 7.16–7.12 (m, 6H, Hi, Hj), 3.89 (s, 6H, Hl); 13C NMR (100 MHz, CDCl3): δ = 166.8, 157.6, 156.7, 156.3, 150.9, 149.3, 146.0, 145.2, 143.2, 141.1, 139.5, 131.3, 130.8, 129.0, 126.5, 124.1, 122.9, 121.7, 116.5, 113.5, 55.20; HRMS (ESI): m/z = 675.2054 [(M + H)+] (calcd for C41H31N4O4S+: m/z = 675.2061).
:1, v/v; 50 mL) solution containing P1c (0.327 g, 0.83 mmol), P1b (0.570 g, 1.00 mmol), K2CO3 (0.881 g, 8.31 mmol) and Pd(PPh3)4 (96 mg, 0.08 mmol) was refluxed for 20 h under N2. The mixture was cooled to room temperature and the solvent removed in vacuo. The residue was purified by column chromatography [SiO2: CH2Cl2–EtOH, 9.5:0.5 v/v Rf = 0.31] to yield 0.196 g (74.3%) of the product as a yellow oil that solidified upon standing. 1H NMR (400 MHz, CDCl3): δ = 8.74 (ddd, 2H, 3J = 4.7, 4J = 2.5, 5J = 0.8 Hz, Ha), 8.69 (s, 2H, He), 8.64 (d, 2H, 3J = 7.9 Hz, Hd), 7.93 (d, 4H, 3J = 6.9 Hz, Hk), 7.86 (ddd, 2H, 3J = 7.7, 7.7, 4J = 1.7 Hz, Hc), 7.75 (d, 1H, 3J = 3.8 Hz, Hf), 7.63 (d, 2H, 3J = 8.7 Hz, Hh), 7.37–7.34 (m, 3H, Hb, Hg), 7.16–7.12 (m, 6H, Hi, Hj), 3.89 (s, 6H, Hl); 13C NMR (100 MHz, CDCl3): δ = 166.8, 157.6, 156.7, 156.3, 150.9, 149.3, 146.0, 145.2, 143.2, 141.1, 139.5, 131.3, 130.8, 129.0, 126.5, 124.1, 122.9, 121.7, 116.5, 113.5, 55.20; HRMS (ESI): m/z = 675.2054 [(M + H)+] (calcd for C41H31N4O4S+: m/z = 675.2061).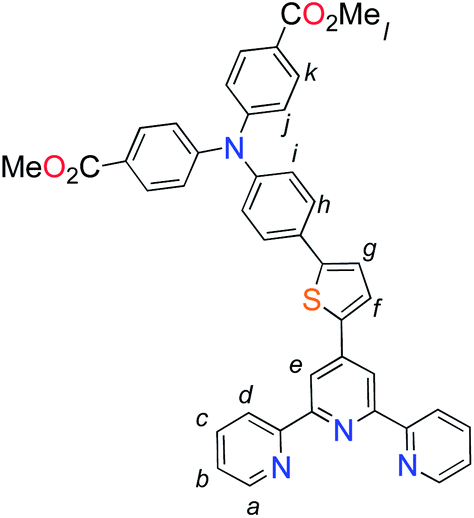 :1:1, v/v/v), Rf = 0.66] to yield 99 mg (58%) of the product as a red solid. 1H NMR (400 MHz, CDCl3): δ = 9.21 (s, 2H, HE), 8.99 (s, 2H, He), 8.88 (d, 2H, 3J = 8.1 Hz, HD), 8.63 (d, 2H, 3J = 8.1 Hz, Hd), 8.50 (d, 1H, 3J = 4.0 Hz, Hf), 7.98–7.93 (m, 8H, HC, Hk, Hc), 7.83 (d, 2H, 3J = 8.6 Hz, Hh), 7.69 (d, 1H, 3J = 3.8 Hz, Hg), 7.49 (d, 2H, 3J = 4.7 Hz, HA), 7.38 (d, 2H, 3J = 4.7 Hz, HB), 7.28 (d, 2H, 3J = 8.6 Hz, Hi), 7.22–7.16 (m, 8H, Hj, Ha, Hb), 3.87 (s, 6H, Hl); HRMS (MALDI-TOF): m/z = 1043.1584 [(M)+] (calcd for C56H40ClN7O4RuS+: m/z = 1043.1600). Anal. calcd for C56H40ClN9O10RuS·3H2O: C, 55.06; H, 3.80; N, 10.32. Found: C, 55.35; H, 3.68; N, 9.97%.:1, v/v, 30 mL) with KOH (79 mg, 1.3 mmol) and refluxed for overnight. The progress of the reaction was monitored by TLC. After the reaction was complete, the solvent was removed in vacuo. The residue was dissolved in 5 mL H2O and acidified with 0.1 M HCl. The precipitate was collected by filtration and washed with H2O and Et2O to yield 52 mg (60%) of the product as a red solid. 1H NMR (400 MHz, MeOD + 1 drop NaOD): δ = 9.12 (s, 2H, HE), 8.82 (d, 2H, 3J = 8.1 Hz, HD), 8.37 (d, 2H, 3J = 8.0 Hz, Hd), 8.23 (d, 1H, 3J = 3.9 Hz, Hf), 7.99 (dt, 2H, 3J = 7.9, 4J = 1.4 Hz, HC), 7.92 (d, 4H, 3J = 8.7 Hz, Hk), 7.85 (dt, 2H, 3J = 7.9 Hz, 4J = 1.4 Hz, Hc), 7.81 (s, 1H, He), 7.78 (d, 2H, 3J = 8.7 Hz, Hh), 7.67 (d, 2H, 3J = 4.9 Hz, HA), 7.61 (d, 1H, 3J = 3.9 Hz, Hg), 7.37–7.32 (m, 4H, HB, Hb), 7.19 (d, 2H, 3J = 8.7 Hz, Hi), 7.11–7.06 (m, 6H, Hj, Ha); HRMS (MALDI-TOF): m/z = 996.1511 [(M)+] (calcd for C54H36N7O5RuS+: m/z = 996.1551.). Anal. calcd for C41H30Cl3N4O4RuS·2H2O: C, 58.75; H, 3.74; N, 8.88. Found: C, 59.12; H, 3.85; N, 8.97%.
:1:1, v/v/v), Rf = 0.66] to yield 99 mg (58%) of the product as a red solid. 1H NMR (400 MHz, CDCl3): δ = 9.21 (s, 2H, HE), 8.99 (s, 2H, He), 8.88 (d, 2H, 3J = 8.1 Hz, HD), 8.63 (d, 2H, 3J = 8.1 Hz, Hd), 8.50 (d, 1H, 3J = 4.0 Hz, Hf), 7.98–7.93 (m, 8H, HC, Hk, Hc), 7.83 (d, 2H, 3J = 8.6 Hz, Hh), 7.69 (d, 1H, 3J = 3.8 Hz, Hg), 7.49 (d, 2H, 3J = 4.7 Hz, HA), 7.38 (d, 2H, 3J = 4.7 Hz, HB), 7.28 (d, 2H, 3J = 8.6 Hz, Hi), 7.22–7.16 (m, 8H, Hj, Ha, Hb), 3.87 (s, 6H, Hl); HRMS (MALDI-TOF): m/z = 1043.1584 [(M)+] (calcd for C56H40ClN7O4RuS+: m/z = 1043.1600). Anal. calcd for C56H40ClN9O10RuS·3H2O: C, 55.06; H, 3.80; N, 10.32. Found: C, 55.35; H, 3.68; N, 9.97%.:1, v/v, 30 mL) with KOH (79 mg, 1.3 mmol) and refluxed for overnight. The progress of the reaction was monitored by TLC. After the reaction was complete, the solvent was removed in vacuo. The residue was dissolved in 5 mL H2O and acidified with 0.1 M HCl. The precipitate was collected by filtration and washed with H2O and Et2O to yield 52 mg (60%) of the product as a red solid. 1H NMR (400 MHz, MeOD + 1 drop NaOD): δ = 9.12 (s, 2H, HE), 8.82 (d, 2H, 3J = 8.1 Hz, HD), 8.37 (d, 2H, 3J = 8.0 Hz, Hd), 8.23 (d, 1H, 3J = 3.9 Hz, Hf), 7.99 (dt, 2H, 3J = 7.9, 4J = 1.4 Hz, HC), 7.92 (d, 4H, 3J = 8.7 Hz, Hk), 7.85 (dt, 2H, 3J = 7.9 Hz, 4J = 1.4 Hz, Hc), 7.81 (s, 1H, He), 7.78 (d, 2H, 3J = 8.7 Hz, Hh), 7.67 (d, 2H, 3J = 4.9 Hz, HA), 7.61 (d, 1H, 3J = 3.9 Hz, Hg), 7.37–7.32 (m, 4H, HB, Hb), 7.19 (d, 2H, 3J = 8.7 Hz, Hi), 7.11–7.06 (m, 6H, Hj, Ha); HRMS (MALDI-TOF): m/z = 996.1511 [(M)+] (calcd for C54H36N7O5RuS+: m/z = 996.1551.). Anal. calcd for C41H30Cl3N4O4RuS·2H2O: C, 58.75; H, 3.74; N, 8.88. Found: C, 59.12; H, 3.85; N, 8.97%.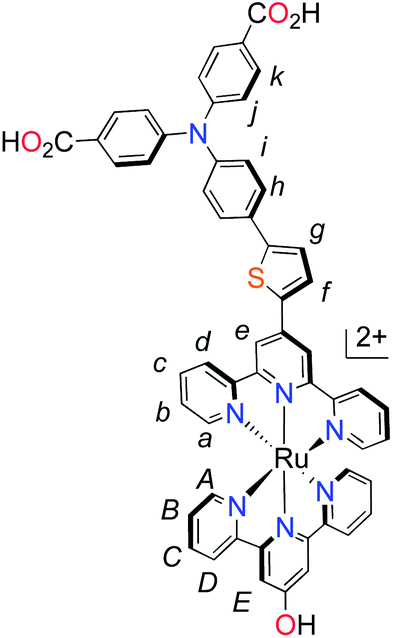 :1), Rf = 0.46] to yield 0.19 g (43%) of the product as a white solid. The product was used immediately used without further purification. 1H NMR (400 MHz, CDCl3): δ = 7.93 (d, 4H, 3J = 8.8 Hz, Hk), 7.76 (d, 2H, 3J = 8.5 Hz, Hh), 7.11–7.08 (m, 6H, Hj, Hi), 1.22 (s, 12H, Hr); HRMS (MALDI-TOF): m/z = 459.1848 [(M)+] (calcd for C26H26BNO6+: m/z = 458.1884).
:1), Rf = 0.46] to yield 0.19 g (43%) of the product as a white solid. The product was used immediately used without further purification. 1H NMR (400 MHz, CDCl3): δ = 7.93 (d, 4H, 3J = 8.8 Hz, Hk), 7.76 (d, 2H, 3J = 8.5 Hz, Hh), 7.11–7.08 (m, 6H, Hj, Hi), 1.22 (s, 12H, Hr); HRMS (MALDI-TOF): m/z = 459.1848 [(M)+] (calcd for C26H26BNO6+: m/z = 458.1884).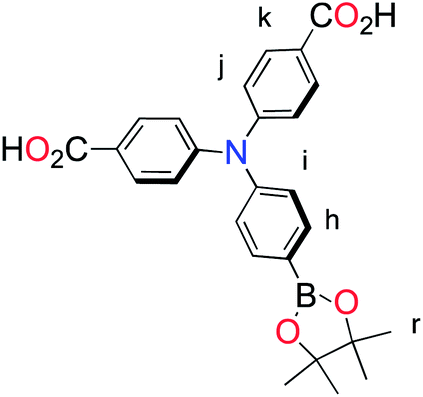 :1, v/v, 52 mL) with n-ethylmorpholine (5 drops). The solution was left to reflux for 16 h. AgNO3 (36 mg, 21 mmol) was added and the solution refluxed for an additional 2 h. The solvent was removed in vacuo and the residue purified by column chromatography [SiO2: CH2Cl2–MeOH, (9:1, v/v), Rf = 0.31] to yield 0.111 g (59%) as a black powder. 1H NMR (400 MHz, CD3OD): δ = 8.90 (s, 2H, HE), 8.77–8.76 (m, 2H, Hd, Hm), 8.60–8.59 (m, 3H, HD, He), 8.22 (s, 1H, Hn), 7.97–7.93 (m, 2H, Hf, Hc), 7.87 (dd, 2H, 3J = 8.2 Hz, 3J = 8.2 Hz, 3J = 8.1 Hz, HC), 7.55 (d, 1H, 3J = 4.7 Hz, Ha), 7.49 (d, 2H, 3J = 5.2 Hz, HA), 7.38 (d, 1H, 3J = 4.0 Hz, Hg), 7.19–7.13 (m, 3H, HB, Hb), 6.76 (d, 1H, 3J = 7.9 Hz, Hp), 5.95 (d, 1H, 3J = 7.8 Hz, Hq). HRMS (ESI): m/z = 829.9353 [(M)+] (calcd for C36H21BrClF3N5O2RuS+: m/z = 829.9372). Anal. calcd for C36H21BrClF3N6O3RuS·2H2O: C, 46.64; H, 2.72; N, 9.06. Found: C, 46.83; H, 2.47; N, 9.18%.
:1, v/v, 52 mL) with n-ethylmorpholine (5 drops). The solution was left to reflux for 16 h. AgNO3 (36 mg, 21 mmol) was added and the solution refluxed for an additional 2 h. The solvent was removed in vacuo and the residue purified by column chromatography [SiO2: CH2Cl2–MeOH, (9:1, v/v), Rf = 0.31] to yield 0.111 g (59%) as a black powder. 1H NMR (400 MHz, CD3OD): δ = 8.90 (s, 2H, HE), 8.77–8.76 (m, 2H, Hd, Hm), 8.60–8.59 (m, 3H, HD, He), 8.22 (s, 1H, Hn), 7.97–7.93 (m, 2H, Hf, Hc), 7.87 (dd, 2H, 3J = 8.2 Hz, 3J = 8.2 Hz, 3J = 8.1 Hz, HC), 7.55 (d, 1H, 3J = 4.7 Hz, Ha), 7.49 (d, 2H, 3J = 5.2 Hz, HA), 7.38 (d, 1H, 3J = 4.0 Hz, Hg), 7.19–7.13 (m, 3H, HB, Hb), 6.76 (d, 1H, 3J = 7.9 Hz, Hp), 5.95 (d, 1H, 3J = 7.8 Hz, Hq). HRMS (ESI): m/z = 829.9353 [(M)+] (calcd for C36H21BrClF3N5O2RuS+: m/z = 829.9372). Anal. calcd for C36H21BrClF3N6O3RuS·2H2O: C, 46.64; H, 2.72; N, 9.06. Found: C, 46.83; H, 2.47; N, 9.18%.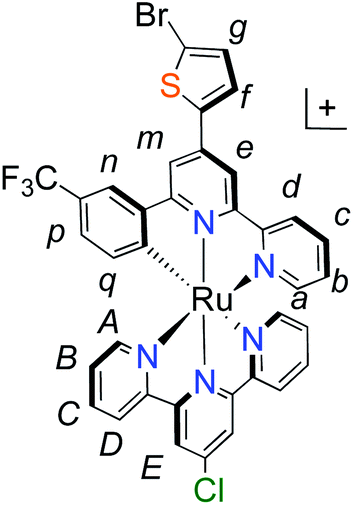 :2 acetone–MeOH, Rf = 0.15] to yield 0.045 g (29.5%) of the product as a dark black solid. 1H NMR (400 MHz, MeOD): δ = 8.90 (s, 2H, HE), 8.83 (s, 1H, He), 8.78 (d, 1H, 3J = 7.6 Hz, Ha), 8.67 (s, 1H, Hd), 8.60 (d, 2H, 3J = 7.6 Hz, HD), 8.22 (s, 1H, Hm), 8.15 (d, 1H, 3J = 3.9 Hz, Hf), 7.97–7.90 (m, 5H, Hc, Hk), 7.82 (t, 2H, 3J = 7.9 Hz, HB), 7.75 (d, 2H, 3J = 8.6 Hz, Hh), 7.58 (d, 1H, 3J = 3.9 Hz, Hg), 7.54–7.53 (m, 3H, Hn, HA), 7.18–7.15 (m, 5H, Hi, Hb, HC), 7.08 (d, 4H, 3J = 8.7 Hz, Hj), 6.75 (d, 1H, 3J = 7.9 Hz, Hp), 5.93 (d, 1H, 3J = 8.0 Hz, Hq). HRMS (ESI): m/z = 1081.11283 [(M)+] (calcd for C56H35ClF3N6O4RuS+: m/z = 1081.11304). Anal. calcd for C56H35ClF9N6O4PRuS·H2O: C, 54.05; H, 3.00; N, 6.75. Found: C, 54.28; H, 2.94; N, 6.60%.
:2 acetone–MeOH, Rf = 0.15] to yield 0.045 g (29.5%) of the product as a dark black solid. 1H NMR (400 MHz, MeOD): δ = 8.90 (s, 2H, HE), 8.83 (s, 1H, He), 8.78 (d, 1H, 3J = 7.6 Hz, Ha), 8.67 (s, 1H, Hd), 8.60 (d, 2H, 3J = 7.6 Hz, HD), 8.22 (s, 1H, Hm), 8.15 (d, 1H, 3J = 3.9 Hz, Hf), 7.97–7.90 (m, 5H, Hc, Hk), 7.82 (t, 2H, 3J = 7.9 Hz, HB), 7.75 (d, 2H, 3J = 8.6 Hz, Hh), 7.58 (d, 1H, 3J = 3.9 Hz, Hg), 7.54–7.53 (m, 3H, Hn, HA), 7.18–7.15 (m, 5H, Hi, Hb, HC), 7.08 (d, 4H, 3J = 8.7 Hz, Hj), 6.75 (d, 1H, 3J = 7.9 Hz, Hp), 5.93 (d, 1H, 3J = 8.0 Hz, Hq). HRMS (ESI): m/z = 1081.11283 [(M)+] (calcd for C56H35ClF3N6O4RuS+: m/z = 1081.11304). Anal. calcd for C56H35ClF9N6O4PRuS·H2O: C, 54.05; H, 3.00; N, 6.75. Found: C, 54.28; H, 2.94; N, 6.60%.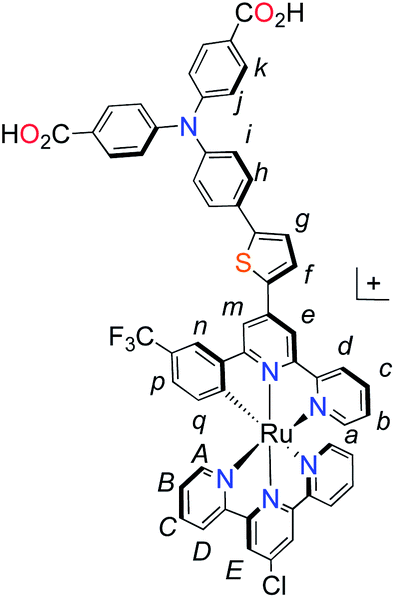
Physical methods
Elemental analysis (EA), electrospray ionization (ESI) and matrix assisted laser desorption ionization time of flight (MALDI-TOF) mass spectrometry (MS) data were collected at the Chemistry Instrumentation Facility of the University of Calgary. Electrochemical measurements were performed under anaerobic conditions with a Princeton Applied Research VersaStat 3 potentiostat using dry solvents, Pt working and counter electrodes, Ag pseudoreference electrode, and 0.1 M NBu4BF4 DMF solutions. Electronic spectroscopic data were collected on CH3OH solutions using a Cary 5000 UV-visible spectrophotometer (Varian).Samples for infrared absorption spectroscopy were prepared as either a KBr pressed disk (dye only), by adsorbing the dye on a mesoporous NiO film deposited on a CaF2 window (Crystran). The NiO films were prepared by spraying a saturated solution of NiCl2 in acetylacetone onto the surface of the CaF2 window which was pre-heated to 450 °C on a hotplate; this was then allowed to cool slowly to room temperature to give a compact film of NiO. The mesoporous layer was then deposited on top of the compact layer using an F108-templated precursor solution containing NiCl2 (1 g), Pluronic® co-polymer F108 (1 g), Milli-Q water (3 g) and ethanol (6 g) and a doctor blade was used to remove the excess. The film was sintered at 450 °C for 30 minutes and an additional layer of precursor solution was applied and sintered to increase the film thickness. Infrared spectra were recorded on a Thermo Nicolet 380 FTIR spectrometer for 16 scans at a resolution of 2 cm−1.
NiO electrodes were made by applying the precursor solution described in previous literature by doctor blade onto conducting glass substrates (Pilkington TEC15, sheet resistance 15 Ω per square) using Scotch tape as a spacer (0.2 cm2 active area), followed by sintering in a Nabertherm B150 oven at 450 °C for 30 min. The NiO electrodes were soaked in a dichloromethane solution of the dye (0.3 mM) for 16 h at room temperature.30 The dyed NiO electrode was assembled face-to-face with a platinized counter electrode (Pilkington TEC8, sheet resistance 8 Ω per square) using a 30 μm thick thermoplastic frame (Surlyn 1702). The electrolyte, containing LiI (1.0 M) and I2 (0.1 M) in acetonitrile, was introduced through the pre-drilled hole in the counter electrode, which was sealed afterwards. The cells were measured 12 hours after assembly. The UV-visible absorption spectra of the dyes adsorbed on NiO films were recorded using an Ocean Optics USB2000+VIS-NIR fibre-optic spectrophotometer. Incident photon-to-current conversion efficiencies were recorded using monochromatic light from a system consisting of a Xenon lamp, a monochromator and appropriate filters and calibrated against a certified reference Si photodiode. Current–voltage measurements were measured using an Ivium CompactStat potentiostat under simulated sunlight from an Oriel 150 W solar simulator, giving light with an intensity of 100 mW cm−2.
Computational details
Geometries of both were optimised in the gas phase by DFT using the B3LYP hybrid functional (20% Hartree–Fock)31 as implemented in the NWChem 6.1 software package.32 Stuttgart–Dresden relativistic small core potential basis set was used for Ru (ref. 33) and 6-311G* basis sets were used for all other atoms.34 Single-point calculations using the same basis sets and functional were carried out using the COSMO solvation model35 for methanol (ε = 32.7, Rsolv = 1.3 Å). The energies and plots of HOMO−10 up to LUMO+10 were calculated. Molecular orbital plots were visualised with the ECCE (Pacific Northwest National Laboratory) software package.36 Time-dependent DFT (TD-DFT) calculations were carried out using the same B3LYP functional, basis sets and COSMO solvation model for methanol on the optimised geometries to calculate electronic absorption spectra. The 50 lowest energy spin-allowed transitions were calculated for each dye. No triplet excited state transitions were calculated.Results
Dye synthesis
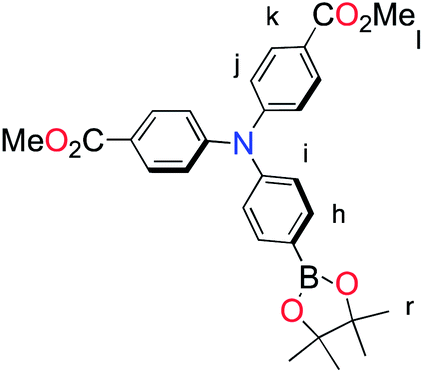
The methyl ester TPA precursor (P1a) was synthesized by a modification of a literature procedure23,24 and subsequently converted to the boronic ester (P1b) using a palladium-catalyzed Miyaura reaction of P1a with bis(pinacolato)diboron (Fig. 2).37 Under Suzuki cross-coupling conditions P1b was reacted with P1c to yield L1 in moderate yields.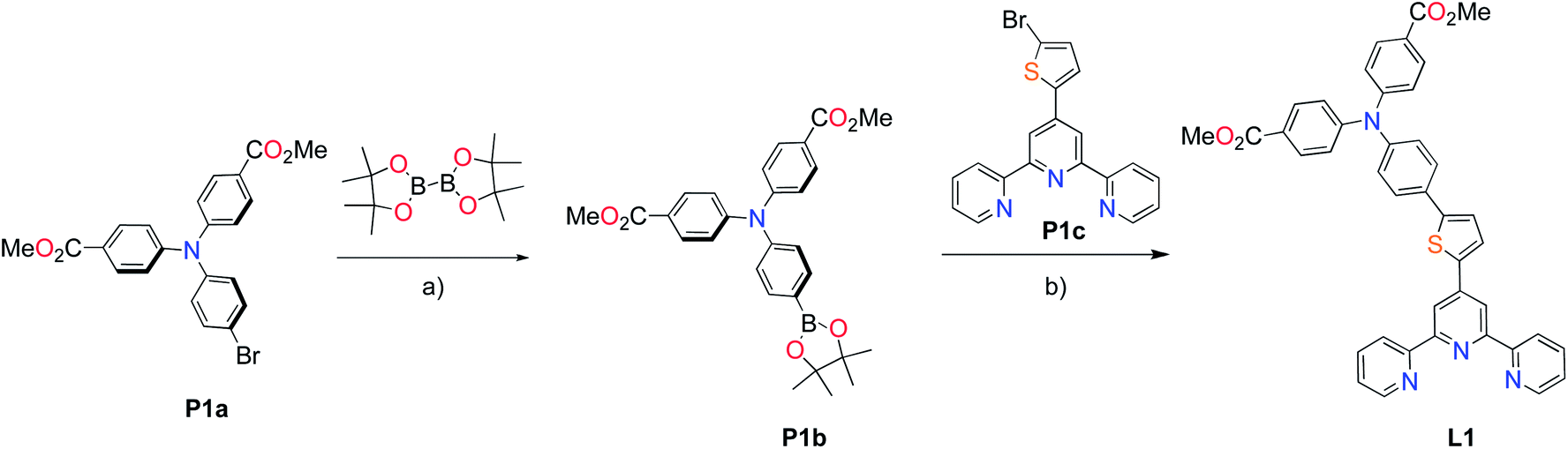 | ||
| Fig. 2 Synthesis of L1. Reaction conditions: (a) Pd(dppf)Cl2, KOAc, DMSO, 80 °C, 6 h, N2; (b) Pd(PPh3)4, K2CO3, THF–H2O (9:1, v/v), reflux, 20 h, N2. | ||
With the ligand L1 in hand, the synthesis of K1 was straight-forward using established synthetic routes involving the subsequent coordination of L1 and L2 to the ruthenium metal center (Fig. 3).28,38,39 An unexpected reaction occurred upon saponification of the methyl ester units to carboxylic acids: chloride substitution by the hydroxide species in solution was observed, most likely occurring via a nucleophilic aromatic substitution reaction.
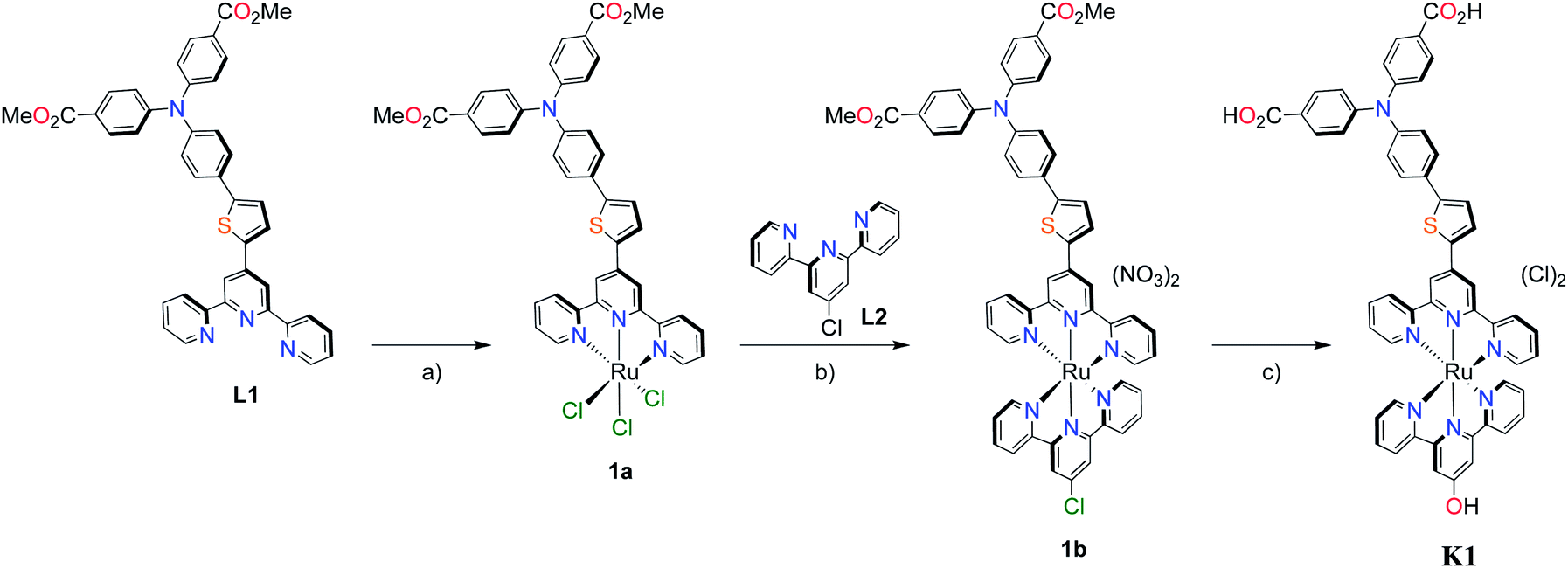 | ||
| Fig. 3 Synthesis of polypyridyl p-type sensitizer (K1). Reaction conditions: (a) RuCl3·3H2O, abs. EtOH, reflux, 4 h; (b) (i) EtOH, n-ethylmorpholine, reflux, 20 h. (ii) AgNO3, reflux, 2 h; (c) KOH, THF–H2O (5:1, v/v). | ||
Unlike the polypyridyl p-type sensitizer (K1), the synthesis of the cyclometalated sensitizer K2 was achieved following a modification of the synthesis previously reported (Fig. 4).40 Cycloruthenation of the precursor Ru(L2)Cl3 with P2a yielded the intermediary compound 2b in moderated yields. The TPA moiety was installed via cross-coupling conditions using P2b to yield 2 after acidic workup.
 | ||
| Fig. 4 Synthesis of cyclometalated p-type sensitizer (K2). Reaction conditions: (a) MeOH–H2O (5:1, v/v), n-ethylmorpholine, reflux, 16 h; (b) Pd(PPh3)4, K2CO3, DMF, 70 °C, 14 h, N2. | ||
Optical and electrochemical characterization
The UV-visible absorption spectra of the dyes, K1 and K2 in methanol solution and adsorbed on NiO films are shown in Fig. 5. The electronic distributions in the frontier orbitals were calculated for K1 and K2 in methanol using DFT and the TDDFT results were compared to the electronic spectra (Fig. 6 and 7). In both complexes the electronic distribution of the HOMO is predicted to be localized over the triphenylamine and thiophene donor. The predominant compositions to the principle electronic transitions with their wavelengths and calculated oscillator strengths (f) are provided in Tables S4 and S5.† Both dyes exhibit one prominent band, appearing at 523 ± 1 nm (ε = 4.73 and 3.34 × 104 L mol−1 cm−1 for K1 and K2 respectively). Results from TD-DFT calculations suggest that this transition may be assigned to a favourable intraligand charge-transfer (ILCT) transition arising principally from the triphenylamine- and thiophene-based HOMO to the terpyridyl-based LUMO in K1 or 3-(trifluoromethyl)phenyl-2,2′-bipyridine-based LUMO in K2.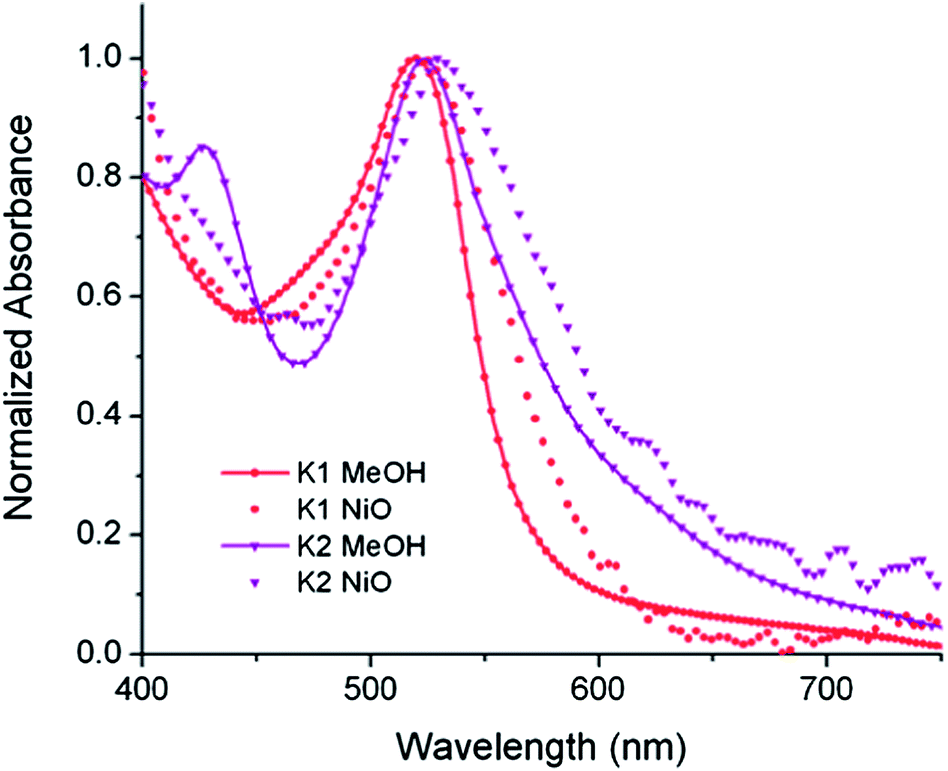 | ||
| Fig. 5 The absorption spectra of K1 (red) and K2 (purple) in MeOH solution (A) and adsorbed onto NiO films on CaF windows (B). | ||
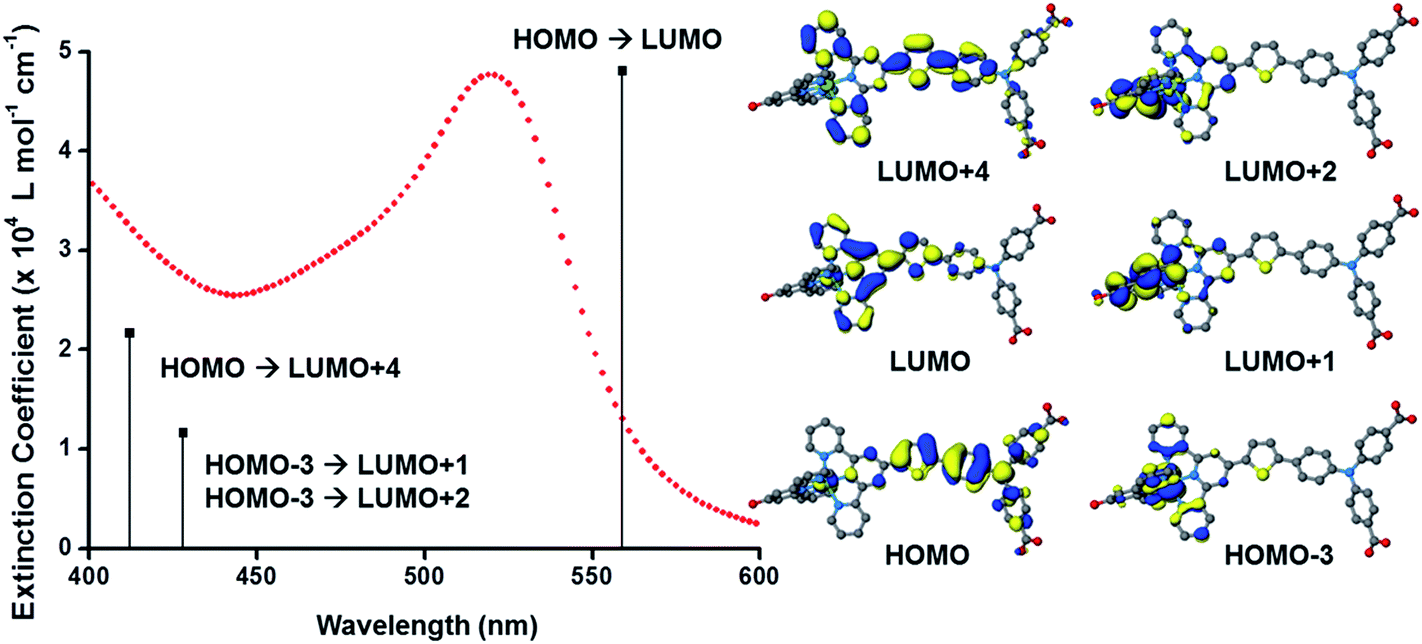 | ||
| Fig. 6 (Left) Plot of the ground state electronic absorption spectrum of K1 in methanol with calculated electronic transitions and their relative intensities; (right) optimised geometries and frontier orbitals of K1 in methanol performed using the B3LYP hybrid functional (20% Hartree–Fock) using the COSMO solvation model. | ||
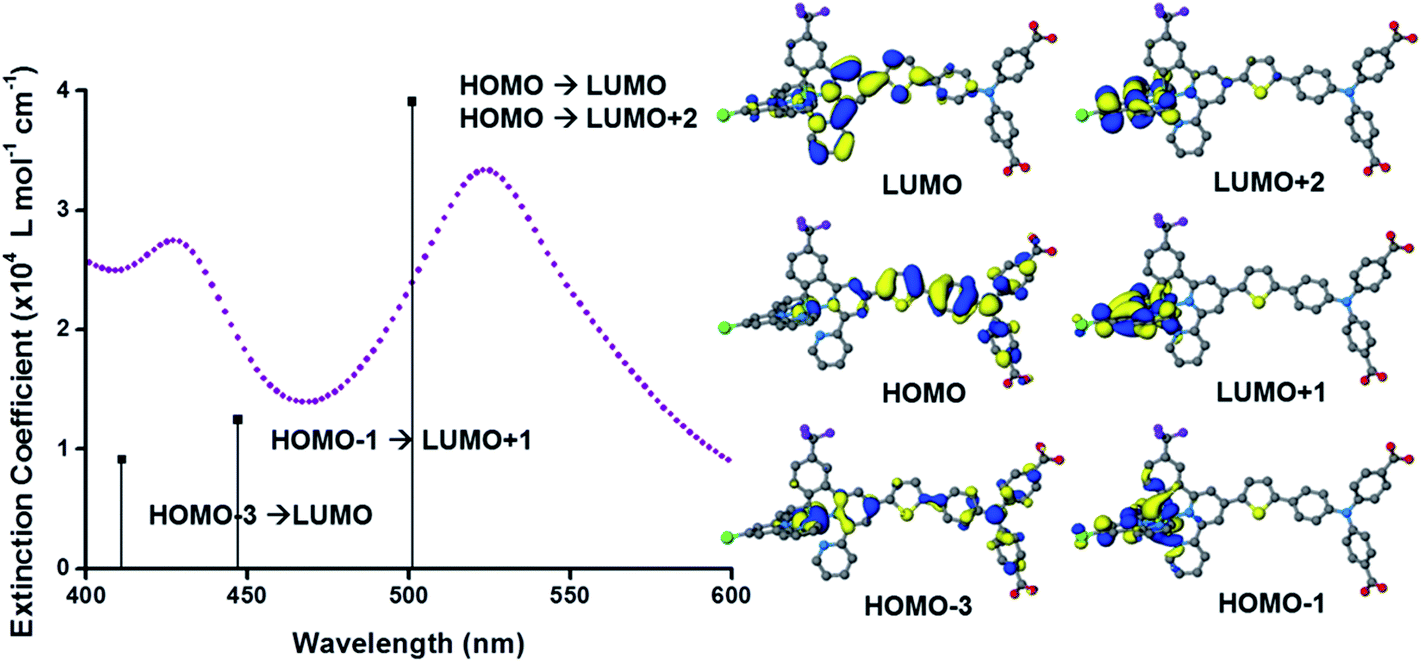 | ||
| Fig. 7 (Left) Plot of the ground state electronic absorption spectrum of K2 in methanol with calculated electronic transitions and their relative intensities; (right) optimised geometries and frontier orbitals of K2 in methanol performed using the B3LYP hybrid functional (20% Hartree–Fock) using the COSMO solvation model. | ||
In addition to the band at 523 nm the spectrum for K1 contains a shoulder at ca. 470 nm, which we have assigned to the MLCT transition involving the HOMO−3 and the due to the lower intensity and higher energy required for excitation. This would be more in agreement with 4′-chloro-2,2′:6′,2′′-terpyridine ruthenium(II) (λmax = 480 nm, ε = 16000 L mol−1 cm−1) and 4′-hydroxy-2,2′:6′,2′-terpyridine ruthenium(II) (λmax = 485 nm ε = 12700 L mol−1 cm−1).41
The absorption spectrum of K2 in solution is even more broadened compared to K1 suggesting a number of overlapping ILCT and MLCT transitions contribute to the spectrum. The large extinction coefficient of the predominant lower energy band for K2 in solution is similar to the intraligand transition at 505 nm (ε = 42000) for the analogous complex “3” in ref. 38 in which the electron withdrawing –CO2H groups are absent. In the reported complex a MLCT transition was assigned to a shoulder at 440 nm (ε = 19400 L mol−1 cm−1). According to our TD-DFT calculations, the lowest energy transition is due to the promotion of an electron from HOMO−2, where the electron density is distributed over the anionic portion of the ligand and the ruthenium centre, to the LUMO+1 where the electron density is predominantly localized on the terpyridine acceptor ligand. The predominant band at 524 nm is a combination of the intraligand charge transfer HOMO → LUMO and the promotion of the electron from the HOMO to the π* orbitals on the terpyridine (LUMO+2). The higher energy band is likely to be due to another intraligand charge transfer (HOMO → LUMO+3). Therefore the predominant electronic transitions contributing to the visible absorption spectrum involve a transfer of electron density away from the donor ligand and therefore the NiO surface when adsorbed through the carboxylate groups which is favorable for sensitization of NiO. The FTIR spectra for the anchored dyes is given in Fig. S3.† Adsorption on NiO did not markedly affect the visible absorption spectrum of the dyes indicating that the electronic structure of the chromophore is unaffected by binding.
Table 1 summarises the optical and electrochemical characterisation data for K1 and K2. The redox potential of L1 here is 1.34 V vs. NHE, 100 mV more positive than the “L3” ligand in ref. 38 in which the electron withdrawing –CO2H groups are absent. The corresponding bis-terpyridyl ruthenium complex had a similar redox potential and so for complex K1 we are confident that the first oxidation at 1.38 V vs. NHE is triphenylamine centred as in agreement with the DFT calculations. We were unable to resolve the Ru(III/II) couple for K1 but we expect it to lie between the redox potential of bis-4′-hydroxy-2,2′:6′,2′-terpyridyl ruthenium (1.36 V vs. NHE) and bis-4′-(p-tolyl)-2,2′:6′,2′-terpyridyl ruthenium (1.5 V vs. NHE).41 For K2 we observed again an oxidation process likely to be oxidation of triphenylamine at 1.40 V vs. NHE in agreement with the DFT calculations which place the HOMO in K2 only 3 eV lower in energy than the HOMO in K1. The first oxidation process, however, was found to be at 0.93 V vs. NHE which is closer to that of the cyclometalated complexes reported by Ji et al. and so we have assigned this to the Ru(III/II) couple.20 The appearance of the Ru(III/II) couple was observed for the analogous complex “4” in ref. 39 which differed from K2 by the substituents on the triphenylamine (electron donating –OMe rather than electron withdrawing –CO2H) and an electron withdrawing trimethyl-4,4′,4′′-tricarboxylate-2,2′:6′,2′-terpyridine ligand. For the previously reported complex the oxidation of the triphenylamine moiety at 1.19 V vs. NHE was accompanied by a reversible Ru(III/II) couple at 1.27 V vs. NHE. Therefore, analogous to similar dyes used for n-type DSCs and in agreement with our DFT studies we have increased the electron density of the dye at the ruthenium centre for K2 compared to the K1. The dramatic shift in the first electrode potential demonstrates that the negatively charged σ-donor ligand stabilises the Ru(III) and therefore ought to stabilise the MLCT compared to the terpyridyl ligand.
| Dye | Absorption | E0–0c (eV) | E’d (V vs. NHE) | ED/D−e (V vs. NHE) | |||
|---|---|---|---|---|---|---|---|
| λmaxa (nm) | ε at λmaxa (×104 L mol−1 cm−1) | λmax on NiOb (nm) | TPA+/0 | RuIII/II | |||
| a Absorption spectra were measured in MeOH at room temperature.b Absorption spectra of sensitized NiO films.c E0–0 was estimated from 10% onset point of absorption spectra in MeOH.d The redox potential of the dyes were measured in DMF with 0.1 M tetrabutylammonium tetrafluoroborate (TBABF4) as a supporting electrolyte (working electrode: glassy carbon; reference electrode: Ag/Ag+ calibrated with ferrocene/ferrocenium (Fc+/0) as an internal reference and converted to NHE by addition of 690 mV, counter electrode: Pt).e First reduction potential of the dyes were estimated from E(TPA+/0) plus E0–0. | |||||||
| K1 | 523 | 4.73 | 525 | 2.1 | 1.38 | ca. 1.5 | −0.73 |
| K2 | 524 | 3.34 | 531 | 1.9 | 1.40 | 0.93 | −0.50 |
The dyes were found to be only weakly emissive and so the E0–0 energies were estimated from the onset of the wavelength at which the absorption was 10% of the maximum. The values for the electrode potential for the TPA+/0 couple is much more positive than the valence band of NiO so there should be a sufficient driving force for hole injection from the dye to the semiconductor. The reduction potential of these dyes adsorbed on NiO are in the range of −0.5 V to −0.75 V vs. NHE which suggests that regeneration of the dyes by I3−/I− is strongly favourable.42
p-Type dye-sensitized solar cells
The photovoltaic performance of solar cells based on these dyes is summarized in Table 2. Sandwich-type solar cells were assembled using K1 and K2 sensitized nanocrystalline NiO as the working electrodes, platinized conducting glass as the counter electrode and 1.0 M LiI, 0.1 M I2 in acetonitrile as the electrolyte. DSCs based on K1 produced a maximum incident photon to current conversion efficiency (IPCE) of 14%, whereas under the same conditions, the devices based on K2 gave a similar maximum IPCE of 9%. This followed the trend in photocurrent measured using simulated sunlight shown in Fig. 8. DSCs using K1 and K2 utilising a cobalt based redox couple (0.1 M Co(dtb)3(ClO4)3(2)) gave a negligible photocurrent (see Fig. S9†). This suggests that, despite our efforts the rate of charge recombination between the reduced dye and the holes in the NiO may still be too fast for the charge on the dye to be intercepted by the cobalt(III) (τ = 1–10 μs).8,43 Nonetheless the results with I3−/I− were significantly better than that typically obtained from ruthenium dyes reported elsewhere14,17,19 (<1 mA cm−2) and the current obtained for K2 was comparable to the cyclometalated ruthenium dye reported by Ji et al.20| Dye | JSC (mA cm−2) | VOC (mV) | FF | η (%) | IPCEmax (%) |
|---|---|---|---|---|---|
| a JSC is the short-circuit current density at the V = 0 intercept; VOC is the open-circuit voltage at the J = 0 intercept; FF is the device fill factor; η is the power conversion efficiency; IPCEmax is the maximum monochromatic incident photon-to-current conversion efficiency. | |||||
| K1 | 2.91 | 96 | 0.32 | 0.09 | 14 |
| K2 | 1.96 | 93 | 0.39 | 0.07 | 9 |
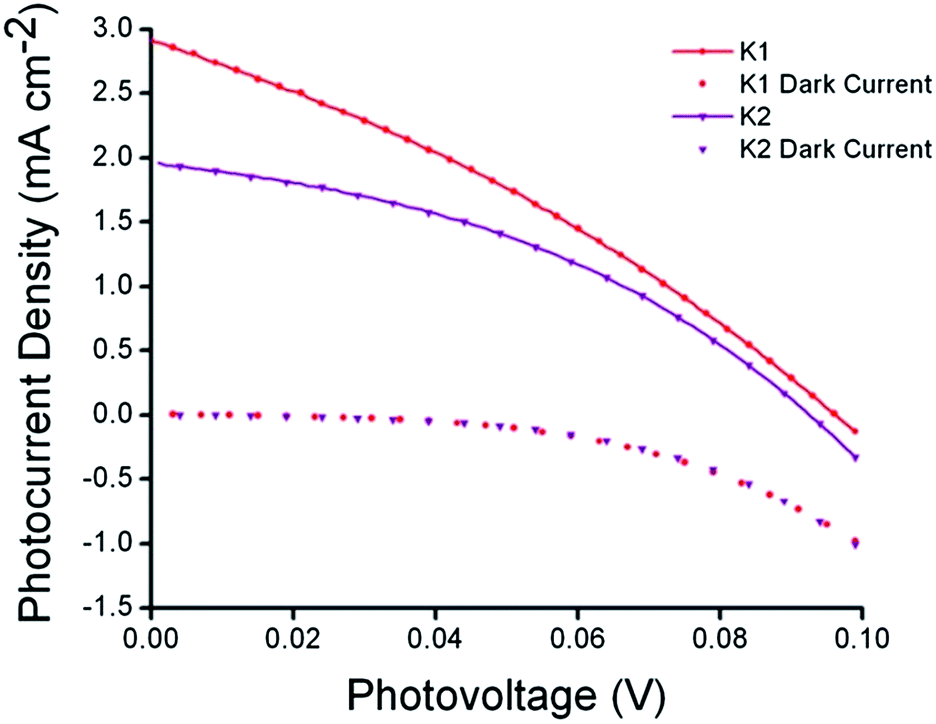 | ||
| Fig. 8 Current–voltage characteristics (right) of K1 (red) and K2 (purple) sensitized NiO solar cells under AM 1.5G (100 mW cm−2) illumination. | ||
Conclusions
We have developed and tested a series of new ruthenium-based sensitizers for NiO based p-DSCs. This work demonstrates that our dye-design is superior to applying “classic” Ru(bpy)32+ derivatives used for TiO2-sensitization in p-DSCs. The complex ligand system dominates the optoelectronic properties of the dye, greatly increasing the extinction coefficients. Whilst the high extinction coefficients are necessary for efficient light collection from NiO photocathodes the efficiency is still limited by recombination reactions preventing the use of alternative cobalt-based redox shuttles. We envisage that better use of triplet states of heavy transition metals could lead to longer lived charge separation that is required for p-DSCs to be as efficient as their TiO2 equivalents, leading to efficient tandem devices that have a theoretical maximum efficiency that surpasses those of their single photo-electrode components and further work towards achieving this goal is ongoing.Acknowledgements
EAG thanks the Royal Society for a Dorothy Hodgkin Research Fellowship. As a member of the UK HPC Materials Chemistry Consortium PIPE acknowledges the EPSRC (grant no. EP/F067496) and the UK's national supercomputing service HECToR as well as the Huddersfield Centre for High Performance Computing for computational resources utilised in this work. The Canadian authors thank National Science & Engineering Research Council (Canada), Canadian Research Chairs and the Alfred P. Sloan Foundation for support.References
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 6595–6663 CrossRef CAS PubMed
.
- A. Hagfeldt, U. B. Cappel, G. Boschloo, L. Sun, L. Kloo, H. Petterson and E. A. Gibson, Dye-Sensitized Photoelectrochemical Cells, in Practical Handbook of Photovoltaics Fundamentals and Applications, ed. A. McEvoy, T. Markvart and L. Castaner, Academic Press/Elsevier Ltd., Oxford, 2012, pp. 479–542 Search PubMed
- M. Wang, P. Chen, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, ChemPhysChem, 2009, 10, 290–299 CrossRef CAS PubMed
- L. Vallejo, A. William, S. Quiñones, A. Cesar and S. J. A. Hernandez, The Chemistry and Physics of Dye-Sensitized Solar Cells, in Solar Cells – Dye-Sensitized Devices, ed. L. A. Kosyachenko, Tech Europe, 2011, pp. 399–418 Search PubMed
- S. Wenger, S. Seyrling, A. N. Tiwari and M. Grätzel, Appl. Phys. Lett., 2009, 94, 173508 CrossRef PubMed
- J. He, H. Lindström, A. Hagfeldt and S. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265–273 CrossRef CAS
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y.-B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef CAS PubMed
- E. A. Gibson, A. L. Smeigh, L. Le Pleux, J. Fortage, G. Boschloo, E. Blart, Y. Pellegrin, F. Odobel, A. Hagfeldt and L. Hammarström, Angew. Chem., Int. Ed. Engl., 2009, 48, 4402–4405 CrossRef CAS PubMed
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414–2423 CrossRef CAS PubMed
- P. Qin, J. Wiberg, E. A. Gibson, M. Linder, L. Li, T. Brinck, A. Hagfeldt, B. Albinsson and L. Sun, J. Phys. Chem. C, 2010, 114, 4738–4748 CAS
- A. Morandeira, G. Boschloo, A. Hagfeldt and L. Hammarström, J. Phys. Chem. B, 2005, 109, 19403–19410 CrossRef CAS PubMed
- A. L. Smeigh, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarström, Chem. Commun., 2012, 48, 678–680 RSC
- M. Borgström, E. Blart, G. Boschloo, E. Mukhtar, A. Hagfeldt, L. Hammarström and F. Odobel, J. Phys. Chem. B, 2005, 109, 22928–22934 CrossRef PubMed
- A. Nattestad, M. Ferguson, R. Kerr, Y.-B. Cheng and U. Bach, Nanotechnology, 2008, 19, 295304 CrossRef PubMed
- P. Qin, H. Zhu, T. Edvinsson, G. Boschloo, A. Hagfeldt and L. Sun, J. Am. Chem. Soc., 2008, 130, 8570–8571 CrossRef CAS PubMed
- Y.-S. Yen, W.-T. Chen, C.-Y. Hsu, H.-H. Chou, J. T. Lin and M.-C. P. Yeh, Org. Lett., 2011, 13, 4930–4933 CrossRef CAS PubMed
- Y. Pellegrin, L. Le Pleux, E. Blart, A. Renaud, B. Chavillon, N. Szuwarski, M. Boujtita, L. Cario, S. Jobic and D. Jacquemin, et al., J. Photochem. Photobiol., A, 2011, 219, 235–242 CrossRef CAS PubMed
- M. Bräutigam, M. Schulz, J. Inglis, J. Popp, J. G. Vos and B. Dietzek, Phys. Chem. Chem. Phys., 2012, 14, 15185–15190 RSC
- J. C. Freys, J. M. Gardner, L. D'Amario, A. M. Brown and L. Hammarström, Dalton Trans., 2012, 41, 13105–13111 RSC
- Z. Ji, G. Natu, Z. Huang, O. Kokhan, X. Zhang and Y. Wu, J. Phys. Chem. C, 2012, 116, 16854–16863 CAS
- K. C. D. Robson, P. G. Bomben and C. P. Berlinguette, Dalton Trans., 2012, 41, 7814–7829 RSC
- P. G. Bomben, T. J. Gordon, E. Schott and C. P. Berlinguette, Angew. Chem., Int. Ed. Engl., 2011, 50, 10682–10685 CrossRef CAS PubMed
- K. Ghosh, G. Masanta and A. P. Chattopadhyay, Eur. J. Org. Chem., 2009, 4515–4524 CrossRef CAS
- K. Ghosh, G. Masanta, R. Fröhlich, I. D. Petsalakis and G. Theodorakopoulos, J. Phys. Chem. B, 2009, 113, 7800–7809 CrossRef CAS PubMed
- M. Beley, D. Delabouglise, G. Houppy, J. Husson and J.-P. Petit, Inorg. Chim. Acta, 2005, 358, 3075–3083 CrossRef CAS PubMed
- R. López, D. Villagra, G. Ferraudi, S. A. Moya and J. Guerrero, Inorg. Chim. Acta, 2004, 357, 3525–3531 CrossRef PubMed
- E. C. Constable and M. D. Ward, J. Chem. Soc., Dalton Trans., 1990, 1405 RSC
- K. C. D. Robson, B. D. Koivisto, A. Yella, B. Sporinova, M. K. Nazeeruddin, T. Baumgartner, M. Grätzel and C. P. Berlinguette, Inorg. Chem., 2011, 50, 5494–5508 CrossRef CAS PubMed
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202–2209 CrossRef CAS PubMed
- S. Sumikura, S. Mori, S. Shimizu, H. Usami and E. Suzuki, J. Photochem. Photobiol., A, 2008, 199, 1–7 CrossRef CAS PubMed
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. van Dam, D. Wang, J. Nieplocha, E. Apra and T. L. Windus, et al., Comput. Phys. Commun., 2010, 181, 1477 CrossRef CAS PubMed
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS
- R. Krishnan, J. S. Binkley, R. Seegar and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS PubMed
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC
- G. Black, K. Schuchardt, D. Gracio, B. Palmer and K. Schuchardt, The Extensible Computational Chemistry Environment: A Problem Solving Environment for High Performance Theoretical Chemistry, 2003, pp. 122–131 Search PubMed
- C. J. Aspley and J. A. G. Williams, New J. Chem., 2001, 25, 1136–1147 RSC
- K.
C. D. Robson, B. D. Koivisto, T. J. Gordon, T. Baumgartner and C. P. Berlinguette, Inorg. Chem., 2010, 49, 5335–5337 CrossRef CAS PubMed
- K. C. D. Robson, B. Sporinova, B. D. Koivisto, E. Schott, D. G. Brown and C. P. Berlinguette, Inorg. Chem., 2011, 50, 6019–6028 CrossRef CAS PubMed
- K. C. D. Robson, B. D. Koivisto and C. P. Berlinguette, Inorg. Chem., 2012, 51, 1501–1507 CrossRef CAS PubMed
- J. Sauvage, J. Collin, J. Chambron, S. Guillerez, C. Coudret, V. Balzani, B. Francesco, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS
- E. A. Gibson, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel, A. Hagfeldt and G. Boschloo, Langmuir, 2012, 28, 6485–6493 CrossRef CAS PubMed
- L. Le Pleux, A. L. Smeigh, E. Gibson, Y. Pellegrin, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarström and F. Odobel, Energy Environ. Sci., 2011, 4, 2075 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra for K1 and K2; FTIR spectra of K1 and K2 adsorbed on NiO; emission decay for K1 in methanol; DFT calculations for K1 and K2 in methanol; J–V and IPCE data for K1 with Co(dtb)3ClO3(2) electrolyte. See DOI: 10.1039/c3ra44690e |
| ‡ Current address: Department of Chemistry, University of British Columbia, Vancouver, V6T 1Z1, BC, Canada. |
| This journal is © The Royal Society of Chemistry 2014 |