Synthesis of highly soluble polymer-coated magnetic nanoparticles using a combination of diazonium salt chemistry and the iniferter method
Nébéwia
Griffete
,
Aazdine
Lamouri
,
Frédéric
Herbst
,
Nordin
Felidj
,
Souad
Ammar
and
Claire
Mangeney
*
ITODYS Laboratory, University Paris Diderot, Paris, France. E-mail: mangeney@univ-paris-diderot.fr; Fax: 0157277263; Tel: 0157276878
First published on 24th November 2011
Abstract
Polymer-coated magnetic nanoparticles were synthesized using an original and simple chemical strategy combining aryl diazonium salt chemistry and the iniferter method. This approach provides individually dispersed, highly soluble and pH-sensitive poly(methacrylic acid)-coated magnetic iron oxide nanoparticles.
Introduction
The grafting of polymer brushes from metal oxide nanoparticles via surface-initiated controlled radical polymerization has been the subject of intense research these last years.1 One of the key steps for obtaining stable hybrid metal oxide cores/polymer shells nanostructures relies on the grafting of an appropriate coupling agent between the nanoparticle surface and the polymer coating. Toward this end, various organic ligands have been used: carboxylates,2 amines or diols,3 phosphates and phosphonates,4 as well as sulfonates and thiols.5 Although effective in several nonaqueous environments, these anchoring groups of the chelate type often fail on metal oxide surfaces in aqueous or protic media due to the hydrolytic instability of the surface attachment and/or the dynamic nature of the interaction. Therefore, the development of versatile and efficient surface modification strategies for obtaining strong and stable linkages in aqueous media, between the metal oxide NPs surface and the polymer coating still remains challenging.We address this issue in the present paper by developing a novel and facile methodology to surface-initiated polymerization (SIP) of vinylic monomers from metal oxide nanoparticles via an aryl diazonium salt-derived initiator. Aryl diazonium salts have been shown recently to be useful coupling agents for the grafting of polymer coatings on carbon-based6 and metallic7 (gold, platinum, palladium, ruthenium and titanium) planar or nanoparticle surfaces, affording strong carbon or metal–carbon linkages. Nevertheless, such unique chemistry using aryl diazonium salts as coupling agents for polymer coatings has never been extended to the spontaneous functionalization of metal oxide nanoparticles, probably due to the poor reducing character of this type of material inhibiting the generation of aryl radicals able to bind to the NPs surface. It is only very recently that our group succeeded in grafting aryl groups derived from diazonium salts on the surface of iron oxide nanoparticles by taking advantage of the transformation of diazonium species to diazoates in basic media.8 This original strategy led to the grafting of a monolayer of functional aryl groups on the surface of the NPs. However, the possibility to grow polymer chains from the NPs surface using this approach has never been investigated so far.
In this paper, we fill this gap by exploring the propensity of aryl diazonium coupling agents to initiate the growth of polymer chains from the iron oxide NPs surface. Our strategy relies on bifunctional initiators, containing (i) a diazonium end group for surface anchoring and (ii) a N,N-diethyldithiocarbamate9 (DEDTC) function able to activate SIP, as schematically showed in Fig. 1. Two kinds of diazonium coupling agents were designed: the first one was a benzylchloridediazonium tetrafluoroborate (BF4,2N–C6H4–CH2–Cl) which required a two-step procedure (grafting of the coupling agent and post-functionalization by DEDTC) in order to provide SIP initiators at the surface of the NPs. The second diazonium compound was a DEDTC-modified benzyldiazonium tetrafluoroborate (BF4,2N–C6H4–CH2–DEDTC) which can be used in a one-step functionalization procedure. We demonstrate this approach by grafting pH-sensitive poly(methacrylic acid) PMAA brushes on magnetic iron oxide nanoparticles. The polymer brushes were grown from the initiator-modified surfaces by surface-initiated photoiniferter-mediated polymerization (SI-PIMP), which is a versatile and robust approach widely used in “graft-from” strategies.10
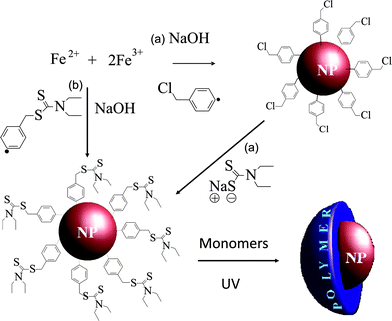 | ||
| Fig. 1 Strategies for surface functionalization of iron oxide NPs; (a) two-steps and (b) one-step grafting of the initiators at the surface. | ||
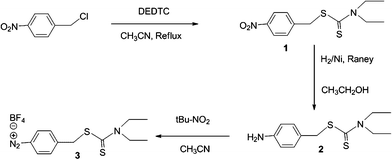 | ||
| Fig. 2 Synthesis of BF4,2N–C6H4–CH2–DEDTC. | ||
The final products of NP-PMAA were washed several times and separated by magnetic separation before being fully characterised by IR, Raman, DRX, TGA and TEM.
Experimental
Materials
Terbutylnitrite, thionyl chloride (SOCl2), tetrafluoroboric acid (HBF4), (4-aminophenyl)methanol, sodium hydroxyde (NaOH), iron chloride (FeCl3), iron sulphate (FeSO4), diethyldithiocarbamate (DEDTC), methacrylic acid (MAA) and ethylene glycol dimethylacrylate (EGDMA) were purchased from Sigma and were used without further purification. All the solvents were obtained from Acros and used as received.Powder X-ray diffraction data were collected on a Siemens D5000 Kevex diffractometer (30 min) using Cu– Ka radiation (l∼1.5405 A°). Electron microscopy and diffraction studies were conducted on a JEOL-100 CX II microscope. Differential thermal and thermogravimetric analyses were carried out on a Setaram TG 92–12 thermal analyser in the temperature range 20–800 °C with a heating rate of 10 °C min−1 under a flow of air at 80 ml min−1 in an alumina crucible. FT-IR spectra were obtained by transmission on an Equinox 55 spectrometer on pressed KBr pellets in the range 400–4000 cm−1. The magnetic measurements on powdered samples were carried out at low temperature (5 K) using a commercial SQUID magnetometer “MPMS–5S” from Quantum Design Corp. Field constant and isothermal dc magnetisation were performed with a field. Raman spectra were obtained with a LABRAM Jobin-Yvon micro-spectrometer using a He-Ne excitation laser (632.8 nm, 1 mW power). All spectra were taken with 1 s integration time in the 250–2500 cm−1 spectral range.
Synthesis of BF4,2N–C6H4–CH2–Cl
This diazonium salt was prepared in two steps using the Tschaen method adapted to our use.4-Aminophenylmethanol (0.5 g, 4 mmol) was added portion wise to 3 ml of thionylchloride. The solution was refluxed for one hour, cooled to room temperature. The excess of thionylchloride was destroyed by addition of water at 0 °C. This aqueous solution was heated to 80 °C for 30 min and then evaporated to give 0.42 g of a viscous green compound which will be used in the next step without further purification.
A cold solution of terbutylnitrite (0.13 mL, 1 mmol) in anhydrous acetonitrile (0.5 mL) was added dropwise to a cold solution (in an ice bath) composed of the chlorinated compound synthesized above (0.15 g, 1 mmol), tetrafluoroboric acid (0.5 mL in ether v/v 1/1) and anhydrous acetonitrile (1.5 mL). The reaction was conducted at −20 °C for 24 h. The viscous diazonium salt was washed 3 times with ether then finally dissolved in acetone and evaporated at room temperature.
Synthesis of BF4,2N–C6H4–CH2–DEDTC
This diazonium salt was prepared in three steps. 1) The 4-nitrobenzyl chloride reacts with the DEDTC to give the thiocarbamate 1 in a good yield. 2) This last intermediate was hydrogenated using RANEY® Nickel and leads to the corresponding primary amine 2 in quantitative yield. 3) A cold solution of terbutylnitrite in anhydrous acetonitrile was added dropwise to a cold solution (in an ice bath) composed of the compound 2, tetrafluoroboric acid and anhydrous acetonitrile. The reaction was conducted at −20 °C for 24 h. The viscous diazonium salt was washed 3 times with ether then finally dissolved in acetone and evaporated at room temperature.Iron oxide NPs synthesis and functionalization
The functionalization of iron oxide nanoparticles by the diazonium salt was done in situ during the nanoparticle synthesis: in a typical reaction, 2.9 mmol of FeCl3 and 1.2 mmol of FeSO4 were dissolved in 5 mL of deionised water. The solution was purged with nitrogen, and the inert atmosphere was maintained for the duration of the synthesis. Then 3 mL of NaOH in water (c = 1 M) was rapidly added under vigorous stirring. The color of the solution changed immediately from yellow to dark, indicating the formation of iron oxide nanoparticles. Then, the diazonium salt (0.5 mmol, c = 0.3 M) synthesized above was added directly into the reaction vessel. The mixture was stirred for 1 h. The mixture was centrifuged many times at 8500 tr/min to eliminate the excess of salts.For the NP–C6H4–CH2–Cl (iron oxide nanoparticles coated with the chlorinated salt), 2 mL of DEDTC in THF (0.15 g, 1 mmol) was added into the solution containing the NPs (0.15 g) in 5 mL of THF and the mixture was warmed at 60 °C during 24 h. The solution was centrifuged many times to eliminate the excess of DEDTC.
Concerning the polymerisation, typically iron oxide NPs containing DEDTC on the surface (0.015 g) were mixed with MAA (0.04 g) and EGDMA (0.04 g) in ethanol (1 mL). The mixture was deoxygenated under argon for 15 min and put under UV light during 4 h. The polymerisation was performed 4 times on the same sample. The purified particles were characterised by IR, Raman, DRX, TGA and TEM.
Results and discussion
Iron oxide cores functionalized with functional aryl groups on the surface were synthesized by one-step coprecipitation of Fe2+/Fe3+ under alkaline conditions in the presence of the diazonium salt. This grafting step was performed within few minutes at room temperature in slightly basic aqueous medium (pH 9). Then, the polymerisation could proceed, mixing the NPs with methacrylic acid (MAA) as the monomer, ethyleneglycoldimethacrylate (EGDMA) as the reticulating agent and irradiating the deoxygenated mixture under UV light for 4 h.X-ray diffraction (XRD) patterns of the aryl-coated iron oxide NPs are characteristic of the spinel phase with broadened peaks (see Fig. 3).
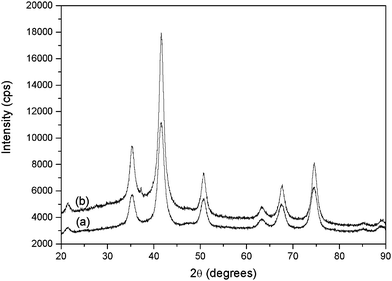 | ||
| Fig. 3 XRD patterns of the synthesized iron oxide nanoparticles (a) NP–C6H4–CH2–Cl and (b) NP–C6H4–CH2–DEDTC. | ||
The cell parameter and the size of coherent diffraction domain (<LXRD>) were determined with the MAUD software11 which is based on the Rietveld method combined with Fourier analysis. The refined agree well with the formation of nanocrystalline spinel iron oxide. The black color of the recovered powders suggests the formation of magnetite. However, the recorded Raman spectra (see Fig. 4) match much more with the maghemite Fe2O3 phase,12 probably due to a subsequent oxidation of the particles in air. Such feature is usual for iron oxide nanoparticles (magnetite and maghemite) due to their high specific area.12
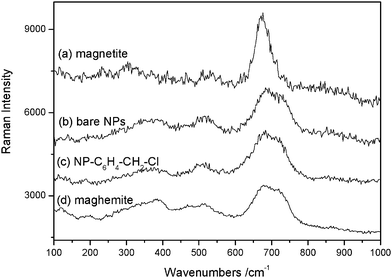 | ||
| Fig. 4 Raman spectra of (a) reference magnetite powders; (b) bare NPs; (c) NP–C6H4–CH2–Cl and (d) reference maghemite powders. | ||
Interestingly, as shown in Table 1, the crystal size appears to be dependent on the presence of the diazonium salt, with lower diameters measured after functionalization (∼30% diameter decrease), compared to bare NPs. This result indicates that the coating limits the crystalline growth, in agreement with previously reported works.13
| Sample | <LXRD> (nm) | <DTEM> (nm) | Org Wt% |
|---|---|---|---|
| a Organic weight percents estimated from SQUID measurements considering the magnetization decrease due to the diamagnetic organic ligand contribution. b Organic weight percents determined from TGA. c Samples obtained after polymerization from NP–C6H4–CH2–DEDTC. | |||
| Bare NPs | 11 | 11 | 4 |
| NP–C6H4–CH2–Cl | 7 | — | 21a, 20b |
| NP–C6H4–CH2–DEDTC | 8 | 8 | 18b |
| NP-PMAA (4h)c | — | — | 43b |
Transmission electron microscopy (TEM) showed faceted particles (see Fig. 5), with different states of aggregation depending on the diazonium salt used: the small BF4,2N–C6H4–CH2–Cl leads to nanoaggregates of particles after functionalization by the initiators and subsequent polymerization while the longer hydrophilic salt BF4,2N–C6H4–CH2–DEDTC produces isolated NPs.
 | ||
| Fig. 5 Transmission electron micrographs showing NP–C6H4–CH2–Cl (a) and NP–C6H4–CH2–DEDTC (c) and their coating with PMAA respectively (b) and (d). | ||
Aryl diazonium salt coupling agents bearing long and hydrophilic side chains should therefore be preferred to smaller ones in order to improve the colloidal stability of the NP-PMAA aqueous suspensions. From TEM images obtained on non-aggregated NPs samples, mean diameters DTEM of ∼8 nm could be estimated for NP–C6H4–CH2–DEDTC. The proximity between sizes inferred from XRD and TEM analysis suggests that the produced particles are single crystals. Fig. 6 shows the modifications of the NPs IR spectra after each organic coating step.
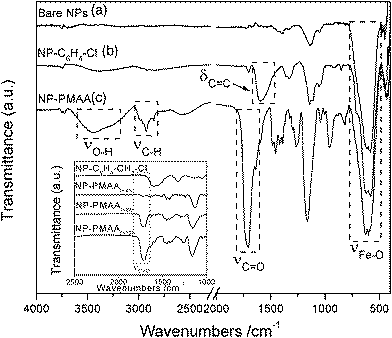 | ||
| Fig. 6 FT-IR spectra of (a) bare iron oxide NPs; (b) NP–C6H4–CH2–Cl; (c) NP-PMAAt=12h. Inset shows the spectra obtained on NP-PMAA samples after various polymerization times. | ||
Bare NPs display one intense band at 550 cm−1, characteristic of the Fe–O stretching vibration. The IR spectra of NP–C6H4–CH2–Cl and NP–C6H4–CH2–DEDTC show the appearance of one main band at 1680 cm−1 due to the phenyl δC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrations. Other bands at 1140 and 1340 cm−1 are also visible in the spectrum of NP–C6H4–CH2–DEDTC, due to the diethyldithiocarbamate unit. It is noteworthy that the N≡N stretching mode near 2280 cm−1 appearing in the IR spectrum of the free diazonium salts is not visible in the spectra of the functionalized NPs, indicating the release of N2 as a consequence of the cleavage of the diazonium moieties during the grafting process. Strong modifications appear in the spectrum of NP-PMAA: a very intense band is seen at ca. 1715 cm−1 due to the CO stretching mode and two medium bands are observed at 3300 cm−1 (νO–H) and 2900 cm−1 (νC–H). These features, characteristic of poly(methacrylic acid), indicate that the polymerization step has been effective. In order to evidence the living character of the polymerisation of MAA from the iron oxide NPs, a kinetic study was carried out by multiplying the polymerization experiments (during 4 h) on a single sample. The samples are abbreviated NP-PMAAt=xh, x standing for the polymerization time. The IR spectra (shown in inset of Fig. 6) show a progressive and strong intensity increase of the νCO vibrational band (characteristic of PMAA) with polymerization time. It is therefore possible to stop the polymerization process and to restart it afterwards, underlining the efficiency of this multistep strategy for obtaining poly(methacrylic acid) coatings of controlled thickness on the surface of iron oxide nanoparticles.
C vibrations. Other bands at 1140 and 1340 cm−1 are also visible in the spectrum of NP–C6H4–CH2–DEDTC, due to the diethyldithiocarbamate unit. It is noteworthy that the N≡N stretching mode near 2280 cm−1 appearing in the IR spectrum of the free diazonium salts is not visible in the spectra of the functionalized NPs, indicating the release of N2 as a consequence of the cleavage of the diazonium moieties during the grafting process. Strong modifications appear in the spectrum of NP-PMAA: a very intense band is seen at ca. 1715 cm−1 due to the CO stretching mode and two medium bands are observed at 3300 cm−1 (νO–H) and 2900 cm−1 (νC–H). These features, characteristic of poly(methacrylic acid), indicate that the polymerization step has been effective. In order to evidence the living character of the polymerisation of MAA from the iron oxide NPs, a kinetic study was carried out by multiplying the polymerization experiments (during 4 h) on a single sample. The samples are abbreviated NP-PMAAt=xh, x standing for the polymerization time. The IR spectra (shown in inset of Fig. 6) show a progressive and strong intensity increase of the νCO vibrational band (characteristic of PMAA) with polymerization time. It is therefore possible to stop the polymerization process and to restart it afterwards, underlining the efficiency of this multistep strategy for obtaining poly(methacrylic acid) coatings of controlled thickness on the surface of iron oxide nanoparticles.
The magnetic properties of the nanohybrids were then examined. As shown in Fig. 7, the saturation magnetization of surface functionalized nanoparticles appeared significantly reduced compared to pure ones.
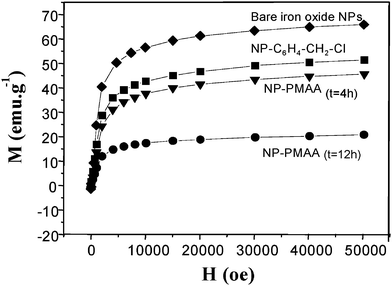 | ||
| Fig. 7 5 K-First magnetization of: bare iron oxide NPs (diamonds); NP–C6H4–CH2–Cl (squares); NP-PMAA after 4 h polymerisation (down triangles) and after 12 h polymerisation (circles). | ||
These measurements showed that the produced hybrids exhibited a non zero magnetization despite their a priori high diamagnetic organic content (about 20 emu.g−1 at 5 K and 50 kOe for NP-PMMA4 h). From the magnetization data obtained at higher field (50 koe), one could afford the diamagnetic contribution due to the polymer coating (x), according to eqn (1):
| Msat(hybrid) = Msat(iron oxide) × (1-x) + Msat(Organic) × x | (1) |
After coating the NPs with PMAA, the weight loss increases significantly, indicating an organic coating of up to 43% after 4 h of polymerization. Although the presence of the comonomer EGDMA as a reticulant prevents us from determining easily the degree of polymerization, this value indicates a high polymer surface coverage.
The stability of the NP-PMAA colloids in water was checked qualitatively. Fig. 8 reveals that the pH strongly influences the dispersion with flocculated NPs in deionized water (pH 5.5) while they appear highly stable in basic medium (pH 8).
 | ||
| Fig. 8 Numerical photographs of aqueous dispersions of NP-PMAA (a) in deionized water; (b) in basic medium (NaOH 1.10−4 M); and (c) in the presence of a magnet. | ||
This pH-sensitive behavior is due to the presence of carboxylic groups all along the PMAA-grafted chains, which are fully protonated or deprotonated one unit below or above their pKa. When deprotonated and negatively charged, the PMAA-coated nanoparticle suspensions are stabilized by electrostatic repulsions between the particles, while at low pH the particles aggregate and precipitate from suspension. Furthermore, the NP-PMAA particles are magnetically responsive and can be spatially manipulated using a permanent magnet, as shown in Fig. 8c.
Conclusions
In summary, we have developed an original and simple route combining aryl diazonium salt chemistry and the iniferter method to elaborate individually dispersed, highly soluble and pH-sensitive PMAA-coated magnetic nanoparticles. Our approach offers several advantages over conventional methods: (i) ease and rapidity of the diazonium-based coupling agent grafting at the NPs surface; (ii) presence of a covalent bonding between the polymer and the NPs surface; (iii) opportunities offered by the living character of the polymerization process. We believe this synthetic approach will not only pave an additional way for the preparation of water-soluble and pH-sensitive PMAA-coated iron oxide nanocrystals but also provide a new general functionalization strategy for magnetic nanoparticles.References
- (a) R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437 CrossRef CAS; (b) F. Hu, K. G. Neoh, L. Cen and E. Kang, Biomacromolecules, 2006, 7, 809 CrossRef CAS.
- (a) R. Tadmor, R. E. Rosensweig, J. Frey and J. Klein, Langmuir, 2000, 16, 9117 CrossRef CAS; (b) R. E. Rosensweig, R. Kaiser and G. Miskolczy, Journal of Colloid and Interface Science, 1969, 29, 680 Search PubMed.
- J. Rockenberger, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc., 1999, 121, 11595 CrossRef CAS.
- C. Yee, G. Kataby and A. Ulman, Langmuir, 1999, 15, 7111 CrossRef CAS.
- (a) J. Lu, J. Fan, R. Xu, S. Roy, N. Ali and Y. Gao, Journal of Colloid and Interface Science, 2003, 258, 427 CrossRef CAS; (b) G. Kataby, A. Ulman, R. Prozorov and A. Gedanken, Langmuir, 1998, 14, 1512 CrossRef.
- (a) S. A. Dahoumane, M. N. Nguyen, A. Thorel, J. P. Boudou, M. M. Chehimi and C. Mangeney, Langmuir, 2009, 25, 9633 CrossRef CAS; (b) T. Matrab, M. N. Nguyen, S. Mahouche, P. Lang, C. Badre, M. Turmine, G. Girard, J. Bai and M. M. Chehimi, J. Adhes., 2008, 84, 684 CrossRef CAS; (c) T. Liu, R. Casado-Portilla, J. Belmont and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4695 CrossRef CAS; (d) S. Mahouche Chergui, N. Abbas, T. Matrab, M. Turmine, E. Bon Nguyen, R. Losno, J. Pinson and M. M. Chehimi, Carbon, 2010, 48, 2106 CrossRef CAS; (e) T. Matrab, M. M. Chehimi, J. Pinson, S. Slomkowski and T. Basinska, Surf. Interface Anal., 2006, 38, 565 CrossRef CAS.
- (a) H. Gehan, L. Fillaud, M. M. Chehimi, J. Aubard, A. Hohenau, N. Felidj and C. Mangeney, ACSNano, 2010 Search PubMed , ASAP; (b) F. Hauquier, T. Matrab, F. Kanoufi and C. Combellas, Electrochim. Acta, 2009, 54, 5127 CrossRef CAS; (c) T. Matrab, M. M. Chehimi, C. Perruchot, A. Adenier, V. Guillez, M. Save, B. Charleux, E. Cabet-Deliry and J. Pinson, Langmuir, 2005, 21, 4686 CrossRef CAS; (d) S. Gam-Derouich, M. N. Nguyen, A. Madani, N. Maouche, P. Lang, C. Perruchot and M. M. Chehimi, Surf. Interface Anal., 2010, 42, 1050 CrossRef CAS; (e) M. N. Nguyen, T. Matrab, C. Badre, M. Turmine and M. M. Chehimi, Surf. Interface Anal., 2008, 40, 412 CrossRef CAS.
- (a) N. Griffete, F. Herbst, J. Pinson, S. Ammar and Claire Mangeney, J. Am. Chem. Soc., 2011, 133, 1646 CrossRef CAS; (b) C. Combellas, F. Kanoufi, J. Pinson and F. I. Podvorica, Electrochim. Acta, 2009, 54, 2164 CrossRef CAS.
- A. M. Imroz Ali and A. G. Mayes, Macromolecules, 2010, 43, 837 CrossRef CAS.
- (a) T. Otsu, T. Ogawa and T. Yamamoto, Macromolecules, 1986, 19, 2087 Search PubMed; (b) S. B. Rahane, S. M. Kilbey and A. T. Metters, Macromolecules, 2005, 38, 8202 CrossRef CAS; (c) B. de Boer, H. K. Simon, M. P. L. Werts, E. W. van der Vegte and G. Hadziioannou, Macromolecules, 2000, 33, 349 CrossRef CAS.
- L. Lutterotti, S. Matthies and H. R. Wenk, Newsletter of the Commission on Powder diffraction, 1999, 21, 14 Search PubMed.
- (a) A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804 Search PubMed; (b) J. Mürbe, A. Rechtenbach and J. Töpfer, Mater. Chem. Phys., 2008, 110, 426 CrossRef.
- P. Dallas, A. B. Bourlinos, D. Niarchos and D. Petridis, J. Mater. Sci., 2007, 42, 4996 CrossRef CAS.
- The specific surface area (Ssp) was calculated using Ssp = 6/ρD where ρ is the ferric oxide density and D is their particle diameter. Then, from the weight % of the aryl organic coating (Waryl) and the magnetic iron oxide core (Wiron oxide) determined by the TGA analysis, the surface coverage of aryl groups, Γ could be computed using: Γ = (NA × Waryl)/(Ssp × M × Wiron oxide), where M is the molar mass of attached benzyl chloride (125.5 g mol−1) and NA is the Avogadro's number.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429 RSC.
| This journal is © The Royal Society of Chemistry 2012 |