Investigating the non-enzymatic methylation of arsenite by methylcobalamin B12 using high-performance liquid chromatography on-line with inductively coupled plasma-mass spectrometry
Spiros A. Pergantis†a, Maria Miguens-Rodrigueza, Nohora P. Velab and Douglas T. Heitkemper*b
aBirkbeck College, University of London, School of Biological and Chemical Sciences, Gordon House, 29 Gordon Square, London, UK WC1H 0PP
bU.S. Food and Drug Administration, Forensic Chemistry Center, 6751 Steger Drive, Cincinnati, OH 45237, USA
First published on 17th November 2003
Abstract
In this study, we have demonstrated the use of HPLC-ICP-MS as a powerful tool for studying the reaction of arsenite with methylcobalamin in the presence of glutathione. Even though this reaction has previously been investigated using radioactive 73As, HPLC-ICP-MS provides considerable advantages relating to sample throughput, sensitivity, and separation efficiency of the arsenic-containing reaction products. As a result of these advantages, kinetic studies were easily conducted, potentially providing valuable insight into the reaction itself. In addition, the HPLC-ICP-MS approach allowed for the tentative identification of methylarsonous acid, an arsenic species that had escaped detection in previous studies of the same reaction. Overall, this study confirmed that methylcobalamin acts as a methylating agent for arsenite in the presence of glutathione in an abiotic environment. A series of methylated arsenic species, identified as methylarsonic acid, dimethylarsinic acid and methylarsonous acid were detected.
Introduction
Arsenic is ubiquitous in the environment, and as a result humans are exposed to it continuously.1,2 In particular, chronic exposure to elevated concentrations of inorganic arsenic in drinking water is believed to cause adverse health effects and contribute to cancers of the lung, bladder and skin.3–7 It is for this reason that the maximum permissible concentration of arsenic in drinking water has been set at 10 µg l−1 by the EPA and WHO.8,9To date, the majority of epidemiological studies on arsenic have assumed that inorganic arsenic is by far the most toxic form of the element, and it is believed to cause the most adverse health effects. Until recently, the process of arsenic methylation, which occurs in certain organisms including humans, was considered to be a means of detoxification as it gives rise to the less toxic methylarsonic acid (MMAsV) and dimethylarsinic acid (DMAsV) species which are readily excreted in urine.10–12 However, this view is now being challenged as a result of several recent studies reporting on the presence of additional methylated arsenic species, i.e. methylarsonous acid (MMAsIII) and dimethylarsinous acid (DMAsIII), in urine samples from people exposed to relatively high levels of arsenic in their drinking water.13–16 In some cases, MMAsIII and DMAsIII have been found to exhibit greater cytotoxicity and genotoxicity than the inorganic forms of the metalloid.17–20
In view of these latest findings, it is of particular interest to revisit the ideas concerning arsenic methylation and biomethylation with the intention of improving our understanding of factors affecting the formation of various methylated forms of arsenic. Numerous studies have demonstrated that S-adenosylmethionine acts as a source of carbocations (CH3+) responsible for the oxidative methylation of inorganic arsenite (iAsIII)1,21via nucleophilic substitution. This view is widely shared amongst chemists investigating the behaviour of arsenic in environmental and biological systems. In contrast, only a few studies have reported on the possibility of vitamin B12 or any of its derivatives acting as methylating agents for arsenic. For example, it has been suggested that methylcobalamin or methylvitamin B12 (CH3B12), a derivative of vitamin B12 in human metabolism, functions as a methyl donor in the biosynthesis of dimethylarsine from arsenate or arsenite in cell extracts of methanobacillus.22 Also, Buchet and Lauwerys suggested that CH3B12 methylates arsenite at a low rate in the presence or absence of rat liver extracts.23 Furthermore, Styblo et al. added 100 µg CH3B12/ml in their arsenite methyltransferase studies, although experimental evidence for its requirement was not presented.24,25 Broader acceptance of CH3B12 acting as a methylating agent for arsenic has been hampered by lack of a theory suitable to explain the mechanism by which it acts. With respect to this, Zakharyan and Aposhian conducted a more fundamental study in which they demonstrated that CH3B12 methylates arsenic in the absence of any enzymes, providing glutathione is present.26
The purpose of the present study was to initially confirm the findings of the Zakharyan and Aposhian study,26 and to subsequently try to achieve higher methylation efficiencies. It was also our intention to improve the analytical methodology that was used in the original study, i.e. avoid using radioactive 73As and open column chromatography and instead use modern state-of-the-art analytical techniques such as high-performance liquid chromatography coupled on-line with inductively coupled plasma mass spectrometry (HPLC-ICP-MS). These analytical techniques have the potential to make arsenic methylation studies simpler and more efficient to conduct. In fact, using HPLC–ICP-MS allows for the efficient separation of various arsenic species and their subsequent very sensitive and selective detection.
Experimental
Reagents
Ultrapure 18 MΩ deionized water, DIW (Millipore, MA, USA) was used in the preparation of all reagents and standards. A stock standard of arsenite (iAsIII, sodium m-arsenite, Sigma, St. Louis, MO, USA) used in the methylation experiments was prepared at a concentration of 360 µg mL−1 As. A 0.1 M solution of Tris buffer (Tris(hydroxymethyl)aminomethane hydrochloride, Sigma) was prepared and the pH was adjusted to 7.8. Methylcobalamin (CH3B12) and glutathione (GSH, γ-Glu-Cys-Gly; reduced form) were also obtained from Sigma and reagent solutions were prepared at concentrations of 1 mM and 0.18 mM, respectively. GSH and CH3B12 solutions were stored at 4 °C in amber-colored HDPE bottles and were prepared weekly because it was observed that freshly prepared reagents were required to achieve optimum as well as reproducible methylation efficiencies. A stock solution of selenite (iSeIV, sodium selenite, Alfa Products, Danvers, MA, USA) at a concentration of 1000 µg mL−1 was also prepared. Hydrogen peroxide used as an oxidizing agent was obtained from J. T. Baker (Ultrex II 30%, Phillipsburg, NJ, USA)Ammonium phosphate, ammonium nitrate and ammonium hydroxide obtained from J. T Baker (Phillipsburg, NJ, USA) were used to prepare and adjust the pH of the chromatographic mobile phase used for Separation 1 (see Procedures section). Ammonium carbonate (J. T. Baker) was used to prepare the chromatographic mobile phase used for Separation 2 (see Procedures section).
Standards for arsenic speciation analysis
Stock standards of arsenite (iAsIII) and arsenate (iAsV) (1000 µg As mL−1 as As2O3 in 2% HCl and H3AsO4·½ H2O in 2% HNO3, respectively) were obtained from Spex Industries (Metuchen, NJ, USA). Disodium methylarsonate (MMAsV, 99%) and dimethylarsinic acid (DMAsV, 98%) were obtained from ChemService (West Chester, PA, USA). Methyloxoarsine originally obtained from Dr. William R. Cullen at the University of British Columbia, was a gift from the US EPA, National Exposure Research Laboratory in Cincinnati, Ohio. Stock standard solutions (∼1000 µg As mL−1) of MMAsIII, MMAsV and DMAsV were prepared in DIW and stored at 4 °C in amber-colored HDPE bottles.Instrumentation
A model PQ3 ICP-MS (VG Elemental, Franklin, MA, USA) was used for element-specific chromatographic detection of arsenic-containing species. A concentric nebulizer and cooled conical spray chamber were used to introduce the chromatographic effluent into the plasma. The operating parameters were as follows: forward power 1350 W; coolant argon flow 12 L min−1; auxiliary argon flow 1.0 L min−1; nebulizer gas flow 0.8 L min−1. Chromatographic data were collected and integrated using PlasmaLab software. Masses 75 and 77 were monitored with a 250 ms dwell time and 10 min data acquisition time. Integrated peak areas were imported into a spreadsheet program for further data reduction.The chromatographic system used consisted of a GP50 gradient pump (Dionex, Sunnyvale, CA, USA) and AS3500 autosampler (Thermo Separations Products, San Jose, CA, USA). The sample injection volume was 25 µL. A non-metallic automated switching valve (Waters, Milford, MA USA) with a 25 µL sample loop was used to introduce a flow injection peak (20 ng As mL−1) post-column at the beginning of each chromatographic run. The flow injection peak was used to correct for instrument drift throughout the day. A length (∼60 cm ) of 0.25 mm id PEEK® tubing was used to interface the chromatographic system to the ICP-MS nebulizer.
Procedures
Tris buffer solutions (pH 7.8) containing GSH and iSeIV were left to stand for 30 min prior to adding iAsIII and CH3B12. The resulting reaction mixtures were incubated at 37 °C for various time intervals. Microlitre aliquots of the reaction mixtures were taken, diluted five-fold with DIW and immediately analyzed using HPLC-ICP-MS. Two different anion exchange HPLC separations were used throughout this study. The main HPLC column used was a PRP-X100 column (Hamilton, Reno, NV, USA, 250 × 4.1 mm id). The eluent consisted of 10 mM NH4H2PO4/10 mM NH4NO3 (pH 6.3) at a flow rate of 1.0 mL min−1 (Separation 1). A second anion-exchange HPLC column was also used to confirm identification of the arsenic products formed. This was an IC-Pak Anion HR column (Waters, Milford, MA, USA, 75 × 4.6 mm id), eluted with 10 mM (NH4)2CO3 (pH 10) at a flow rate of 1 mL min−1 (Separation 2).The arsenic methylation reaction was monitored using HPLC–ICP-MS as described above. Methylation efficiency was defined according to the following equation using integrated chromatographic peak areas:
| 100%(DMAsV + MMAsV + MMAsIII)/(iAsIII + iAsV+ DMAsV + MMAsV + MMAsIII) |
Results and discussion
Identification of reaction products by using HPLC–ICP-MS
During the reaction of CH3B12, GSH, iSeIV and iAsIII, samples of the reaction mixture were taken at various time intervals, diluted five-fold and analyzed immediately by using HPLC–ICP-MS with Separation 1 (Fig. 1). For the chromatograms shown in Fig. 1B, the starting reaction mixture consisted of 3.4 µg mL−1 iAsIII, in 0.07 M Tris buffer with 0.17 mM CH3B12, 20 µg mL−1 iSeIV, and 22 µM GSH. After approximately 4 hours incubation at 37 °C, the resulting chromatogram revealed the presence of four arsenic species in addition to the starting material, iAsIII. Upon comparing the retention times of various arsenic standards (Fig. 1a) with those of the reaction products, it was possible to identify the presence of MMAsV, DMAsV and iAsV. Identification of these arsenic species through the use of HPLC–ICP-MS is currently considered routine as numerous publications have repeatedly described a variety of HPLC methods and applications suitable for this purpose.27–32 However, only recently have HPLC methods been developed that are suitable for the identification of methylated arsenic species in which arsenic exists in the three oxidation state, i.e. MMAsIII and DMAsIII.13–15,33 Several studies have recently used this approach to determine MMAsIII in human urine13–15 and DMAsIII and MMAsIII in wool from sheep exposed to arsenosugars.34 One of the reasons for the delayed development of this methodology has been the lack of pure and well-characterized standards for both of these species. Overcoming this problem to some extent has made it possible to use HPLC retention times for the identification of these species in “real” samples, especially urine samples. In the present study, MMAsIII was identified to be one of the reaction products. MMAsIII eluted between DMAsV and MMAsV when using Separation 1 (Fig. 1a).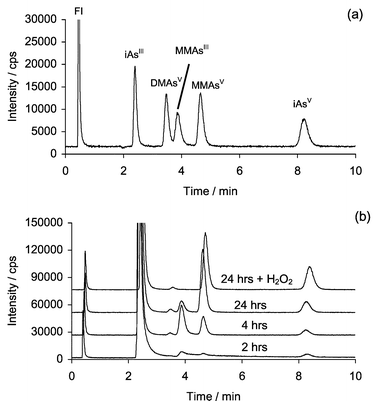 | ||
| Fig. 1 HPLC–ICP-MS chromatograms showing arsenic species detected at m/z 75 using Separation 1. (a) Standard chromatogram, each peak approximately 20 ng As mL−1; (b) sample chromatograms taken after 2, 4, and 24 hours incubation of 3.4 µg mL−1 iAsIII with 0.17 mM CH3B12, 22 µM GSH, 20 µg mL−1 iSeIV in 0.07 M TRIS buffer (note: iAsIII peak is off scale), also shown is the chromatogram obtained for 24 h incubation after the addition of H2O2. | ||
In order to further support the validity of these identifications, a second HPLC method (Separation 2) was used to analyze the reaction mixture. Both of the separations make use of resin-based anion exchange columns with quaternary ammonium fuctional groups. The PRP-X100 column (Separation 1) utilizes a poly(styrene-divinyl)benzene polymeric support while the IC-Pak A HR column (Separation 2) is based on a polymethacrylate resin. These stationary phase differences resulted in significant selectivity differences for the arsenic species studied, and so agreement in the speciation findings of the two methods increased the validity of our identifications. With Separation 1 the order of elution was: iAsIII, DMAsV, MMAsIII, MMAsV and iAsV (Fig. 1a), while with Separation 2 the order of elution was: MMAsIII, DMAsV, iAsIII, MMAsV and iAsV (Fig. 2a).
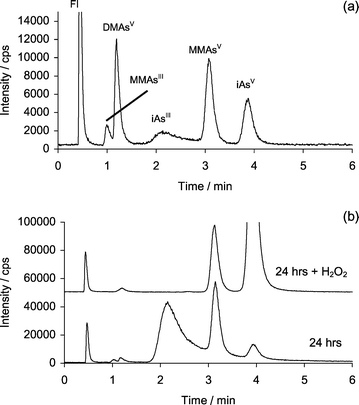 | ||
| Fig. 2 HPLC–ICP-MS chromatograms showing arsenic species detected at m/z 75 using Separation 2. (a) Standard chromatogram, original concentrations were approximately 20 ng As mL−1 each peak (partial oxidation of MMAsIII and iAsIII species in the standard is noted); (b) sample chromatograms taken after 24 h incubation of 3.4 µg mL−1 iAsIII with 0.17 mM CH3B12, 22 µM GSH, 20 µg mL−1 iSeIV in 0.07 M TRIS buffer, also shown is the chromatogram obtained for 24 h incubation after the addition of H2O2. (Note: iAsV peak is off scale.) | ||
Complimentary evidence supporting the presence of MMAsIII was obtained when adding 20 µL of 30% H2O2 to the 1∶5 diluted reaction mixture and subsequently analyzing it (Fig. 1B). Upon doing so it was observed that the peak assigned to be MMAsIII completely disappeared, while the peak area corresponding to MMAsV increased. At the same time, the iAsIII peak decreased in size and the iAsV peak area increased as expected. Similar results were obtained using Separation 2. In this case, both the MMAsIII and iAsIII species were completely oxidized as more time had passed between the addition of H2O2 and the analysis. Unfortunately, an analytical method capable of a more detailed identification of MMAsIII is currently unavailable. Attempts in our laboratory to identify this species using electrospray mass spectrometry have not been successful to date; however, efforts in this area are continuing.
Zakharyan and Aposhian26 did not report the presence of MMAsIII in their report describing the non-enzymatic methylation of arsenic, even though reaction conditions were almost identical with those of the present study. The reason for this is probably because H2O2 was used to quench the reaction prior to analysis. This would have prevented detection of MMAsIII as it would have oxidized it to MMAsV.
In the present study, it is possible that an arsenic-containing complex was observed during the early stages of the reaction. It was noted that sample aliquots taken early in the reaction exhibited excessive tailing of the peak at the retention time of iAsIII. Fig. 3 shows the chromatograms obtained after 2 h incubation of a mixture containing 7.2 µg mL−1 iAsIII, in 0.06 M Tris buffer with 0.18 mM CH3B12, and 18 µM GSH. The tailing was no longer observed upon further reaction or if H2O2 was added to the sample. After addition of H2O2 considerable amounts of the methylated species appeared almost immediately, mainly MMAsV (Fig. 3b). It is possible that a GSH-MMAsIII and/or GSH-iAsIII complex forms early in the reaction which later hydrolyses releasing MMAsIII. Styblo et al. reported on the use of H2O2 for the improved quantification of methylated arsenic species using thin layer chromatography.35 It is possible that the improvement seen resulted from the release of MMAsIII bound to GSH and subsequent oxidation to MMAsV. The detection and identification of arsenic–glutathione species has been reported.36–38
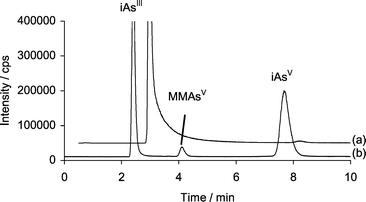 | ||
| Fig. 3 HPLC–ICP-MS chromatograms showing arsenic species detected at m/z 75 using Separation 1. (a) Chromatogram for sample taken after 2 h incubation of 7.2 µg mL−1 iAsIII with 0.18mM CH3B12, and 18 µM GSH in 0.06 M TRIS buffer. (b) Chromatogram for same sample after addition of H2O2 (note: iAsIII is off scale). | ||
Reaction kinetics
The nonenzymatic methylation of iAsIII using two different GSH concentrations, each in duplicate, was monitored over a 24 h period. The average kinetic profiles of the resulting methylated arsenic species are presented in Fig. 4. When a 22 µM GSH solution was used (Fig. 4a), it was observed that the overall average methylation efficiency was significantly greater than that observed when an 11 µM GSH solution was used (Fig. 4b), i.e., 17% vs. 5.5%. At both GSH levels examined, the efficiency of arsenic methylation reached a relatively constant level after 3–4 h. In both cases DMAsV remained relatively constant for the remainder of the reaction. However, MMAV levels continuously increased, while MMAsIII levels continuously decreased for the remainder of the reaction. Based on this data it seems that MMAsIII is being continuously oxidized to MMAsV during the reaction.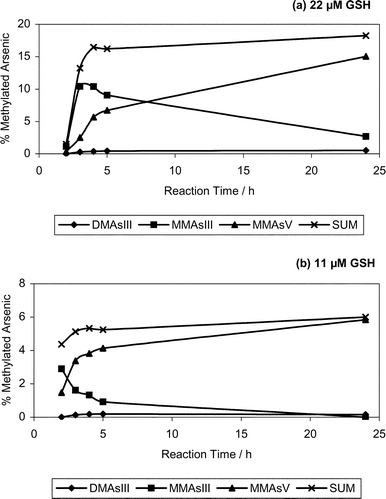 | ||
| Fig. 4 Kinetic profiles of individual methylated arsenic species and sum of all methylated species (methylation efficiency). Methylation efficiency was defined according to the following equation using integrated chromatographic peak areas: 100%(DMAsV + MMAsV + MMAsIII)/(iAsIII + iAsV+ DMAsV + MMAsV + MMAsIII). Experimental conditions: a) mixture containing 3.4 µg mL−1 iAsIII, 0.17mM CH3B12, 22 µM GSH, 20 µg mL−1 iSeIV in 0.07 M TRIS buffer incubated at 37 °C for various time intervals, (b) 3.6 µg mL−1 iAsIII with 0.18 mM CH3B12, 11 µM GSH, 20 µg mL−1 iSeIV in 0.07 M TRIS buffer incubated at 37 °C for various time intervals. | ||
Comparison with previous studies
It was observed that more reproducible results and higher methylation efficiencies were obtained by mixing the TRIS buffer initially with iSeIV and GSH and allowing the mixture to stand for 30–60 minutes prior to the addition of iAsIII and CH3B12. Using peak areas and Separation 1, the methylation efficiency obtained for 11 µM GSH increased from 3% (n = 2) to 5.6% (n = 5, RSD 6%) when allowing 30–60 min mixing time (reaction conditions as in Fig. 4b). The maximum arsenic methylation efficiency recorded after 24 hours reaction time for duplicate preparations was 15% and 18% (reaction conditions as in Fig. 4a). If measured after the addition of H2O2, the calculated methylation efficiency was 18% and 20%.Methylation efficiency could also be calculated based on the concentration of DMAsV and MMAV determined after addition of H2O2, thereby eliminating the need to use a MMAIII standard in the calculation. This would also eliminate any potential problems with the calculation of methylation efficiency using peak areas that could arise from slight differences in HPLC-ICP-MS response for each of the arsenic species. In this case, methylation efficiency is defined as the following:
| 100%([DMAsV] + [MMAsV])/[iAsIII]starting |
These numbers compare reasonably well to the 12% methylation reported by Zakharian and Aposhian.26 However, the concentrations of the reagents used in the two studies were different. In agreement with the Zakharyan and Aposhian26 study, we observed that in the absence of GSH, arsenic methylation did not occur.
As suggested previously,26 it seems that the most likely explanation for the non-enzymatic methylation of arsenic by CH3B12 is that under the highly reducing conditions of this reaction, methylation occurs via nucleophilic attack of an arsenite-GSH complex on the Co–C bond. In support of this is previous work by Hogenkamp et al. demonstrating the heterolytic cleavage of the C–Co bond of methylcobalamin by the nucleophilic thiolate anion under relatively mild conditions.39,40
Conclusions
In this study we have confirmed that CH3B12 acts as a methylating agent for iAsIII in the presence of GSH. This study has been carried out without the use of radioactive chemicals. Instead what are now considered to be routine analytical techniques were used, i.e. HPLC–ICP-MS. This analytical approach is rapid, extremely sensitive, and thus allows for detailed kinetic studies to be carried out without any requirements for radioactive arsenic.However, even though the HPLC–ICP-MS approach has been shown to be extremely powerful for the speciation of arsenic, it is desirable to further confirm the presence of MMAsIII using other independent analytical techniques. In addition, it is our intention to study further the methylation of arsenic by CH3B12, in particular, the mechanism by which it occurs. This will be carried out by employing HPLC–ICP-MS methods that also allow for the speciation of selenium and Co in the various forms of vitamin B12. It is also our intention to monitor the relative amounts of the reduced and oxidized GSH throughout the reaction.
Acknowledgements
This work was supported in part under IAG DW75939361 between the US FDA and the US EPA. S.A.P. and M.M.-R. would like to thank the FDA for funding. M.M.-R. would also like to thank the Engineering and Physical Sciences Research Council (EPSRC) of the United Kingdom for funding (Grant numbers GR/N08974). Finally, we would like to thank Dr John Creed, USEPA for the gift of methyloxoarsine.References
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713 CrossRef.
- B. K. Mandal and K. T. Suzuki, Talanta, 2002, 58, 201 CrossRef CAS.
- National Research Council, Arsenic in Drinking Water, National Academy Press, Washington, DC, 1999 Search PubMed.
- C. Hopenhayn-Rich, M. L. Biggs, A. Fuchs, R. Bergoglio, E. E. Tello, H. Nicolli and A. H. Smith, Epidemiology, 1996, 7, 117 CAS.
- C.-J. Chen, Y.-C. Chuang, T.-M. Lin and H.-Y. Wu, Cancer Res., 1985, 45, 5895 CAS.
- M. E. Cebrian, A. Albores, M. Aguilar and E. Blakely, Human Toxicol., 1983, 2, 121 Search PubMed.
- P. E. Enterline, R. Day and G. M. Marsh, Occup. Environ. Med., 1995, 52, 28 CAS.
- WHO Arsenic compounds, Environmental Health Criteria 224, World Health Organisation, Geneva, 2nd edn., 2001.
- U.S. Environmental Protection Agency, National Primary Drinking Water Regulations; Arsenic and Clarifications to Compliance and New Source Contaminants Monitoring: Final Rule, Fed. Reg. 66, 6976 (January 22, 2001) Search PubMed.
- R. A. Goyer, in The Basic Science of Poisons, ed. C.D. Klaassen, McGraw-Hill, New York, 1996, pp. 696–697 Search PubMed.
- A. B. Fischer, J. P. Buchet and R. R. Lauwerys, Arch. Toxicol., 1985, 57, 168 CAS.
- M. Vahter and E. Marafante, Chem.-Biol. Interact., 1983, 47, 29 Search PubMed.
- X. C. Le, X. Lu, M. Ma, W. R. Cullen, H. V. Aposhian and B. Zheng, Anal. Chem., 2000, 72, 5172 CrossRef CAS.
- X. C. Le, M. Ma, X. Lu, W. R. Cullen, H. V. Aposhian and B. Zheng, Environ. Health Perspect., 2000, 108, 1015 CAS.
- B. K. Mandal, Y. Ogra and K. T. Suzuki, Chem. Res. Toxicol., 2001, 14, 371 CrossRef CAS.
- H. V. Aposhian, E. S. Gurzau, X. C. Le, A. Gurzau, S. M. Healy, X. Lu, M. Ma, L. Yip, R. Zakharyan, R. M. Malorino, R. C. Dart, M. C. Tircus, D. Gonzalez-Ramirez, D. L. Morgan, D. Avram and M. M. Aposhian, Chem. Res. Toxicol., 2000, 13, 693 CrossRef.
- M. J. Mass, A. Tennant, B. C. Roop, W. R. Cullen, M. Styblo, D. J. Thomas and A. D. Kligerman, Chem. Res. Toxicol., 2001, 14, 355 CrossRef CAS.
- J. S. Petrick, B. Jagadish, E. A. Mash and H. V. Aposhian, Chem. Res. Toxicol., 2001, 14, 651 CAS.
- S. M. Cohen, L. L. Arnold, E. Uzvolgyi, M. Cano, M. S. John, S. Yamamoto, X. Lu and X. C. Le, Chem. Res. Toxicol., 2001, 15, 1150 CrossRef CAS.
- M. Styblo, Z. Drobna, I. Jaspers, S. Lin and D. J. Thomas, Env. Health. Perspect., 2002, 110, 767 Search PubMed.
- F. Challenger, Adv. Enzymol., 1951, 12, 429 Search PubMed.
- B. C. McBride and R. S. Wolfe, Biochemistry, 1971, 10, 4312 CrossRef CAS.
- J. P. Buchet and R. Lauwerys, Arch. Toxicol., 1985, 57, 125 CAS.
- M. Styblo, H. Yamauchi and D. J. Thomas, Toxicol. Appl. Pharmacol., 1995, 135, 172 CrossRef CAS.
- M. Styblo, M. Delnomdedieu and D. J. Thomas, Chem.-Biol. Interact., 1996, 99, 147 Search PubMed.
- R. Zakharyan and H. V. Aposhian, Toxicol. Appl. Pharmacol., 1999, 154, 287 CrossRef CAS.
- N. P. Vela, D. T. Heitkemper and K. R. Stewart, Analyst, 2001, 126, 1011 RSC.
- X. C. Le, M. Ma and N. A. Wong, Anal. Chem., 1996, 68, 4501 CrossRef CAS.
- X. C. Le and M. Ma, J. Chromatogr. A., 1997, 764, 55 CrossRef CAS.
- J. Lintschinger, P. Schramel, A. Hatalak-Rauscher, I. Wendler and B. Michalke, Fresenius’ J. Anal. Chem., 1998, 362, 313 CrossRef CAS.
- S. Wangkarn and S. A. Pergantis, J. Anal. At. Spectrom., 2000, 15, 627 RSC.
- C. J. Hwang and S.-J. Jiang, Anal. Chim. Acta, 1994, 289, 205 CrossRef CAS.
- J. Gailer, S. Madden, W. R. Cullen and M. B. Denton, Appl. Organomet. Chem., 1999, 13, 837 CrossRef CAS.
- A. Raab, H. R. Hansen, L. Zhuang and J. Feldmann, Talanta, 2002, 58, 67 CrossRef CAS.
- M. Styblo, M. Delnomdedieu, M. F. Hughes and D. J. Thomas, J. Chromatogr. B, 1995, 668, 21 CrossRef CAS.
- N. Scott, K. M. Hatlelid, N. E. MacKenzie and D. E. Carter, Chem. Res. Toxicol., 1993, 6, 102 CrossRef CAS.
- J. Gailer and W. Lindner, J. Chromatogr. B, 1998, 716, 83 CrossRef CAS.
- S. V. Kala, M. W. Neely, G. Kala, C. I. Prater, D. W. Atwood, J. S. Rice and M. W. Lieberman, J. Biol. Chem., 2000, 275, 33404 CrossRef CAS.
- H. P. Hogenkamp, G. T. Bratt and A. T. Kotchevar, Biochemistry, 1987, 26, 4723 CrossRef CAS.
- H. P. Hogenkamp, G. T. Bratt and S.-Z. Sun, Biochemistry, 1985, 24, 6428 CrossRef CAS.
Footnote |
| † Current address: Department of Chemistry, Environmental Chemical Processes Laboratory, University of Crete, PO Box 1470, 71409, Heraklion, Greece. E-mail: spergantis@chemistry.uoc.gr |
| This journal is © The Royal Society of Chemistry 2004 |