Arsenic–glutathione complexes—their stability in solution and during separation by different HPLC modes
Andrea Raaba, Andrew A. Mehargb, Marcel Jasparsa, David R. Genneyb and Jörg Feldmann*ab
aDepartment of Chemistry, College of Physical Sciences, University of Aberdeen, Meston Walk, Old Aberdeen, Scotland, UK AB24 3UE. E-mail: j. feldmann@abdn.ac.uk
bSchool of Biosciences, University of Aberdeen, St Machar Drive, Aberdeen, Scotland, UK AB24 3UE
First published on 21st October 2003
Abstract
Complexes of arsenic compounds and glutathione are believed to play an essential part in the metabolism and transport of inorganic arsenic and its methylated species. Up to now, the evidence of their presence is mostly indirect. We studied the stability and chromatographic behaviour of glutathione complexes with trivalent arsenic: i.e. AsIII(GS)3, MAIII(GS)2 and DMAIII(GS) under different conditions. Standard ion chromatography using PRP X-100 and carbonate or formic acid buffer disintegrated the complexes, while all three complexes are stable and separable by reversed phase chromatography (0.1% formic acid/acetonitrile gradient). AsIII(GS)3 and MAIII(GS)2 were more stable than DMAIII(GS), which even under optimal conditions tended to degrade on the column at 25 °C. Chromatography at 6 °C can retain the integrity of the samples. These results shed more light on the interpretation of a vast number of previously published arsenic speciation studies, which have used chromatographic separation techniques with the assumption that the integrity of the arsenic species is guaranteed.
Introduction
Arsenic and its species are of great interest today due to their wide occurrence in the environment and their health impact. Oxygen and sulfur compounds of arsenic (inorganic arsenic species) without arsenic–carbon bonds are the most wide-spread in the abiotic environment. The uptake of these compounds into living cells often results in synthesis of organic arsenic species which contain between one and four arsenic–carbon bonds. This synthesis of organo-arsenicals results from the reduction of pentavalent to trivalent arsenic species followed by oxidative formation of an arsenic–carbon bond. For the formation of the next arsenic–carbon bond the arsenic species has to be reduced again to its trivalent form. The formation of dimethylarsinic acid (DMA(V)), the main excretion product of arsenic by humans, requires for example two reduction and two oxidative methylation steps. The exact mechanism of the reduction step is not yet known, but it is thought that in each case, sulfur-containing molecules, like glutathione (GSH) or thioredoxin (TR), and specific enzymes are involved. In mammalian metabolism, glutathione, the most dominant sulfhydryl-containing species in cells, is essential for the reduction of methylarsonic acid (MA(V)). The complex of GSH and arsenite (As(III)) may be required for enzymatic methylation of As(III) to MA(V).1,2 Oxidative formation of arsenic–carbon bonds, normally by methylation, is enzymatically controlled.1The uptake mechanism of inorganic arsenic depends on its valence. Arsenate is taken up via phosphate transporters and As(OH)3via glycerol transporters.3 Arsenic export from cells is known to take place via specific As(III) exporters.4 These exporters might transport arsenite as arsenic–glutathione complexes.4 Mono- and dimethylated arsenic species are probably excreted in a similar way. Biliary excretion studies of arsenic species in rats with and without down regulated GSH production have demonstrated the essential role of GSH for arsenic excretion by bile.5–7 Other studies demonstrate that bile contains mostly trivalent arsenicals and conclude that these must be bound to glutathione because of the high affinity of trivalent arsenicals to sulfur.8 Experiments in cell culture using synthesised arsenic–glutathione complexes show that their toxicity is comparable to or higher than inorganic arsenic and that they are potent inhibitors of certain enzymes.1
Up to now, evidence for the occurrence of arsenic–glutathione complexes in biological tissues is mostly derived indirectly by comparison of results from experiments with and without GSH inhibitors.5,6 In arsenic resistant earthworms (Lumbricus rubellus), arsenic speciation determined by standard methods for extraction (methanol–water) and separation (anion exchange chromatography) show that arsenic is present mainly as As(III); arsenobetaine and As(V) are minor compounds. X-ray absorption spectrometric analysis of these worms shows, however, that arsenic is bound in vivo to sulfur containing species, which are not detectable by HPLC-ICP-MS after extraction.9 The question of correct in vivo identification of arsenic species arising from the earthworm studies led us to investigate the stability of arsenic–glutathione complexes under different separation, extraction and storage conditions using HPLC-ESI-MS and HPLC-ICP-MS in an attempt to find conditions that enable identification and quantification of these complexes.
Over the years, anion and cation exchange chromatography, in addition to ion-pair chromatography, have evolved as the standard separation methods for inorganic and organic arsenicals and are usually used with an ICP-MS as an element specific detector. Since it is necessary to use both an element-specific and a molecular-specific detector for the correct identification of the arsenic–glutathione complexes, the mobile phase of the chosen separation method must be compatible with both ICP-MS and ESI-MS. Since ICP-MS is better suited for non-organic mobile phases and ESI-MS does not give good results using high salt concentrations or non-volatile mobile phases, compromise conditions have to be found and certain techniques can not be used. For these reasons, ion-pair chromatography was incompatible with ESI-MS and therefore not tested further as a separation method for the complexes, despite successful use in combination with ICP-MS for determination of a whole range of arsenicals.10 For the same reason, phosphate buffers must also be avoided. Mobile phases found to be compatible with both detectors consist of carbonate, ammonia, formic (or acetic) acid or pyridine, with as little organic modifier (preferable methanol) as possible. Glutathione with pKa values of 2.1, 3.5 and 9.6 can be expected to present as a mono- and/or divalent anion in solutions between pH 2 and 9. The formation of complexes does not involve peptide carbonyl groups and should consequently not vary the pKa values to a great extent, which allows the use of anion exchange chromatography for the separation of the arsenic–glutathione complexes. Furthermore, cation exchange and more gentle separation techniques like size exclusion and reverse phase separation were also tested in our study.
Here, we describe the stability of three different arsenic glutathione complexes in solution at different pH values, temperatures, in the presence of matrix and under different chromatographic conditions.
Experimental
Chemicals
All reagents used were analytical grade or better quality. Deionised water (18 MΩ Elga, UK) was used throughout the experiments. Arsenic trioxide (As2O3) (BDH, UK), sodium arsenate (Na2HAsO3*7 H2O) (BDH, UK), disodium monomethyl arsonate (MA(V)) (ChemService, USA), sodium cacodylate (DMA(V)) (Strem, USA) and reduced glutathione (GSH) (Sigma) were used for the preparation of standard solutions and for synthesis of the complexes. Monomethylarsine oxide (MA(III)) and dimethylarsinous iodide (DMA(III)) were synthesised according to published procedures from MA(V) and DMA(V) by reduction with sulfur dioxide and, in the case of DMA(III), potassium iodide.11 Methanol and ethanol (both BDH, UK) were used during synthesis and clean-up of complexes. D2O used as solvent for the 1H experiments was purchased from Sigma (USA). For the preparation of the mobile phase solutions ortho-phosphoric acid (85% p.a. BDH, UK), ammonia (25% p.a. BDH, UK), citric acid (p.a. BDH, UK), ammonium carbonate (90% BDH, UK), pyridine (99+% BDH, UK), formic acid (100% p.a. BDH, UK), methanol (Fisher, UK) and acetonitrile (HPLC grade, Rathburn, UK) were used.1H-NMR
A Varian Unity Inova 400 was used for the 1H-NMR measurements with the parameters: relaxation delay 1 s, pulse 44.3 degrees, spectral width 5602.2 Hz at 400 MHz using a line broadening of 0.2 Hz at 25 °C. For the NMR measurements 38–48 mg substance was dissolved in degassed D2O under nitrogen, the tubes were capped immediately and measurements took place within 2 h of preparation.HPLC
The HP1100 HPLC system (Agilent Technologies, USA) with cooled autosampler and peltier controlled column-compartment was used throughout the experiments. For separations using columns other than the reversed phase column the column-compartment was set to ambient temperature, for reversed phase chromatography it was varied between 6 and 30 °C. The autosampler was cooled to 4 °C except where stated otherwise. The HPLC parameters used throughout the experiments were 1 mL min−1 mobile phase flow and 100 µL sample volume. For flow injection measurements the sample volume was 20 µL. Post-column the flow was split in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶4 (1 part into the ICP-MS, 4 parts into the ESI-MS) using a microsplitter (Upchurch, UK).
∶4 (1 part into the ICP-MS, 4 parts into the ESI-MS) using a microsplitter (Upchurch, UK).ESI-MS
An HP1100 series LC/MSD (Agilent Technologies, USA) was used as a molecule-specific detector for post-column detection of the arsenic–glutathione complexes by their molecular peaks (M+H+) or (M+2H)2+. The MSD was used in the positive or negative ionization mode from m/z 120 to m/z 1000 or in the single ion mode with an API-electrospray head. The settings were: capillary voltage 4000 V, nebulizer pressure 40 psi, drying gas flow 12 L min−1 at 350 °C, quadrupole temperature 100 °C and fragmentor voltage 100 V for positive ionization mode. For negative ionization mode the capillary voltage was 3000 V and the nebulizer pressure 35 psi, the other parameters remained the same. These parameters were optimised using a mixture of the glutathione complexes in the FIA mode of the instrument. The mass calibration was controlled regularly and when necessary optimised using the calibration solution supplied by Agilent (m/z 112 to 2233).ICP-MS
An ICP-MS 7500 (Agilent Technologies, USA) was used for element specific detection of arsenic. The instrument was equipped with either a Babington nebulizer or a micro-concentric nebulizer (flow rate < 100 µL min−1), a peltier cooled spray chamber and the option to use oxygen as an additional plasma gas. The instrument was mostly used in the soft extraction mode without oxygen, 3% oxygen was mixed into the nebulizer gas when acetonitrile was used in the mobile phase of the HPLC. The instrument settings were checked daily for arsenic sensitivity and optimised when necessary. The elements monitored were arsenic (m/z 75), tellurium (m/z 130), sulfur (m/z 34) and sulfur-oxide (m/z 48).Synthesis of arsenic–glutathione complexes
Complexes were synthesised according to previously published procedures.12 Briefly, the synthesis involved stirring the reagents for 12 h at room temperature under nitrogen. Tri(glutamylcysteinylglycinyl)trithioarsenite (AsIII(GS)3) was synthesised by dissolving 117 mg Na2HAsO4·7H2O and 0.5 g GSH in 1 mL water (0.38 mmol As to 1.6 mmol GSH). The reaction mixture was then mixed with 3 mL methanol and the resulting precipitate was filtered and dried at room temperature. For di(glutamylcysteinyl-glycinyl)methyl-dithio-arsonite (MAIII(GS)2) 56 mg MA(V) and 0.6 g GSH were dissolved in 2 mL H2O (0.4 mmol As to 1.95 mmol GSH), after which the solution was mixed with 10 mL ethanol. The precipitated product was filtered and dried at room temperature. For γ-glutamylcysteinyl-glycinyl dimethyl thioarsinite (DMAIIIGS) 224 mg DMA(V) were mixed in 18 mL water with 1.6 g GSH (1.39 mmol As to 5.21 mmol GSH), after which the solvent was evaporated at room temperature in a rotor-evaporator. The resulting solid was extracted with methanol and the extract evaporated under reduced pressure in the rotor-evaporator. For the formation of MAIII(GS)2 and DMAIII(GS) complexes, a surplus of glutathione was used since the pentavalent arsenicals must be reduced before complex formation.The reactions follow the equations:
| AsIII(GS)3: 3H+ + AsO33− + 3GSH → AsIII(GS)3 + 3H2O |
| MAIII(GS)2: (CH3)AsO(OH)2 + 4GSH → GS-SG + (CH3)AsIII(GS)2 + 3H2O |
| DMAIII(GS): (CH3)2As(O)OH + 3GSH → GS-SG + (CH3)2AsIIISG + 2H2O |
Stability of arsenic–glutathione complexes in solution
In order to test the stability of AsIII(GS)3, MAIII(GS)2 and DMAIII(GS) under pH conditions of possible mobile phases, their stability was studied in deionised water, 0.1% (v/v) formic acid pH 2.5 and 15 mM ammonium carbonate pH 8.3 using FIA-ESI-MS at room temperature. The mobile phase for FIA was the same as the solvent for the complexes. The solutions were stored in the autosampler at 4 °C or 25 °C and measured every 6 min.Stability of arsenic–glutathione complexes under different chromatographic conditions
The synthesised arsenic complexes were separated using either a strong anion exchange PRP X 100 Hamilton (triethylammonium groups) (150 × 4.1 mm), a cation exchange Supelcosil LC-SCX (sulfonate groups) (250 × 4.1 mm), a silica based size exclusion column TSK G2000 SW (250 × 7.6 mm) or a Waters Spherisorb S5 ODS2 (250 × 4.6 mm) reversed phase column. The separation conditions tested are shown in Table 1. Each column was equilibrated with at least 300 mL mobile phase and the sample was injected three times for determination of retention times and reproducibility. Stock solutions of the complexes (ca. 1 mg mL−1) were prepared every 3 d under nitrogen in 1% formic acid and stored at 1 °C. The working solutions were prepared daily from these stock solutions in 0.1% formic acid except where stated otherwise.| Reversed phase chromatography | column: Spherisorb S5 ODS2 (250 × 4.6 mm) |
| eluent A: 1% formic acid in water | |
| eluent B: methanol | |
| gradient: 0–20 min up to 13% B, then 10 min 100% A | |
| or | |
| eluent A: 0.1% formic acid in water | |
| eluent B: acetonitrile | |
| gradient: 0–20 min up to 5–30% B, then 10 min 5% B | |
| Strong anion exchange chromatography | column: PRP X 100 Hamilton (150 × 4.1 mm) |
| mobile phase: 10 mM citric acid pH 2.0 or 20 mM ammonium carbonate pH 8.0 | |
| Strong cation exchange chromatography | column: Supelcosil LC-SCX (250 × 4.1 mm) |
| mobile phase: 20 mM pyridine pH 2.5 | |
| Size exclusion chromatography | column: TSK G 2000 SW (300 × 7.6 mm) |
| mobile phase: 20 mM pyridine pH 2.5 or 20 mM ammonium carbonate pH 8.0 |
Stability of the arsenic–glutathione complexes during extraction
Since the determination of arsenic species in most biological matrices requires an extraction step before the separation of the species, the stability of arsenic–glutathione complexes under the extraction conditions needs to be evaluated. Methanol/water mixtures are one of the most often used solvents for the extraction of arsenicals from biological samples, in addition a mixture of water/acetonitrile (1∶1 v/v) was tested. Due to the increased stability of the complexes in acidic solution, we also tested 1%
(v/v) formic acid and 2%
(v/v) trifluoroacetic acid (TFA) as extraction media. A non-contaminated sample of the grass Holcus lanatus
(freeze-dried and powdered) was used as the test matrix, which was first mixed thoroughly with a mixture of the synthesized complexes (16–50 µg per 10 mg grass). These spiked samples were than extracted using 1%
(v/v) formic acid, water/methanol (1∶1 v/v), water/acetonitrile (1∶1) or 2%
(v/v) TFA (1 mL solvent per 10 mg sample). The samples were either stored at 4 °C for 24 h and then filtered or extracted using an ultrasonic bath three times for 10 min each; the samples were cooled to 4 °C for 30 min between each ultrasonic step.Results and discussion
Synthesis of arsenic–glutathione complexes
To verify the success of the synthesis 1H-NMR spectra of solutions of the arsenic–glutathione complexes in D2O were measured (Table 2). The spectra were compared to GSH, DMA(V) and MA(V) spectra measured under the same conditions and to previously reported results.12,15 The main shift in the 1H-NMR occurs at the cysteine Hβ protons, the diastereomeric protons split into two multiplets and are shifted to a lower field when compared to GSH, as can be expected when an electronegative atom like arsenic is bound to the neighbouring sulfur. The protons at the methyl groups from DMA bound to GSH were shifted to a higher field and became diastereomeric due to the influence of the GSH. The chemical shifts found by Scott et al. for AsIII(GS)3 and MAIII(GS)2 using 0.5 M phosphate buffer at pH 7.0 are similar to those found by us.12| AsIII(GS)3 | MAIII(GS)2 | DMAIII(GS) |
|---|---|---|
| δ1H(ppm), mult, J(Hz) | δ1H(ppm), mult, J(Hz) | δ1H(ppm), mult, J(Hz) |
| 1.53 s | 1.19 s 1.18 s | |
| 3.07 dd 14.1, 5.1 | 2.98 dd 14.0, 4.4 | 3.00 dd 14.4, 4.8 |
| 3.21 dd 14.4, 7.9 | 3.11 dd 14.0, 7.9 | 3.12 dd 15.0, 7.2 |
From these NMR-data the amount of complex in the solution was calculated from the 1H-signals of unbound glutathione and the complexed glutathione as 83% As(GS)3, 74% MAIII(GS)2 and 50% DMAIII(GS).
For additional verification of the complex formation, solutions of the complexes were injected into the ESI-MS using the FIA-mode. Mass spectra of the complexes are shown in Fig. 1a–f. Using the positive scan mode (Fig. 1a–c), AsIII(GS)3 showed a strong molecular mass peak at m/z 994 [M + H]+ in addition to a signal at m/z 497.5 [M + 2H] 2+. DMAIII(GS) showed signals at m/z 412 [M + H]+, m/z 823 [2M + H]+ and unbound DMA(V) at m/z 139 [M + H]+. The molecular signals of MAIII(GS)2 were at m/z 703 [M + H]+ and m/z 352 [M + 2H] 2+ and, in addition, unbound MA(V) was detected at m/z 141 [M + H]+. Unbound, reduced glutathione (GSH) at m/z 308 [M + H]+ and oxidised glutathione (GSSG) m/z 613 [M + H]+ were detected in all solutions. Using the ESI-MS in negative mode (Fig. 1d–f) AsIII(GS)3 showed the expected molecular peak at m/z 992 [M-H]− in addition to GSH (m/z 306 [M-H]−) and GSSG (m/z 611 [M-H]−); MAIII(GS)2 had a molecular peak at m/z 701[M-H]− in addition to unbound MA(V) at m/z 139 [M-H]− and DMAIII(GS) had a molecular peak at m/z 410 [M-H]− and unbound DMA(V) at m/z 137 [M-H]−. As(III) and As(V) were not detected by ESI-MS under the chosen conditions. In formic acid solutions (pH ≤ 3), AsIII(GS)3 and MAIII(GS)2 preferably formed double charged molecule ions (Fig. 1a–c) while using water or methanol as mobile phase and solvent the single charged molecule ion was the dominant form (data not shown). Positive ionization was used throughout the rest of the experiments, since the negative mode was less sensitive. Neither the intermediates with one GSH attached to MA(III) as MAIIIGS or two GSH attached to As(III) were detected nor the pentavalent arsenic glutathione complexes as proposed by Cullen and coworkers.13,14 Delnomdedieu et al. described the formation of the arsenic–glutathione complex as a two step process, where the first step is the reduction of pentavalent arsenical to its trivalent form, before complex formation takes place.15 They followed the complex formation with NMR at a glutathione concentration of 0.3 mol L−1.
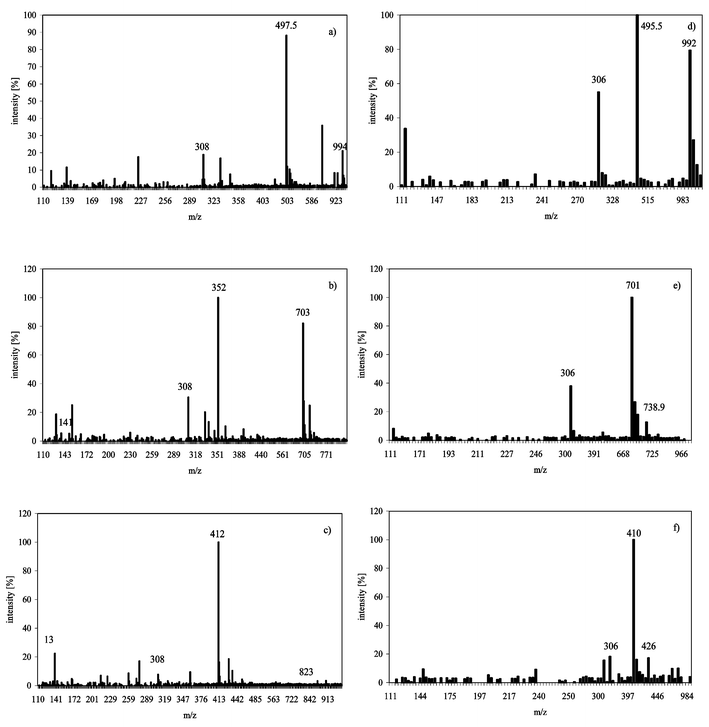 | ||
| Fig. 1 Mass spectra using flow injection ESI-MS of AsIII(GS)3, MAIII(GS)2 and DMAIII(GS) in 0.1% formic acid pH 2.5; positive ionization mode (a) AsIII(GS)3, (b) MAIII(GS)2, (c) DMAIII(GS); negative mode, (d) AsIII(GS)3, (e) MAIII(GS)2, (f) DMAIII(GS). | ||
Stability of arsenic–glutathione complexes in solution
The recovery rates of the different arsenic glutathione complexes at two temperatures in three different mobile phases/pHs are illustrated in Fig. 2a–c. All three complexes dissolved in ammonium carbonate buffer at pH 8.3 started to degrade in less than 5 min. After 10 min, less than 30% of the original signal was still detectable and complexes were completely degraded after 20 min at both 4 °C and 25 °C. No significant difference in the stability of the three complexes was detected.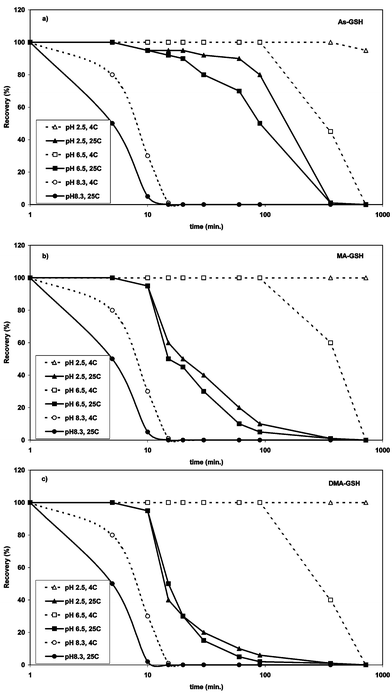 | ||
| Fig. 2 Recovery of (a) AsIII(GS)3, (b) MAIII(GS)2 and DMAIII(GS) (c) in solutions of different pH versus time in dependence of storage temperature. Signals (peak area of m/z 412 (DMAIII(GS)), 703 (MAIII(GS)2) and 994 (AsIII(GS)3)) was normalized to the measurement after 1 min, standard error ca. ± 3%. | ||
When dissolved in deionised water (pH 6.5) and stored at 4 °C for 6 h, DMAIII(GS) was the least stable complex with 60% loss, followed by AsIII(GS)3 with ca. 55% loss and MAIII(GS)2 with ca. 40% loss. At 25 °C, the stability of the complexes was drastically reduced with a loss of ca. 50% of MAIII(GS)2 and DMAIII(GS) after 15 min. AsIII(GS)3 was somewhat more stable at 25 °C, but the loss after 90 min was also more than 50% and after 6 h ca. 99% of all complexes were disintegrated.
At pH 2.5 in 0.1% (v/v) formic acid, the recovery of AsIII(GS)3 was 95% and for DMAIII(GS) and MAIII(GS)2 100% after 12 h. At 25 °C the stability of the complexes was much less. MAIII(GS)2 and DMAIII(GS) seem to be less stable at higher temperatures in acidic solution than AsIII(GS)3 of which ca. 80% was still present after 90 min. After 6 h storage at 25 °C at pH 2.5, less than 1% of the complexes were still detectable. This confirms the results of Delnomdedieu et al.15 who found that AsIII(GS)3 and DMAIII(GS) are stable in solutions with a pH ranging from 1.5 to 7.7 at ambient temperature using NMR measurements. The dissociation of the complexes calculated by them was identical for DMAIII(GS) and AsIII(GS)3. However, no indication was given of how long the complexes were stable. Kala et al.16 studied the stability of the arsenic–glutathione complexes in bile (pH 8.0) over time with HPLC-ESI-MS and found that AsIII(GS)3 is the least stable with a half-life of 20 min, while MAIII(GS)2 and DMAIII(GS) were much more stable with a half-life of approximately 40 min. We found that the half-life of the complexes in ammonium carbonate solution at pH 8.3 was slightly shorter (between 5 to 10 min), one of the reasons might be that bile contains other molecules which can stabilise the complexes.
Stability of arsenic–glutathione complexes under different chromatographic conditions
A comparison of the different separation conditions and their ability to separate AsIII(GS)3, MAIII(GS)2 and DMAIII(GS) is given in Table 1 and Table 3.| GSH m/z 308 | AsIII(GS)3m/z 994 | MAIII(GS)2m/z 703 | DMAIII(GS) m/z 412 | DMA(V) m/z 139 | MA(V) m/z 141 | recovery of total As in % of injected | |
|---|---|---|---|---|---|---|---|
| RP ODS2 C18 acetonitrile gradient | 4.5 min | 9.6 min | 11.3 min | 14.3 min | 3.5/13.4 min | 2.8 min | 100 ± 3% total, 50% as complexes, 50% as DMAIII(GS) , MAIII(GS)2 and As(GS)3 |
| RP ODS2 C18 methanol gradient 1% | 5.2 min | 16.8 min | 21.1 min | no (17.8 min DMA m/z 139) | 3.9 min | 3.0 min | 97 ± 5% total; 57% unbound As, 30% as As(GS)3, 11% as MAIII(GS)2 |
| PRP X 100 pH 8 | no | no | no | no | 3.0 min | 4.7 min | 99 ± 2% as As(III), DMA(V), MA(V) not bound to GSH |
| PRP X 100 pH 2.0 | 3.0 min | no | no | no | 1.7 min | 1.8 min | 101 ± 3% as As(III), DMA(V), MA(V) not bound to GSH |
| Supelcosil pH 2.5 | 3.8 min | 3.9 min | 4.0 min | 4.4 min | 4.2 min | 4.0 min | 100 ± 5%, complexes coelute with unbound arsenic species |
| TSK G2000 SW pH 8 | 8.4 min | no | no | no | 8.8 min | 8.5 min | 92 ± 5% as As(III), DMA(V), MA(V) not bound to GSH |
| TSK G2000 SW pH 2.5 | 10.7 min | 10.2 min | 10.4 min | 11.1 min | 11.2 min | n.d. | 90 ± 5%, complexes in part coelute with unbound arsenic species |
We tested the commonly used strong anion exchange column PRP X 100 from Hamilton with an alkaline and an acidic mobile phase (Fig. 3a and 3b). The use of 15 mM ammonium carbonate (pH 8.0) resulted in a total retardation of GSH, only unbound DMA(V), MA(V), As(III) and As(V) were detectable (Fig. 3a). Using 10 mM citric acid at pH 2.0, GSH, DMA(V), MA(V), As(III) and As(V) eluted from the column, but again there were no detectable complexes (Fig. 3b). The fact that MA(III) was not detectable by ICP-MS or ESI-MS and that the dissociation of the AsIII(GS)3 complex resulted in partial oxidation of trivalent arsenic shows that strong, probably oxidising, interactions occur between the column matrix and the sample molecules. Suzuki et al.17 used 10 mM citric buffer pH 2.0 with a Shodex Asahipak ES-502N 7C column (anion exchange with diethylaminoethyl groups) in combination with ICP-MS detection. They found a slight shift in the retention time of the arsenic species when comparing the retention times of the “complexes” with the retention times of the unbound arsenic species. They did not use a molecule-specific detector so shifts in retention time can never be excluded, especially in a retention time window of only 4 min. Therefore the difference between their results and ours might either not exist because they did not detect the complexes themselves or the different ion exchange groups of the two columns might have a significant influence on the stability of the complexes during separation. Although slight tailing of the peaks indicates disintegration on the column, this tailing is minimal, which indicates that the complexes disintegrated very quickly at the very start of the separation.
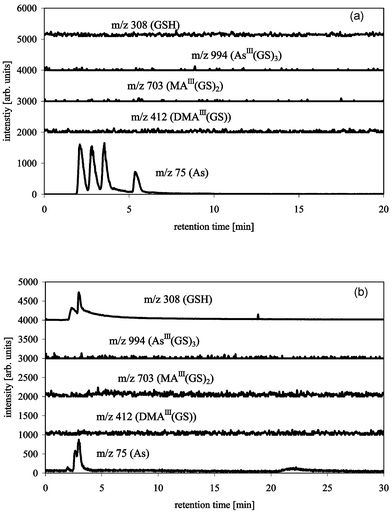 | ||
| Fig. 3 HPLC-ICP-MS/ESI-MS chromatograms of AsIII(GS)3, DMAIII(GS) and MAIII(GS)2 with PRP X 100 Hamilton (150 × 4.1 mm), buffer: 20 mM ammonium carbonate pH 8.0 (a), or 10 mM citric acid pH 2.0 (b), flow 1 mL min−1, m/z 75 (As) measured by ICP-MS, m/z 412 (DMAIII(GS)), 703 (MAIII(GS)2), 994 (AsIII(GS)3) and 308 (GSH) measured by ESI-MS. Peaks 1–4 for m/z 75 are As(III), DMA(V), MA(V) and As(V). | ||
Cation exchange chromatography was tested using a 20 mmol L−1 pyridine buffer at pH 2.5 with a silica-base Supelcosil LC-SCX column (Fig. 4). The complexes seem to elute intact from the column, but as expected, only a slight separation of AsIII(GS)3 and MAIII(GS)2 from DMAIII(GS) was achieved since GSH is negatively charged at the pH used for the separation. However, the complexes co-eluted with the unbound arsenicals, therefore no information about the amount of disintegration of the complexes during separation can be given.
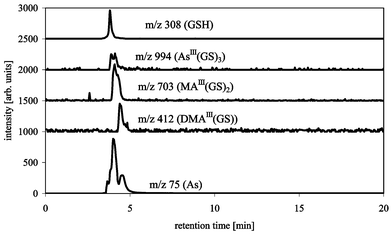 | ||
| Fig. 4 HPLC-ICP-MS/ESI-MS chromatograms of AsIII(GS)3, DMAIII(GS) and MAIII(GS)2 with Supelcosil LC-SCX (250 × 4.6 mm), buffer: 20 mM pyridine pH 2.5, flow 1 mL min−1, m/z 75 (As) measured by ICP-MS, m/z 412 (DMAIII(GS)), 703 (MAIII(GS)2), 994 (AsIII(GS)3) and 308 (GSH) measured by ESI-MS. | ||
Size exclusion chromatography is theoretically the chromatographic technique with the least interactions between sample and column matrix. A TSK G2000 SW column was tested for the separation of the complexes using mobile phases of ammonium carbonate (pH 8.0) or pyridine (pH 2.5). Using a mobile phase of pH 8.0, the complexes were not stable; GSH, DMA(V) and MA(V) coelute and As(III) and As(V) elute later as can be seen from the ICP-MS data (Fig. 5a). Using acidic conditions the complexes elute from the column with a slight separation in the order AsIII(GS)3, MAIII(GS)2, DMAIII(GS) ≈ GSH/GSSG, and inorganic arsenic (Fig. 5b). The same experiments were performed using a polymer based TSK G2000 PW with identical results (data not shown). The column matrix, whether it is silica or polymer, has obviously no influence on the stability of the complexes.
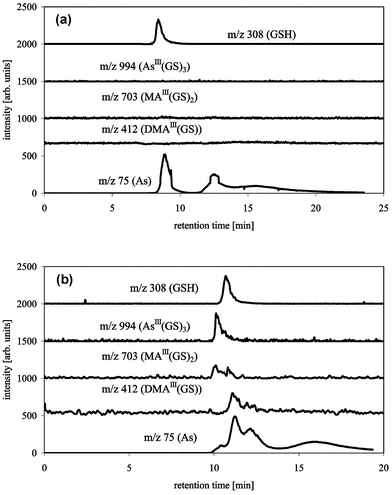 | ||
| Fig. 5 HPLC-ICP-MS/ESI-MS chromatograms of AsIII(GS)3, DMAIII(GS) and MAIII(GS)2 with TSK G2000 SW (300 × 7.6 mm), buffer: 15 mM ammonium carbonate pH 8.0 (a), or 20 mM pyridine pH 2.5 (b), flow 1 mL min−1, m/z 75 (As) measured by ICP-MS, m/z 412 (DMAIII(GS)), 703 (MAIII(GS)2), 994 (AsIII(GS)3) and 308 (GSH) measured by ESI-MS. | ||
Using reverse phase chromatography AsIII(GS)3 and MAIII(GS)2 were separable on a C18 ODS2 column using a methanol–1% formic acid gradient (column-compartment at 30 °C) (Fig. 6a). DMAIII(GS) disintegrated on the column using these separation conditions. This disintegration on the column can be followed by observing DMA(V) on m/z 139, which shows an additional very broad peak that more or less coelutes with AsIII(GS)3 and MAIII(GS)2. This can also be seen in the 75As trace of the ICP-MS (Fig. 6a) as a broad hump under the signals of AsIII(GS)3 and MAIII(GS)2. Changing the gradient and/or the concentration of formic acid (0.1% and 2% were also tested) did not improve recovery of DMAIII(GS) on m/z 412. The only separation condition, so far, which allows separation and detection of all three arsenic–glutathione complexes, is an acetonitrile–0.1% formic acid gradient on a C18 column (Fig. 6b) similar to that described previously.16 The use of a gradient from 5–30% acetonitrile made the addition of 3% oxygen to the plasma gas necessary. The change from methanol to acetonitrile as the organic solvent improved the recovery of DMAIII(GS), but even with acetonitrile DMAIII(GS) partially disintegrated on the column, as can be seen on m/z 139 (DMA(V)) and the 75As trace of the ICP-MS. The stability of DMAIII(GS) during the separation was improved by cooling the column-compartment to 6 °C (Fig. 7). Furthermore, a signal at the mass of the oxidised form of DMAIII(GS) was detectable, whether this oxidation takes place in the interface region of the ESI-MS or on the column needs to be studied further. The instability of trivalent DMA compounds seems to confirm the results from others, e.g., Gong et al.18 found that DMA(III) was less stable than MA(III) in urine. Overall, the chromatographic recovery of arsenic is reasonably good. However, under certain conditions, either the arsenic complexes disintegrate on the column giving non-Gaussian peaks or simply fall apart immediately, so that the complexes elute as different arsenic species, which can lead to misidentification and misinterpretation of the species.
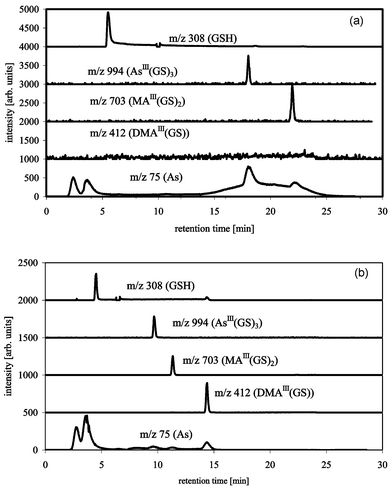 | ||
| Fig. 6 HPLC-ICP-MS/ESI-MS chromatograms of AsIII(GS)3, DMAIII(GS) and MAIII(GS)2 with Waters ODS2 C18, buffer A: 1% formic acid, buffer B: methanol, flow 1 mL min−1, gradient 0–20 min 0–13% B, 20–30 min 0% B (a), or buffer A: 0.1% formic acid, buffer B: acetonitrile, flow 1 mL min−1, gradient 0–20 min 5–30% B, 20–30 min 5% B, column-oven at 6 °C (b); m/z 75 (As) measured by ICP-MS, m/z 412 (DMAIII(GS)), 703 (MAIII(GS)2), 994 (AsIII(GS)3) and 308 (GSH) measured by ESI-MS. | ||
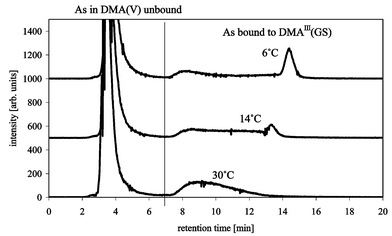 | ||
| Fig. 7 Effect of column temperature on the separation/recovery of DMAIII(GS) with Waters ODS 2 C18, buffer A: 0.1% formic acid, buffer B: acetonitrile, flow 1 mL min−1, gradient 0–20 min 5–30% B, 20–30 min 5% B, m/z 75 (As) measured by ICP-MS. | ||
Detection limits
All experiments to date have used arsenic concentrations of the different arsenic species between 16–8000 µg mL−1. The detection limits for ESI-MS in the positive scan mode using an acetonitrile gradient are 30 µg mL−1 for AsIII(GS)3, 10 µg mL−1 for MAIII(GS)2 and 40 µg mL−1 for DMAIII(GS) based on three times the standard deviation of the blank signal of the biggest molecular peak. Detection limits for the ESI-MS in SIM mode (single ion monitoring) are about a factor of five better than with the scan mode and detection limits in the negative mode are worse than in the positive mode (factor of 5 to 10). Detection limits for arsenic using the ICP-MS in the on-line mode with a flow splitter in the ratio 1∶4 were around 1 ng mL−1.Stability of the complexes during extraction
The different extraction methods produced different results. Stability of the complexes was quantified by using the molecular peaks of the different complexes using ESI-MS. When TFA was used, neither method allowed successful identification of any of the complexes. Using the ultrasonic bath method at ambient temperature with the previously cooled sample, all other extractions (water–methanol, water–acetonitrile or 1% formic acid) show disintegration of DMAIII(GS), confirming again the instability of this complex. However, AsIII(GS)3, MAIII(GS)2 and DMAIII(GS) were all detectable in the extracts made by storing the extracts (water–methanol, water–acetonitrile or 1% formic acid) at 4 °C. The recovery of the spiked DMAIII(GS) was 75%, while AsIII(GS)3 had a recovery of 80% and MAIII(GS)2 of 95% using methanol–water or water–acetonitrile as solvent. The recovery of the complexes is slightly lower with 60% of the spiked MAIII(GS)2 and 50% of AsIII(GS)3 and DMAIII(GS) in 1% formic acid solution.Conclusion
All these experiments about the formation, stability, separation and detection of AsIII(GS)3, MAIII(GS)2 and DMAIII(GS) show that biological samples that may contain these complexes must be treated very carefully. They must be kept permanently cooled in an acidic environment and measured as soon as possible if their detection is expected to be successful. Many papers have published studies on arsenic speciation in which the integrity of the arsenic species has been assumed or tested. Our results indicate that many previously published results must be reinterpreted, since possible arsenic glutathione complexes have not successfully been detected due to the harsh conditions used in “routine” analysis of arsenic species in biological extracts.Acknowledgements
This project has been supported by BBSRC (REI 2002,18479) and AR thanks the College of Physical Sciences for their financial support.References
- R. A. Zakharyan and H. V. Aposhian, Chem. Res. Toxicol., 1999, 12, 1278 CrossRef CAS.
- E. Wildfang, R. A. Zakharyan and H. V. Aposhian, Toxicol. Appl. Pharmacol., 1998, 152, 366 CrossRef CAS.
- O. I. Sanders, C. Rensing, M. Kuroda, B. Mitra and B. P. Rosen, J. Bacteriol., 1997, 179, 3365 CAS.
- B. P. Rosen, Trends Microbiol., 1999, 7, 207 CrossRef CAS.
- Z. Gregus and A. Gyurasics, Biochem. Pharmacol., 2000, 59, 1375 CrossRef CAS.
- A. Gyurasics, F. Varga and Z. Gregus, Biochem. Pharmacol., 1991, 42, 465 CrossRef CAS.
- A. Gyurasics, F. Varga and Z. Gregus, Biochem. Pharmacol., 1991, 41, 937 CrossRef CAS.
- L. Csanaky and Z. Gregus, Comp. Biochem. Physiol., C, 2002, 131, 355 Search PubMed.
- C. J. Langdon, A. A. Meharg, J. Feldmann, T. Balgar, J. Charnock, M. Farquhar, T. G. Piearce, K. T. Semple and J. Cotter-Howells, J. Environ. Monit., 2002, 4, 603 Search PubMed.
- X. C. Le, X. F. Lu, M. S. Ma, W. R. Cullen, H. V. Aposhian and B. S. Zheng, Anal. Chem., 2000, 72, 5172 CrossRef CAS.
- M. J. Mass, A. Tennant, B. C. Roop, W. R. Cullen, M. Styblo, D. J. Thomas and A. D. Kligerman, Chem. Res. Toxicol., 2001, 14, 355 CrossRef CAS.
- N. Scott, K. M. Hatlelid, N. E. Mackenzie and D. E. Carter, Chem. Res. Toxicol., 1993, 6, 102 CrossRef CAS.
- W. R. Cullen, B. C. McBride and J. Reglinski, J. Inorg. Biochem., 1984, 21, 45 CrossRef CAS.
- W. R. Cullen, B. C. McBride and J. Reglinski, J. Inorg. Biochem., 1984, 21, 179 CrossRef CAS.
- M. Delnomdedieu, M. M. Basti, J. D. Otvos and D. J. Thomas, Chem.-Biol. Interact., 1994, 90, 139 Search PubMed.
- S. V. Kala, M. W. Neely, G. Kala, C. I. Prater, D. W. Atwood, J. S. Rice and M. W. Lieberman, J. Biol. Chem., 2000, 275, 33404 CrossRef CAS.
- K. T. Suzuki, T. Tomita, Y. Ogra and M. Ohmichi, Chem. Res. Toxicol., 2001, 14, 1604 CrossRef CAS.
- Z. L. Gong, X. F. Lu, W. R. Cullen and X. C. Le, J. Anal. At. Spectrom., 2001, 16, 1409 RSC.
| This journal is © The Royal Society of Chemistry 2004 |