
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-enabled ortho-C–H olefination of phenylacetic amides with unactivated alkenes†
Ming-Zhu
Lu
a,
Xing-Rong
Chen
a,
Hui
Xu
a,
Hui-Xiong
Dai
 *ab and
Jin-Quan
Yu
*ac
*ab and
Jin-Quan
Yu
*ac
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 LingLing Road, Shanghai 20032, China
bDepartment of Medicinal Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Road Zu Chong Zhi, Zhangjiang Hi-Tech Park, Shanghai, 201203, China. E-mail: hxdai@simm.ac.cn
cDepartment of Chemistry, The Scripps Research Institute, 10550N. Torrey Pines Road, La Jolla, California 92037, USA. E-mail: yu200@scripps.edu
First published on 8th December 2017
Abstract
Although chelation-assisted C–H olefination has been intensely investigated, Pd(II)-catalyzed C–H olefination reactions are largely restricted to acrylates and styrenes. Here we report a quinoline-derived ligand that enables the Pd(II)-catalyzed olefination of the C(sp2)–H bond with simple aliphatic alkenes using a weakly coordinating monodentate amide auxiliary. Oxygen is used as the terminal oxidant with catalytic copper as the co-oxidant. A variety of functional groups in the aliphatic alkenes are tolerated. Upon hydrogenation, the ortho-alkylated product can be accessed. The utility of this reaction is also demonstrated by the late-stage diversification of drug molecules.
Introduction
The use of inert C–H bonds as coupling partners in diverse carbon–carbon and carbon–heteroatom bond-forming reactions has undergone substantial progress in recent years as it provides a straightforward tool to access a variety of valuable molecules from simple hydrocarbon derivatives.1 Most notably, Pd(II)-catalyzed C(sp2)–H bond olefination reactions have been extensively investigated in terms of both catalyst development and mechanistic understanding.2 Despite these significant advances, chelation-assisted C–H olefination reactions have been largely restricted to electronically activated alkenes such as acrylates and styrenes.3 Catalytic C–H olefination reactions with abundant unactivated, aliphatic alkenes remain an unsolved problem because of their intrinsic poor reactivity.4 It is of great synthetic value to develop robust catalytic systems to address these long-standing issues.5–7Our group has focused on the development of weakly coordinating, monodentate directing groups for a diverse range of C–H activation reactions (e.g., alcohols, ethers, amides and carboxylic acids),8 for the following reasons: monodentate substrates have the potential to match a wide range of mono and bidentate ligands to achieve ligand-acceleration; secondly, a wide range of readily available chemicals contain monodentate functional groups that could serve as native directing groups. In 2010, we reported an example of the Pd(II)-catalyzed ortho-C–H activation of phenylacetic acid with linear 1-hexene using a mono-N-protected amino acid (MPAA) ligand.9 However, this process predominantly afforded the non-conjugated allylation product allyl benzene (eqn (1)). The successful example of C–H olefination with unactivated 1-hexene was also reported by our group using a weakly monodentate hydroxyl group (eqn (2)).10 In this case, a mono-N-protected amino acid (MPAA) was used as the ligand to promote this transformation, yet the unsatisfactory efficiency as well as the limited substrate scope substantially hampered the utility of this reaction. Recently, a single example of C–H olefination with unactivated olefins using the Daugulis’ bidentate directing group was disclosed (eqn (3)).11 To reach our goal of using versatile ligands and potentially a broad range of monodentate substrates, we pursued further development of the Pd(II)-catalyzed C–H olefination reaction directed by weakly monodentate auxiliaries with unactivated alkenes by identifying an effective ligand. The success of pyridine ligands in C–H activation inspired us to revisit the C(sp2)–H olefination with unbiased alkenes.12 Herein, we report the Pd(II)-catalyzed ortho-C–H olefination of phenylacetic amides assisted by a weak amide group (eqn (4)). The identified monodentate quinoline-based ligand is found to be crucial for this olefination to proceed smoothly. This protocol is compatible with a wide range of simple as well as functionalized aliphatic alkenes, leading to diverse synthetically valuable β-alkylated styrenes that are frequently encountered in complex natural products (Scheme 1A).13 Importantly, the use of molecular oxygen with a catalytic Cu(II) salt as the co-oxidant is a practical advantage compared to the costly silver oxidants required in related palladium chemistry.14 The utility of this protocol was also demonstrated by the late-stage diversification of drug molecules.
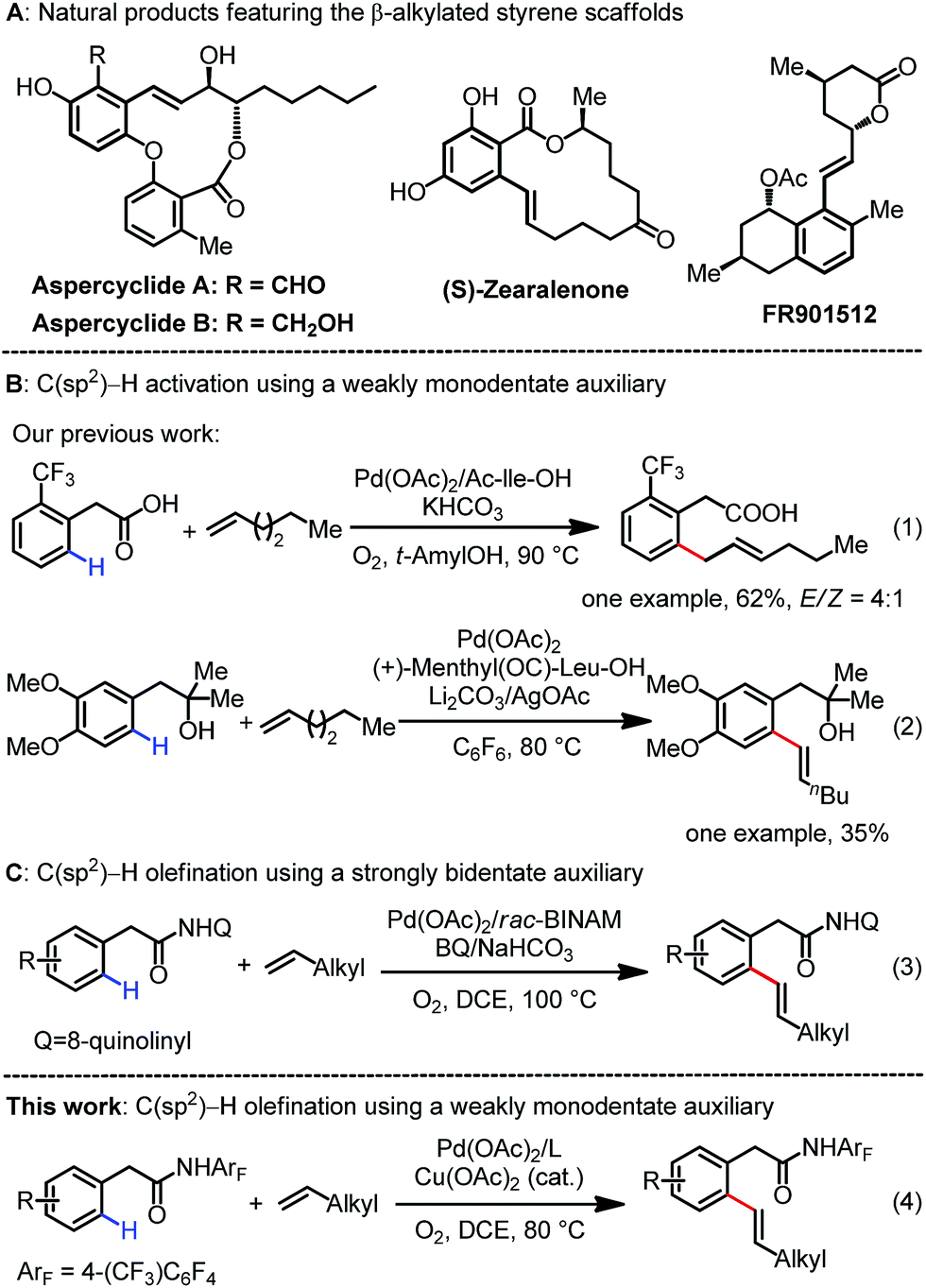 | ||
| Scheme 1 Pd(II)-catalyzed C–H functionalization with unactivated alkenes. | ||
Results and discussion
We began our initial investigation by screening various weakly coordinating groups derived from phenylacetic acid that can facilitate the distal C(sp2)–H activation via six-membered cyclopalladation. Encouragingly, we found that phenylacetic-acid-derived amide 1 is the most promising substrate for this reaction (see the ESI†). After extensive screening of the various parameters, we established that the treatment of amide 1 with 3 equiv. of linear 1-octene, 2.0 equiv. of Cu(OAc)2, 10 mol% Pd(OAc)2, and 20 mol% of the quinoline-based ligand 21 in DCE at 80 °C for 6 h provides the desired product 3a in 85% isolated yield (linear/branched = 3.8/1) (Table S6†). A control experiment showed that the ligand is indispensable for this reaction, as only a trace amount of the olefination product was detected without addition of the ligand (Table 1). This clearly demonstrates the importance of the interaction between ligand acceleration and the weak coordination substrates. With the preliminary conditions established, we proceeded to systematically re-examine the pyridine- and quinoline-based ligands in an effort to improve the efficiency of this reaction. This study revealed that both pyridine L1 and 2-picoline L2 could furnish the olefinated product in good yield, whereas the pyridine ligand bearing an electron-withdrawing group at the 2-position (L3) was found to be inactive, probably due to its poor coordinating ability. Pyridines bearing one electron-donating substituent at other positions (L4–L6) delivered the product in moderate yields (55–64%). Among the di- and trisubstituted pyridine ligands tested (L7–L12), ligands containing a methyl group at the 2-position performed well with good yields (up to 83%). However, the sterically bulky 2,6-di-tert-butylpyridine ligand (L11) has a fatal effect on this cross-coupling reaction. Inspired by our recent progress in developing different types of quinoline-based ligand for C–H functionalization reactions,15 we further turned to survey this class of ligand. Gratifyingly, the simple quinoline (L13) was effective in giving the best result (79%), while other substituted quinoline-based ligands were detrimental to this reaction (L14–L18). Acridine (L19) and phenanthridine (L20) failed to increase the efficiency, and the product was obtained in 70% and 46% yields, respectively. In terms of electronic effects, the more electron-rich tricyclic quinoline ligand (L22) with a tert-butyl group installed at the 6-position was further identified as the optimal ligand to accelerate this reaction, allowing the formation of the olefinated product in 88% yield. Conversely, bidentate ligands such as 2,2′-bipyridine (L23) and 1,10-phenanthroline (L24) completely inhibited this reaction. Furthermore, we continued to optimize the parameters to search for more practical conditions. We were pleased to find that this olefination reaction can also be conducted with a catalytic amount of Cu(OAc)2 (0.2 equiv.) under an oxygen atmosphere without any decrease in the isolated yield (Table S7†). Finally, reducing the catalyst loading to 5 mol% did not affect the reactivity, affording the product in 87% yield.| a Reaction conditions: phenylacetic amide 1 (0.1 mmol), 1-octene 2a (3.0 equiv.), Pd(OAc)2 (10 mol%), ligand (20 mol%), Cu(OAc)2 (2.0 equiv.), DCE (2.0 mL), 80 °C, 6 h. b Isolated yields. c The data in parentheses is the ratio of linear and branched isomers determined using 1H NMR analysis. d Pd(OAc)2 (5 mol%), ligand (10 mol%), and Cu(OAc)2 (0.2 equiv.) were used under an O2 atmosphere. |
|---|
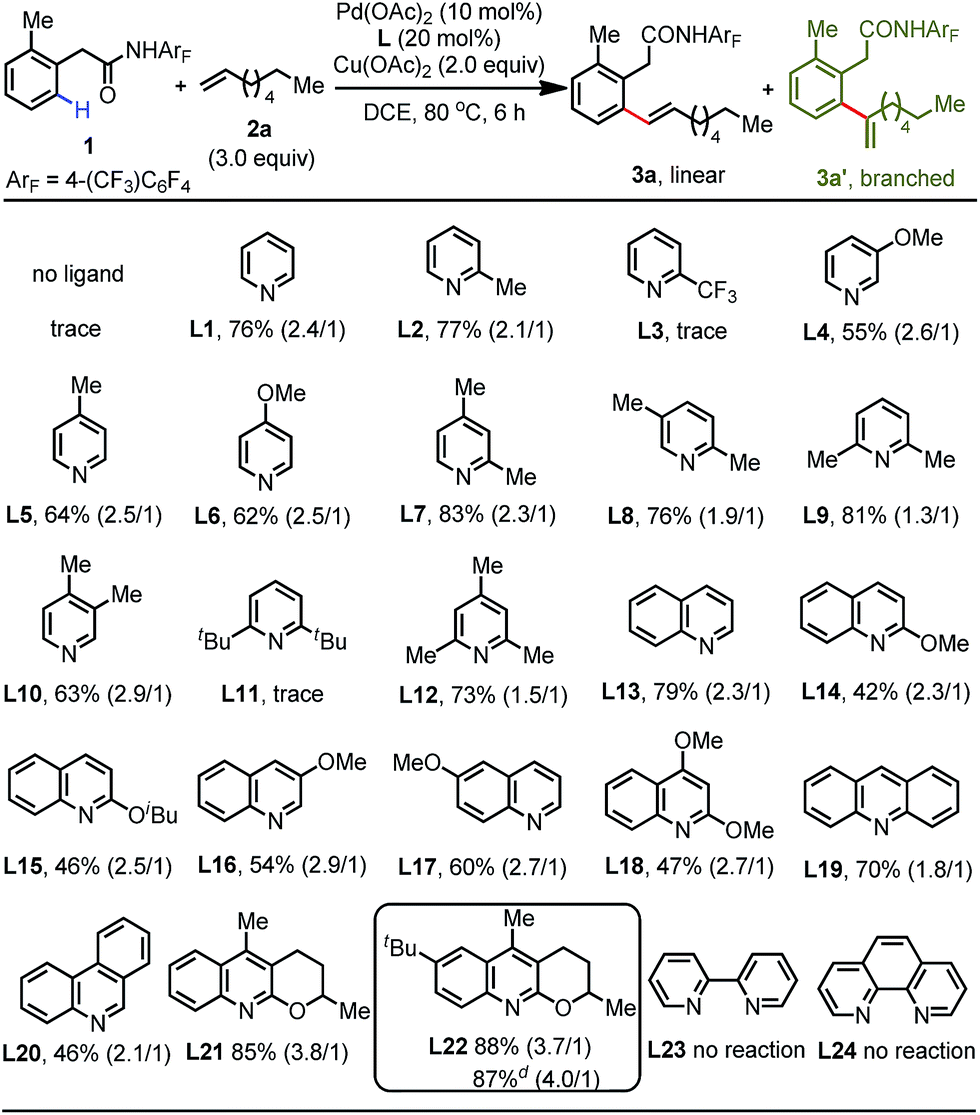
|
With the optimal conditions established, we next proceeded to explore the substrate scope of this reaction with respect to simple aliphatic alkenes (Table 2). This protocol is compatible with a variety of terminal unactivated alkenes, affording the corresponding olefinated products in excellent yields (83–92%) with moderate linear/branched selectivity (3a–3d). Alkenes bearing branched alkanes are also efficient coupling partners for this process (3e–3h). It is noteworthy that the regioselectivity improved with the increased steric hindrance of branched alkenes. For instance, the sterically bulky 3,3-dimethylbut-1-ene reacted smoothly under standard conditions to give the single isomer olefination product, albeit in moderate yield (3i).
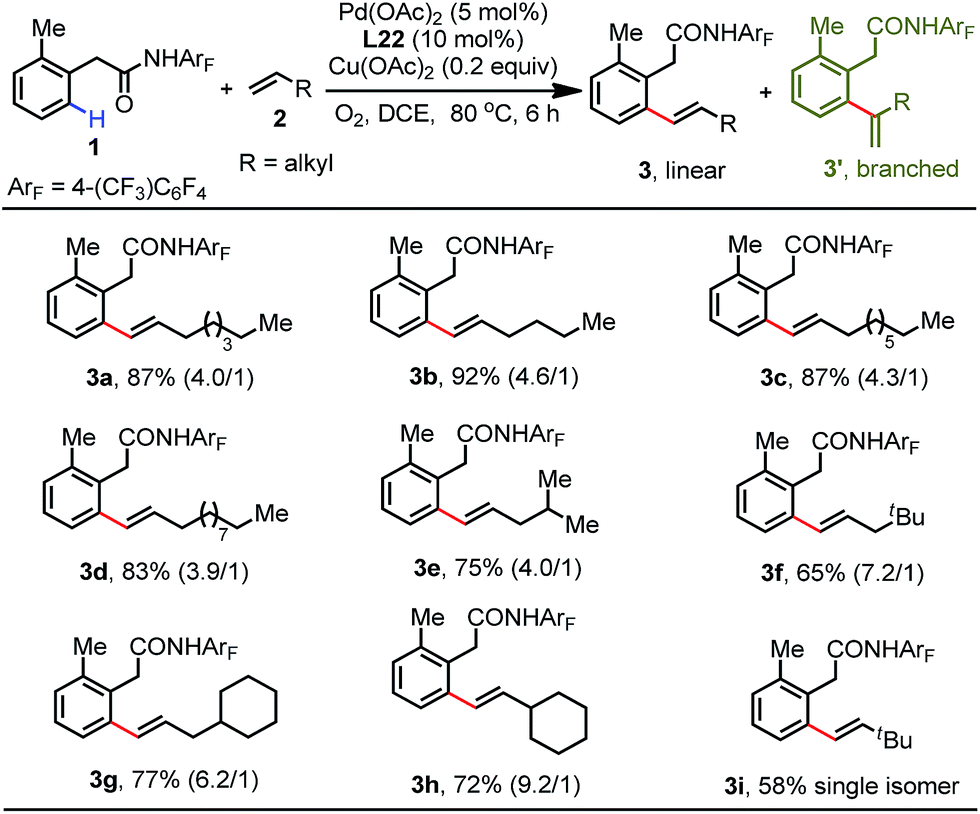
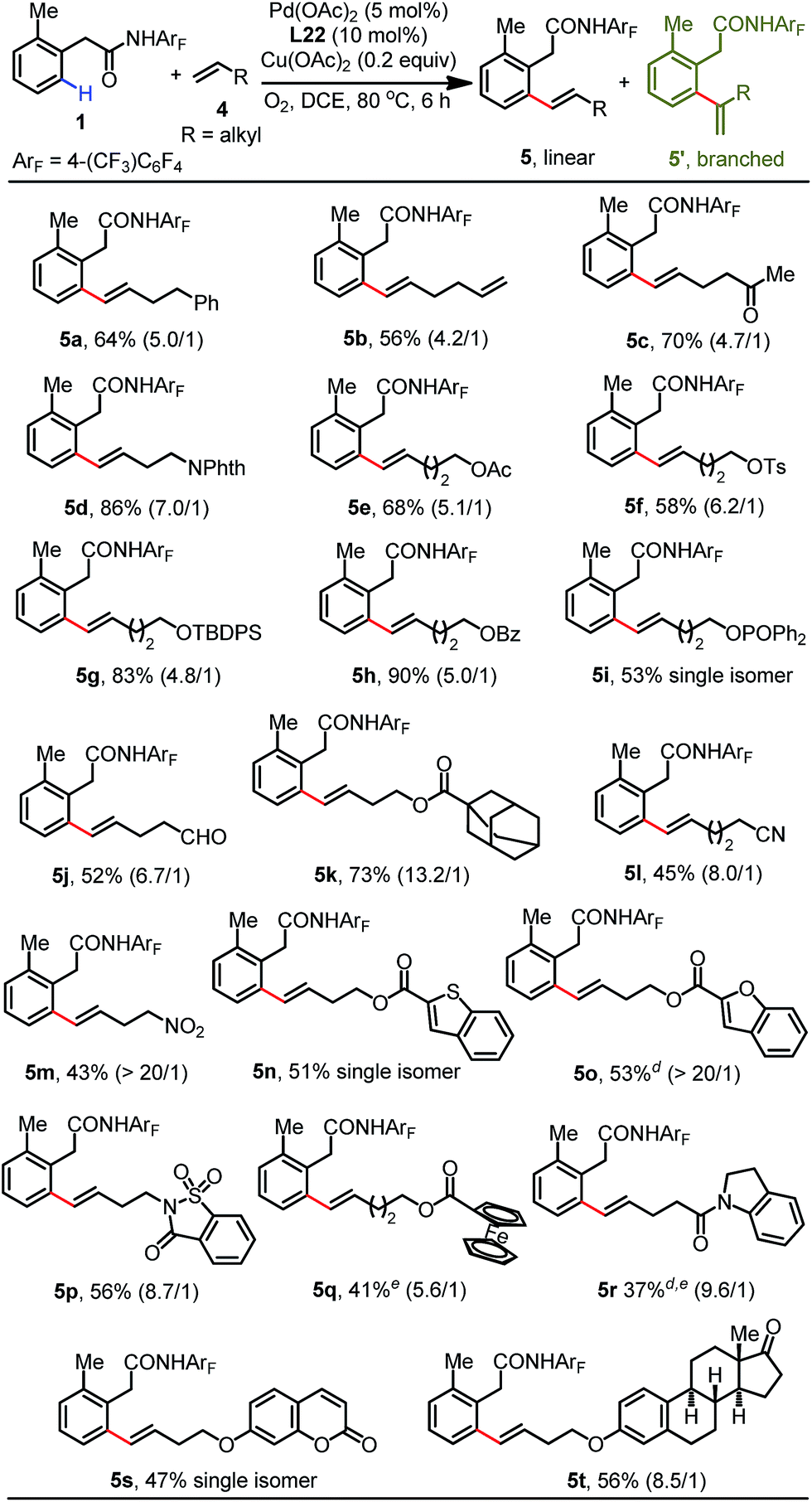
We subsequently proceeded to investigate the generality of this protocol with a wide range of functionalized aliphatic alkenes (Table 3). A variety of aliphatic alkenes can be converted into the corresponding olefination products in modest to excellent yields (5a–5k). Excitingly, the mild conditions allowed for high functional group compatibility, and many synthetically useful functionalities were well-tolerated with these conditions without any compromise (5c–5k). However, substrates containing electron-deficient coordinating groups such as nitrile (5l) and nitro (5m) groups exhibited lower reactivity to produce the products in 45% and 43% yields, respectively. Substrates containing heterocycles were also suitable to generate products in moderate yields (5n–5r). Moreover, olefins derived from coumarin and estrone proceeded smoothly to produce the olefinated products in acceptable yields with excellent regioselectivity (5s and 5t). It should be noted that the ortho-C–H allylation products were detected in trace amounts for most of the substrates.
| a Reaction conditions: phenylacetic amide 1 (0.1 mmol), alkene 4 (3.0 equiv.), Pd(OAc)2 (5 mol%), L22 (10 mol%), Cu(OAc)2 (0.2 equiv.), DCE (2.0 mL), 80 °C, O2, 6 h. b Isolated yields. c The data in parentheses is the ratio of linear and branched isomers determined using 1H NMR analysis. d At 100 °C. e 12 h. |
|---|
|
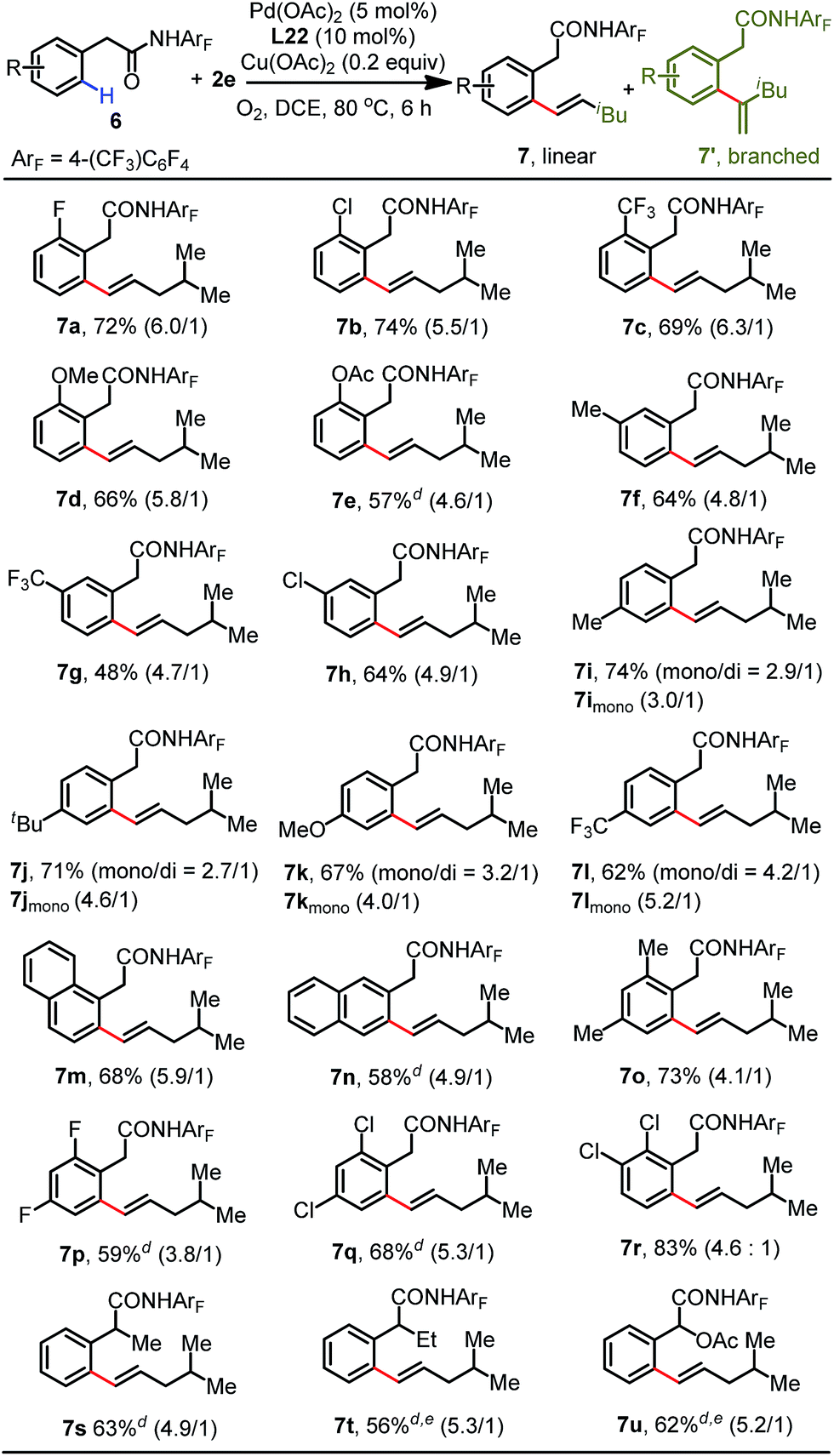
Various phenylacetic amide substrates were next subjected to the standard conditions to further evaluate the scope of this protocol (Table 4). A variety of phenylacetic amides bearing both electron-donating and -withdrawing substituents at the ortho-position were successfully olefinated to give the corresponding products in yields from 57% to 74% (7a–7e). Substrates bearing methyl (6f), trifluoromethyl (6g) and chloro (6h) groups at the meta-position were well-tolerated with this protocol, providing the less sterically hindered olefination products. The reaction of para-substituted phenylacetic amides proceeded smoothly, and the olefination products (7i–7l) were isolated in good yields (62–74%). Moreover, the reaction of a 1-naphthalene substrate with the aliphatic olefin occurred to generate 7m in 68% yield. It is worth noting that the 2-naphthalene substrate showed high site-selectivity in this process, exclusively delivering the β-olefinated product 7n. Disubstituted phenylacetic amides were also proven to be favorable for this transformation, enabling the formation of desired products 7o–7r in good yields (68–83%). Lastly, α-substituted substrates underwent this olefination reaction to give the corresponding products in modest yields (7s–7u).16
| a Reaction conditions: phenylacetic amide 6 (0.1 mmol), 2e (3.0 equiv.), Pd(OAc)2 (5 mol%), L22 (10 mol%), Cu(OAc)2 (0.2 equiv.), DCE (2.0 mL), 80 °C, O2, 6 h. b Isolated yields. c The data in parentheses is the ratio of linear and branched isomers determined using 1H NMR analysis. d At 100 °C. e 12 h. |
|---|
|
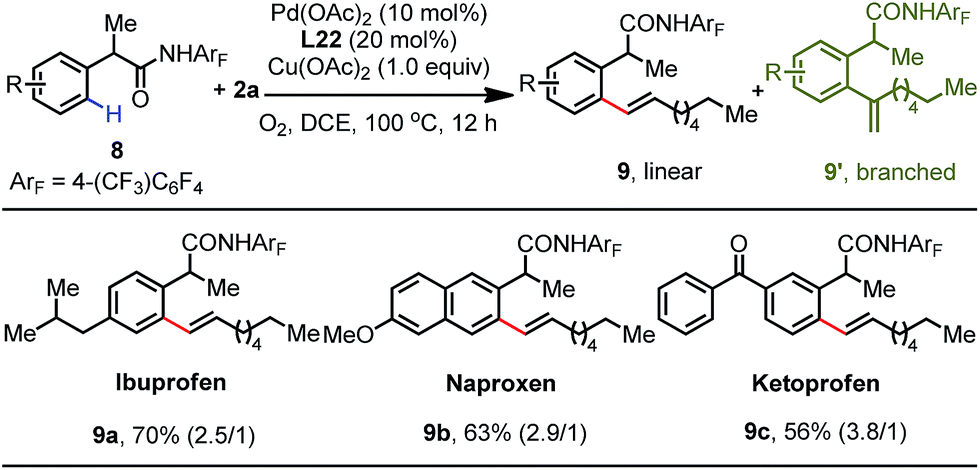
To showcase the synthetic utility of this protocol, we sought to apply this method to the late-stage diversification of drug molecules (Table 5). Gratifyingly, phenylacetic amides derived from ibuprofen 8a, naproxen 8b and ketoprofen 8c were smoothly olefinated to give the corresponding olefination products 9a–9c in 70%, 63% and 56% yields, respectively.16
To further investigate the feasibility of this transformation, we carried out a gram scale ortho-C(sp2)–H olefination reaction of substrate 1 with 1-octene 2a. The desired olefination product was isolated in 77% yield when the reaction was performed on a 3.0 mmol scale (Scheme 2). Furthermore, hydrogenation and deprotection reactions were conducted. The hydrogenation of the linear olefination product 3a at room temperature provided the ortho alkylation product 10 in a quantitative yield (Scheme 3). Moreover, the amide auxiliary could be readily removed by treatment with BF3·E2O in MeOH, affording the methyl ester 11 in 83% yield (Scheme 3).
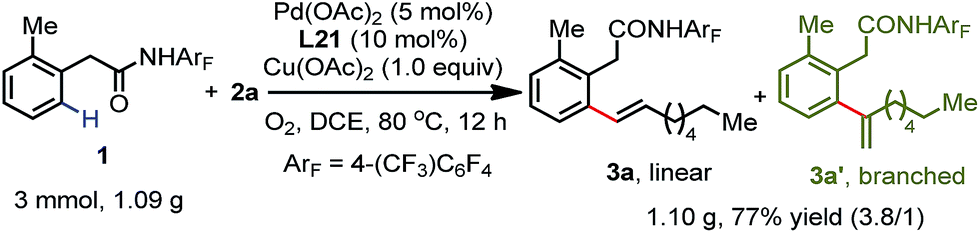 | ||
| Scheme 2 Gram-scale synthesis. | ||
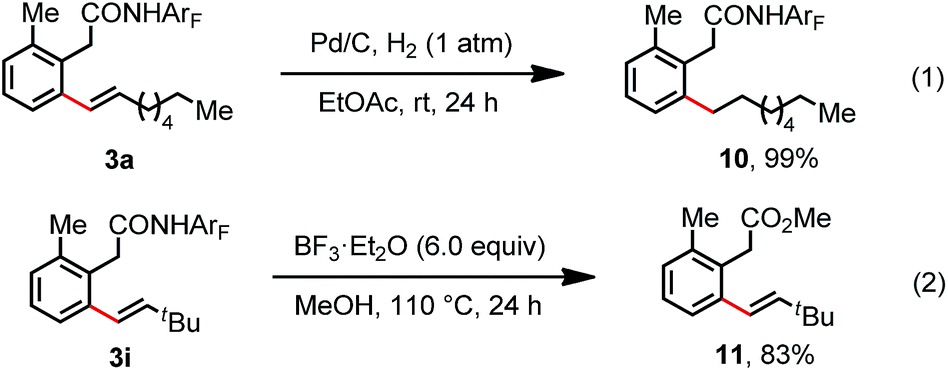 | ||
| Scheme 3 Hydrogenation and deprotection. | ||
A plausible mechanism for this Pd(II)-catalyzed ortho-C(sp2)–H olefination with aliphatic alkenes is proposed (Scheme 4).4 The C–H activation and further olefin coordination steps form intermediate A, which undergoes subsequent 1,2-migratory insertion with the aliphatic olefin to produce the eight-membered palladacycle intermediate B. In contrast to the coordination of the carboxylate directing group to generate the allylated product, the amide group may be likely to undergo fast dissociation to afford alkylpalladium species C, which makes bond rotation necessary for syn elimination. β-Hydride elimination with the benzylic hydrogen atom leads to the formation of kinetically and thermodynamically favored olefinated products.
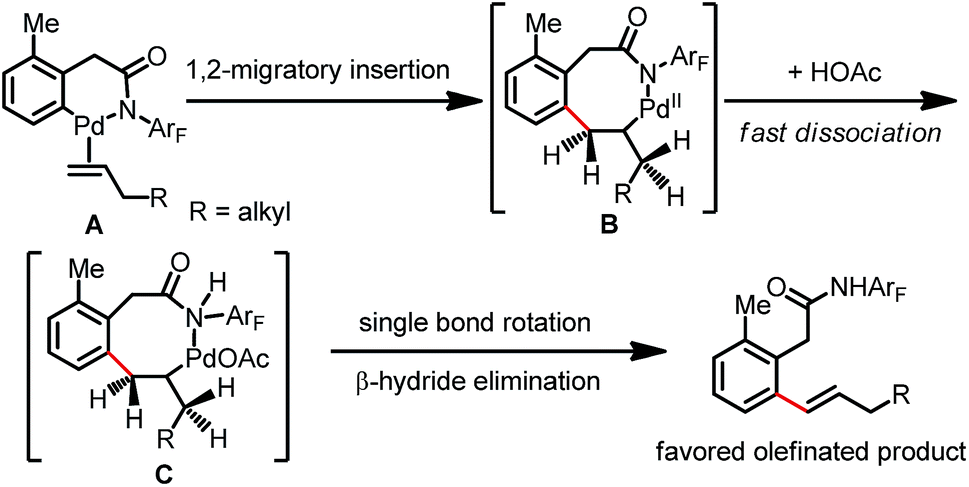 | ||
| Scheme 4 Plausible mechanism for the formation of olefinated products. | ||
Conclusions
In summary, we have developed a Pd(II)-catalyzed ortho-C(sp2)–H olefination of phenylacetic acid derivatives with unactivated aliphatic alkenes using a monodentate amide coordinating group. The identified quinoline-type ligand is found to be crucial for enabling this transformation. Both simple and functionalized aliphatic alkenes can be converted into the valuable β-alkylated styrene derivatives. Importantly, this catalytic system employs molecular oxygen as the terminal oxidant. We anticipate that the use of monodentate directing groups and ligands will lead to further development of this transformation to include a wide range of readily available monodentate substrates.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the Shanghai Institute of Organic Chemistry, the Chinese Academy of Sciences, the CAS/SAFEA International Partnership Program for Creative Research Teams, NSFC-21121062, NSFC-21472211, NSFC-21421091, the Youth Innovation Promotion Association CAS (No. 2014229), and The Recruitment Program of Global Experts for financial support. We also acknowledge The Scripps Research Institute and NSF under the CCI Center for Selective C–H Functionalization, CHE-1205646 for their generous financial support.References
- For selected reviews on C–H functionalization, see: (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) S. D. Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef; (e) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (f) G. Shi and Y. Zhang, Adv. Synth. Catal., 2014, 356, 1419 CrossRef CAS; (g) Z. Huang, H. M. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC; (h) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS PubMed; (i) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (j) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635 CrossRef CAS PubMed; (k) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003 CrossRef CAS PubMed; (l) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (m) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed; (n) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed.
- For examples of Pd(II)-catalyzed C–H olefination with activated alkenes, see: (a) X. Ye and X. Shi, Org. Lett., 2014, 16, 4448 CrossRef CAS PubMed; (b) N. Dastbaravardeh, T. Toba, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 9877 CrossRef CAS PubMed; (c) Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617 CrossRef CAS PubMed; (d) C. He and M. J. Gaunt, Chem. Sci., 2017, 8, 3586 RSC; (e) S.-X. Li, Y.-N. Ma and S.-D. Yang, Org. Lett., 2017, 19, 1842 CrossRef CAS PubMed.
- For selected examples, see: (a) Z.-K. Wen, Y.-H. Xu and T.-P. Loh, Chem. Sci., 2013, 4, 4520 RSC; (b) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760 CrossRef CAS PubMed; (c) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515 CrossRef CAS PubMed; (d) S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed; (e) S. Maity, E. Hoque, U. Dhawa and D. Maiti, Chem. Commun., 2016, 52, 14003 RSC; (f) H.-J. Xu, Y. Lu, M. E. Farmer, H.-W. Wang, D. Zhao, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 2200 CrossRef CAS PubMed; (g) S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888 CrossRef CAS PubMed.
- For C–H olefination with aliphatic alkenes, see: (a) K.-H. Kwon, D. W. Lee and C. S. Yi, Organometallics, 2009, 28, 4266 CrossRef PubMed; (b) A. S. Tsai, M. Brasse, R. G. Bergman and J. A. Ellman, Org. Lett., 2011, 13, 540 CrossRef CAS PubMed; (c) X. Li, X. Gong, M. Zhao, G. Song, J. Deng and X. Li, Org. Lett., 2011, 13, 5808 CrossRef CAS PubMed; (d) C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 10625 CrossRef CAS PubMed; (e) N. Gigant and J.-E. Bäckvall, Org. Lett., 2014, 16, 4432 CrossRef CAS PubMed; (f) R. Manoharan, G. Sivakumar and M. Jeganmohan, Chem. Commun., 2016, 52, 10533 RSC; (g) X. Xue, J. Xu, L. Zhang, C. Xu, Y. Pan, L. Xu, H. Li and W. Zhang, Adv. Synth. Catal., 2016, 358, 573 CrossRef CAS; (h) Y. Takahama, Y. Shibata and K. Tanaka, Chem.–Eur. J., 2015, 21, 9053 CrossRef CAS PubMed.
- For C–H allylation with aliphatic alkenes, see: (a) Y. Takahama, Y. Shibata and K. Tanaka, Org. Lett., 2016, 18, 2934 CrossRef CAS PubMed; (b) T. Yamaguchi, Y. Kommagalla, Y. Aihara and N. Chatani, Chem. Commun., 2016, 52, 10129 RSC; (c) S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparkar and D. Maiti, ACS Catal., 2016, 6, 5493 CrossRef CAS; (d) S. Maity, P. Dolui, R. Kancherla and D. Maiti, Chem. Sci., 2017, 8, 5181 RSC.
- For selected examples of hydroarylation with aliphatic alkenes via C–H functionalization, see: (a) N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935 CrossRef CAS PubMed; (b) M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977 CrossRef CAS PubMed; (c) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098 CrossRef CAS PubMed; (d) D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 10378 CrossRef CAS PubMed; (e) A. T. Tran and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10530 CrossRef CAS PubMed.
- For a recent review on unactivated alkenes, see: A. Deb and D. Maiti, Eur. J. Org. Chem., 2017, 1239 CrossRef CAS.
- For selected examples, see: (a) X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203 CrossRef CAS PubMed; (b) G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS PubMed; (c) X.-G. Zhang, H.-X. Dai, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 11948 CrossRef CAS PubMed.
- (a) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed; (b) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137 CrossRef CAS PubMed; (c) K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 6169 CrossRef CAS PubMed.
- Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed.
- (a) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602 CrossRef CAS PubMed; (b) A. Deb, A. Hazra, Q. Peng, R. S. Paton and D. Maiti, J. Am. Chem. Soc., 2017, 139, 763 CrossRef CAS PubMed; (c) K. Seth, M. Bera, M. Brochetta, S. Agasti, A. Das, A. Gandini, A. Porta, G. Zanoni and D. Maiti, ACS Catal., 2017, 7, 7732 CrossRef CAS.
- (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (b) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed.
- (a) J. Pospíšil, C. Müller and A. Fürstner, Chem.–Eur. J., 2009, 15, 5956 CrossRef PubMed; (b) T. Yoshino, I. Sato and M. Hirama, Org. Lett., 2012, 14, 4290 CrossRef CAS PubMed; (c) M. Inoue and M. Nakada, J. Am. Chem. Soc., 2007, 129, 4164 CrossRef CAS PubMed; (d) B. Cramer, M. Bretz and H.-U. Humpf, J. Agric. Food Chem., 2007, 55, 8353 CrossRef CAS PubMed.
- (a) Y. Deng and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888 CrossRef CAS PubMed; (b) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS PubMed; (c) H. Jiang, J. He, T. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 2055 CrossRef CAS PubMed.
- (a) R.-Y. Zhu, K. Tanaka, G.-C. Li, J. He, H.-Y. Fu, S.-H. Li and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 7067 CrossRef CAS PubMed; (b) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed; (c) R.-Y. Zhu, T. G. Saint-Denis, Y. Shao, J. He, J. D. Sieber, C. H. Senanayke and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 5724 CrossRef CAS PubMed.
- For substrates 6f–6h, 6s–6u and 8a, the corresponding diolefinated products were detected (<5% yield).
Footnote |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. See DOI: 10.1039/c7sc04827k |
| This journal is © The Royal Society of Chemistry 2018 |