
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Imine-based [2]catenanes in water†
Kenji
Caprice
 a,
Marion
Pupier
a,
Anneli
Kruve
b,
Christoph A.
Schalley
b and
Fabien B. L.
Cougnon
*a
a,
Marion
Pupier
a,
Anneli
Kruve
b,
Christoph A.
Schalley
b and
Fabien B. L.
Cougnon
*a
aDepartment of Organic Chemistry, University of Geneva, 30 Quai Ernest Ansermet, 1211 Geneva 4, Switzerland. E-mail: Fabien.cougnon@unige.ch
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustraße 3, 14195 Berlin, Germany
First published on 18th December 2017
Abstract
We report the efficient condensation of imine-based macrocycles from dialdehyde A and aliphatic diamines Bn in pure water. Within the libraries, we identified a family of homologous amphiphilic [2]catenanes, whose self-assembly is primarily driven by the hydrophobic effect. The length and odd-even character of the diamine alkyl linker dictate both the yield and the conformation of the [2]catenanes, whose particular thermodynamic stability further shifts the overall equilibrium in favour of imine condensation. These findings highlight the role played by solvophobic effects in the self-assembly of complex architectures.
Introduction
The synthesis of topologically complex molecules continues to challenge chemists,1,2 prompting the need to develop new efficient synthetic strategies. In the literature, a few examples suggest that solvophobic effects could provide a powerful driving force to direct the assembly of catenanes and knots.3–7 Most notably, it was serendipitously discovered that amphiphilic macrocycles can adopt non-trivial topologies in water3–5 in order to minimize their hydrophobic surface area exposed to the solvent. The role of the hydrophobic effect in this process is poorly understood and it is not yet possible to predict the formation of complex topologies in such systems.We describe here the dynamic combinatorial8 synthesis of imine-based [2]catenanes in water from a dialdehyde (A, Fig. 1) and a series of homologous aliphatic diamines Bn (n = 4 to 9 denotes the number of CH2 groups). Building block A is composed of two water-soluble, electron-deficient aromatic surfaces (isoquinolinium bromide) connected by a p-xylylene linker and terminated by two aldehydes. Closing A with a hydrophobic aliphatic diamine provides amphiphilic macrocycles, wherein permanent positive charges alternate with lipophilic units. Our system was specifically designed to probe whether the hydrophobicity of a purely aliphatic chain is sufficient to drive the formation of interlocked structures in the absence of motifs that favour more traditional supramolecular interactions (e.g. metal coordination, π–π donor–acceptor interactions or cooperative hydrogen bonds) and in spite of the charge repulsion between the isoquinonium moieties of A. In this context, the use of imine condensation presents several advantages. First, aliphatic diamines of various lengths are commercially available, allowing us to study easily the effect of chain length on the product distribution in the dynamic combinatorial libraries. In addition, aliphatic diamines display minimum hydrophilicity after condensation. The reversibility of the imine bond has been extensively used to produce complex supramolecular architectures.9 In water, the condensation of an aldehyde and an amine is generally disfavoured10,11 but can be high yielding in some cases. In nature, a mechanism central to our visual cycle involves the formation of a Schiff base between the retinal chromophore and a lysine residue positioned in the hydrophobic cavity of opsin.12 In the laboratory, imines can also be stabilized in water by metal coordination,13 within the core of micelles,14 or in the presence of an adjacent boronate.15 In most other situations, chemists have preferred the related, more stable acyl hydrazones and oximes16 over imines. With isoquinolinium-based aldehydes, the imine bond can form rather efficiently in water (condensation yield >60%, Fig. S3–S10†). However, the equilibrium is very sensitive to the experimental conditions. In agreement with the literature,11a we preliminarily assessed that imine condensation was most efficient at higher concentrations of building blocks, higher pH and lower temperatures.
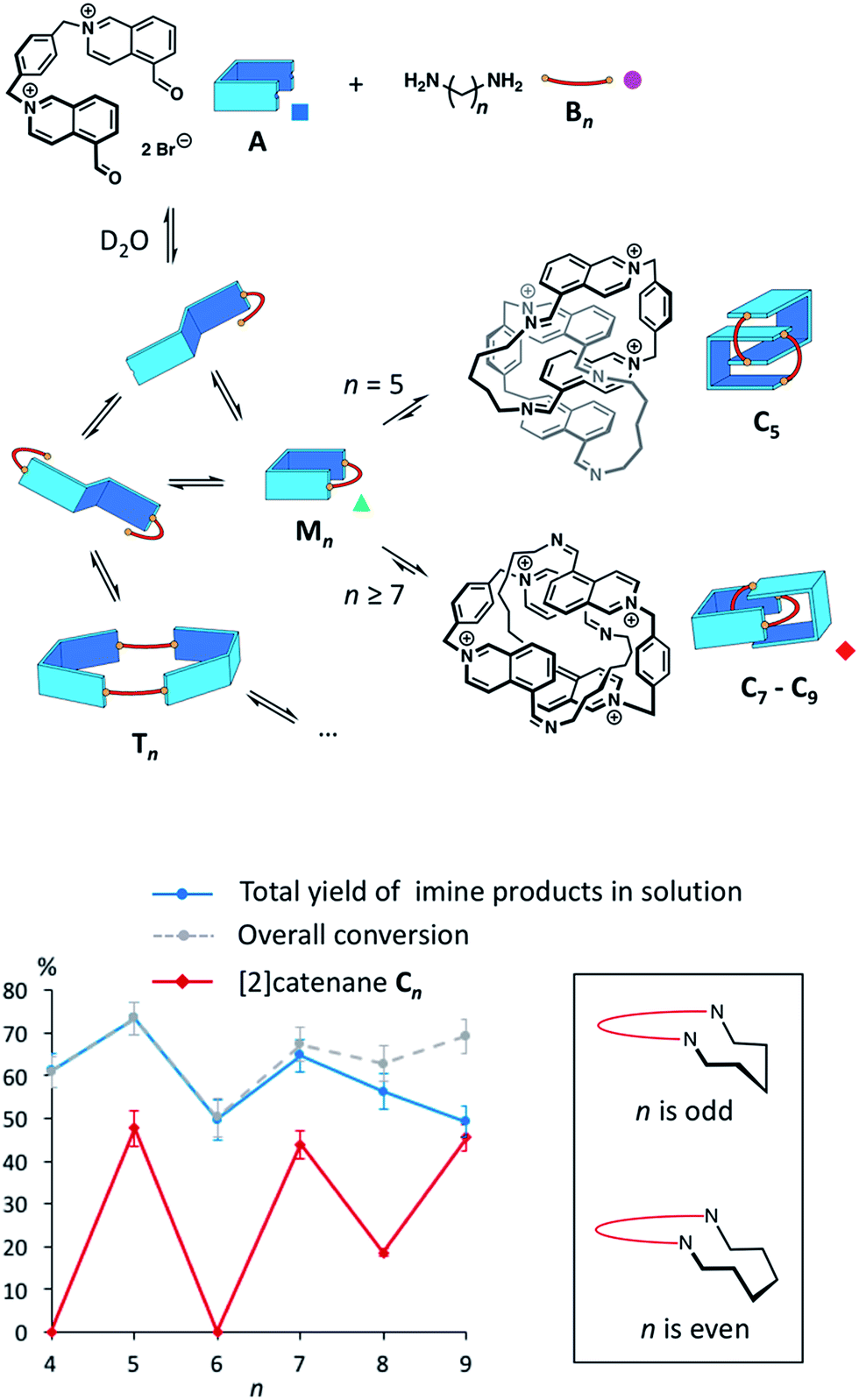 | ||
| Fig. 1 Top: Imine-based dynamic combinatorial libraries in water of dialdehyde A and aliphatic diamine Bn (10 mM each, n = 4–9). Bottom: Overall conversion (grey dotted line), total yield of imine products (blue line) and of [2]catenanes Cn (red line) plotted as a function of n. These values were measured from the integration of the 1H NMR signals (internal standard: hydroquinone). Insert: zigzag representation of odd- and even-numbered aliphatic chains within a macrocycle. | ||
Results and discussion
Dynamic combinatorial libraries were prepared from A and each one of the diamines Bn (20 mM total, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in water under optimum conditions for imine condensation (room temperature, pH 9.6). The libraries re-organised within a few minutes when conditions were changed (Fig. S7–S10†), preventing us from analysing the libraries by conventional chromatography and to isolate the products formed. Clean imine reduction also proved challenging, mostly due to the presence of the reactive isoquinolinium unit. Therefore, we characterised the products formed directly within the libraries, using 1H NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS), including traveling wave ion-mobility spectrometry (TWIMS).
:1) in water under optimum conditions for imine condensation (room temperature, pH 9.6). The libraries re-organised within a few minutes when conditions were changed (Fig. S7–S10†), preventing us from analysing the libraries by conventional chromatography and to isolate the products formed. Clean imine reduction also proved challenging, mostly due to the presence of the reactive isoquinolinium unit. Therefore, we characterised the products formed directly within the libraries, using 1H NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS), including traveling wave ion-mobility spectrometry (TWIMS).
The libraries reached thermodynamic equilibrium within five minutes and their composition did not evolve any further over a few days. At equilibrium, a significant amount of the initial A and Bn remained. Nevertheless, the total yield of imine-based products, measured from the integration of the NMR signals using hydroquinone as an internal standard, ranged between 49% and 74% (Fig. 1).
Overall, the yields of imine-based products displayed an odd-even effect17 with respect to n. Imine yields were lower if n was even (61% and 49% for n = 4 and 6) and higher if n was odd (74% and 65% for n = 5 and 7). The odd-even effect was attenuated for longer diamines and the yields slightly decreased (56% and 49% for n = 8 and 9). With these longer diamines, a fine precipitate was observed, leading to the disappearance of imine oligomers from the solution. This behaviour explains why conversion and total yield of imine diverge when n ≥ 7.
The odd-even effect was also apparent in the library compositions. If n was even, the 1H NMR spectra were rather complex (Fig. S11†) suggesting the formation of mixtures of multiple oligomers. Several species were observable in the ESI mass spectra of these libraries.
The main species corresponded to the closed [1 + 1] macrocycles (Mn). Only traces of the larger [2 + 2] species (I[2+2]/(I[1+1] + I[2+2]) < 7%, Fig. 2a) were observed. In contrast, the 1H NMR spectra were much simpler if n was odd and were clearly dominated by the presence of one major product. In the mass spectra, Mn was still observed, but the abundance of Cn had significantly increased (I[2+2]/(I[1+1] + I[2+2]) > 31%, Fig. 2a). From an entropic point of view, the formation of the relatively large [2 + 2] species was intriguing. Therefore, we investigated whether the it could be a [2]catenane (Cn), composed of two interlocked macrocycles Mn, rather than a trivial macrocycle (Tn). The in-source formation of a non-specific complex of two Mn was immediately ruled out, as charge repulsion would promote dissociation rather than unspecific association, especially at the concentration at which the mass spectra were recorded (20 μM). For clarity, the results of this investigation are only described here when n = 7 as a representative example.
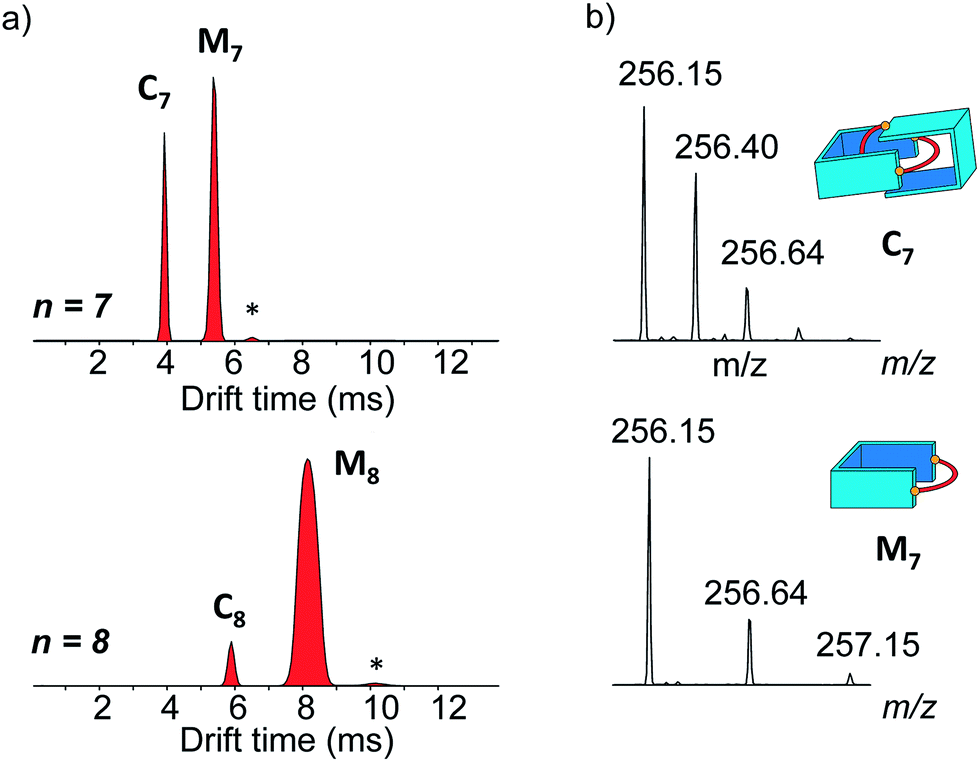 | ||
| Fig. 2 (a) Arrival time distributions for the ions at m/z 256.15 (n = 7) and 263.16 (n = 8). The relative abundances of Mn and Cn between the two cases illustrate the occurrence of an odd-even effect. The asterisk labels a trace of the open form of M7 (top) or M8 (bottom). (b) Isotope patterns of the ion-mobility-separated parent ions C7 and M7 with peak spacings of Δm/z = 0.25 and 0.5 in line with the assigned charge states. | ||
Narrow arrival time distributions (ATDs) for both M7 and the [2 + 2] species were consistent with single molecular structures. Both species were independently subjected to collision-induced decay experiments resulting in virtually identical spectra (Fig. 3a). This result strongly suggested that the [2 + 2] species was the [2]catenane C7. Initial bond cleavage of C7 led to charge-separation-driven dissociation into one closed and one open [1 + 1] macrocycles, which subsequently fragmented like the simpler M7 and thus gave rise to identical spectra. Tn should fragment in a non-symmetrical fashion and its collision-induced decay spectrum should be markedly different from that of Mn. The collision energy needed to induce the fragmentation of C7 (Fig. S38†) also confirmed its interlocked nature. The acceleration voltages at which 50% of the parent ions have fragmented is significantly lower for C7 (15.5 V) than for M7 (23.0 V) even though the larger C7 ions can store more internal energy before they fragment. This agrees only with a catenated structure, which merely requires a single bond cleavage for the initial fragmentation. Charge repulsion within the quadruply charged [2]catenane further reduces the energetic barrier that must be overcome for cleavage. Tn would only fragment when two covalent bonds are broken and would require a higher voltage than Mn.
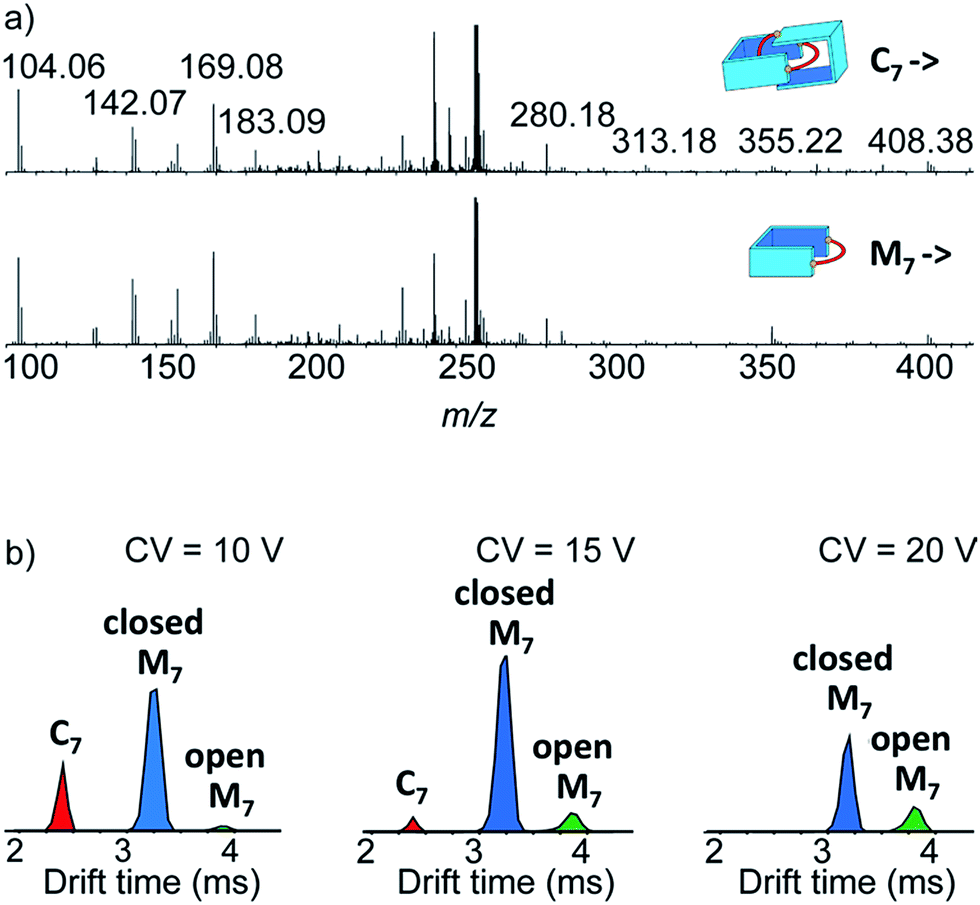 | ||
| Fig. 3 (a) Collision-induced decay spectra of the mass-selected and IMS-separated parent ions C7 and M7 (m/z 256). (b) Arrival time distribution of the species formed at low, medium and high collision voltage (CV) in the collision-induced decay of the non-separated parent ions C7 and M7 (m/z 256). | ||
Another experiment was independently conducted to confirm our assignment without ambiguity. On the mass spectra of the full library, we selected the molecular ion m/z = 256, corresponding to both C7 and M7. We first performed on this specific molecular ion collision-induced decay (CID) at various collision energies. The species produced were subsequently analysed with TWIMS and MS. Three peaks were observed in the mobilogram (Fig. 3b). The first two peaks corresponded to C7 and M7. The third peak corresponded to another isomer of M7, as identified by its isotope pattern, which exactly matched that of M7. Its longer drift time indicated a less compact structure and it could thus be assigned to the open form of M7, produced by cleavage of one bond via in-source fragmentation. Only traces of this species were observed in the previous experiments. As the collision energy increased from 10 V to 20 V, we observed that C7 disappeared while the amount of the open and closed M7 increased (Fig. 3b and S37†). At higher collision energies, M7 also underwent bond cleavage, forming the open M7 and smaller fragments. The cumulated increase of both M7 peaks was directly proportional to the decrease of C7 peak, showing that the fragmentation of C7 cleanly produced the open and closed M7.
Similarly, we demonstrated by ESI-MS that the main products formed from odd-numbered diamines were the [2]catenanes C5, C7 and C9 (Fig. S42†). Detailed 1H NMR analysis of the libraries confirmed the catenated structure of these main products and provided more detailed insight into their binding mode. The spectrum of [2]catenane C5 (Fig. 4b) reflected its interlocked nature. The signals of the aromatic protons b and d were substantially shifted upfield compared to the parent dialdehyde (Δδ ∼ 1.5 ppm). Proton c exhibited the largest shift (Δδ ∼ 3 ppm) and was considerably broadened. These shifts were consistent with a conformation in which the four isoquinolinium units stack in an antiparallel way in order to maximise stacking and minimise charge repulsion. The [2]catenane C5 must be confined into this compact conformation by the short length of the aliphatic chain. The high symmetry reflected in the 1H NMR spectrum implied that the inner and outer isoquinolinium units rapidly exchange on the NMR time-scale by circumrotation of the two wheels. NOE cross-peaks (Fig. 5a) between the stacking isoquinoliniums (g ↔ d) and their neighbouring xylylene (b, c ↔ i) and aliphatic (g ↔ j) moieties correlated well with the proposed structure.
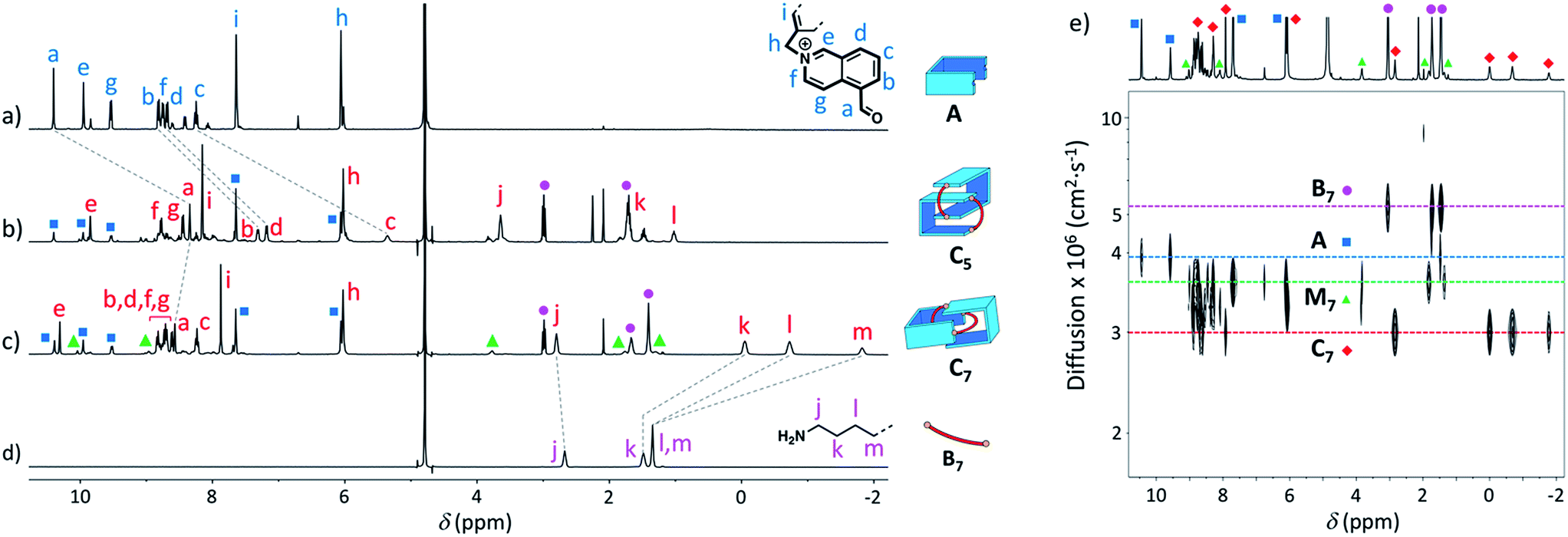 | ||
| Fig. 4 1H NMR spectra in D2O of (a) A (non-labelled signals correspond to the hydrate), (b) library formed from A and B5 (10 mM each), (c) library formed from A and B7 (10 mM each), and (d) B7. Only the signals corresponding to the [2]catenanes are assigned in spectra (b and c). (e) DOSY of the library formed from A and B7 (5 mM each) in D2O. Squares, diamonds and circles refer to the labelling scheme defined in Fig. 1. | ||
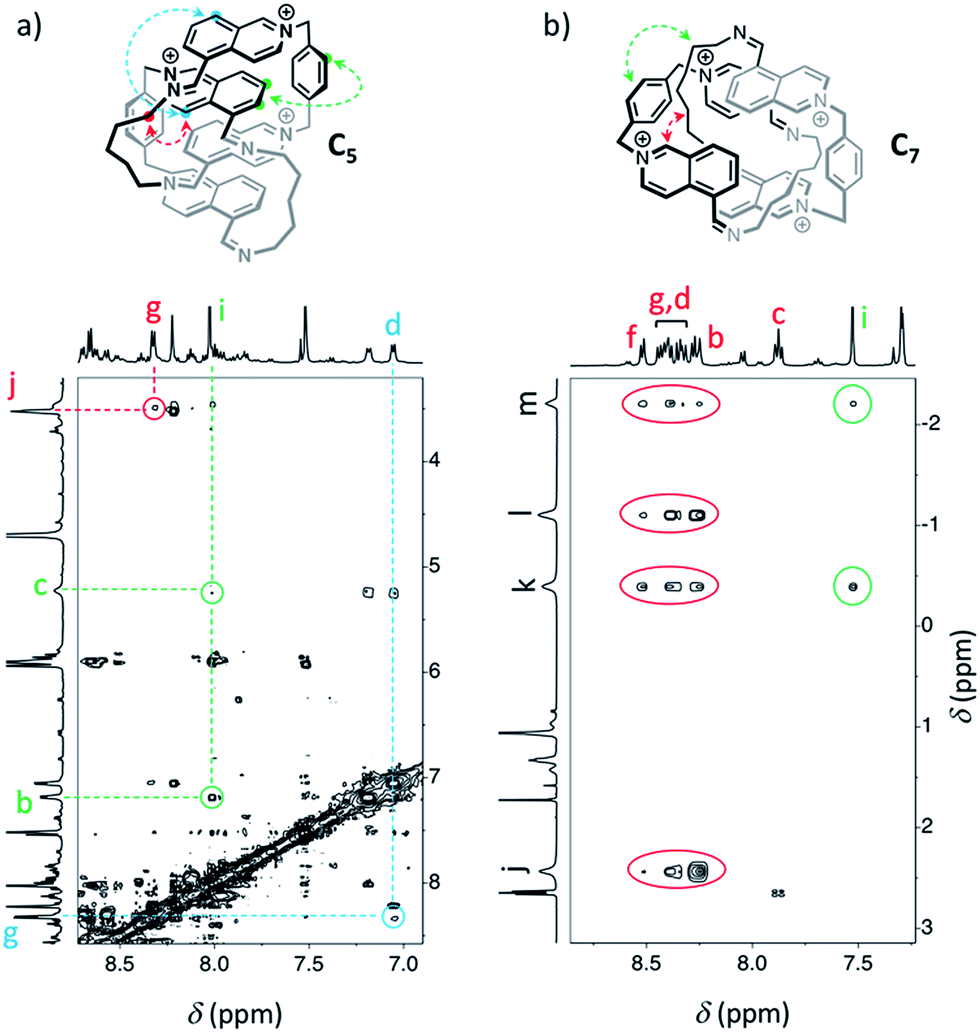 | ||
| Fig. 5 NOESY (D2O, 500 MHz, 298 K, d8 = 500 ms) of the libraries composed of (a) A and B5 (5 mM each), and (b) A and B7 (5 mM each). Correlations in agreement with the proposed structure of [2]catenanes are highlighted. | ||
The [2]catenanes bearing longer aliphatic chains (n ≥ 7) displayed significantly different spectra. The [2]catenane C7 (Fig. 4c) is described here as a characteristic example. All the aromatic protons of C7 resonated in the same range as that of the parent dialdehyde A (7.5–10.3 ppm). However, the aliphatic protons k, l and m, located around 1.3–1.5 ppm in the parent diamine, were significantly shifted upfield, even to negative ppm values (0 to −2 ppm). The [2]catenane C7 must therefore adopt a different conformation, wherein the aliphatic chain is threaded between the isoquinolinium units of the other wheel. This conformation is thermodynamically more stable because the four permanent positive charges are located further apart in space, thus minimizing their repulsion. In agreement with this proposed structure, NOE correlations confirmed the close proximity in space of the inner aliphatic and the outer aromatic protons (Fig. 5b). In comparison, the topologically trivial [1 + 1] macrocycle M7, clearly identifiable in the 1H NMR of the same library (triangles, Fig. 4c), did not present any deviations from the expected chemical shift values. Diffusion-ordered spectroscopy (DOSY, Fig. 4e) further confirmed our assignment of the library members. The smallest species of the library was B7 (diffusion coefficient D = 5.4 × 10−6 cm2 s−1), followed by A (3.9 × 10−6 cm2 s−1), M7 (3.3 × 10−6 cm2 s−1), and finally the larger C7 (3.0 × 10−6 cm2 s−1). As expected, the size of C7 was comparable to that of C5 (3.0 × 10−6 cm2 s−1, Fig. S20†).
With a purely aliphatic chain threaded between the aromatic units, C7 exhibits a rather uncommon structure.4 The formation of such a [2]catenane appears to be mostly driven by the hydrophobic effect, even if the contribution of other supramolecular interactions cannot be excluded. To confirm this hypothesis, we performed an incremental addition of CD3CN3,5 from 0% to 30% to the aqueous library containing C7 (Fig. 6). The presence of a co-solvent with a lower dielectric constant (DCH3CN = 37.5) than water (DH2O = 78.4) alters the hydrophobic effect and shifts the equilibrium in favour of M7. The kinetics of equilibration were also affected by the presence of a co-solvent and equilibration time significantly increased with the proportion of acetonitrile (>2 h in 30% CD3CN). Similarly, all the [2]catenanes disappeared upon incremental addition of CD3CN or CD3OD (Fig. S54–S60†), a solvent closer to D2O in term of its hydrogen bonding ability (DCH3OH = 32.7). This latter experiment highlights the specific properties of water in the context of this study. Finally, the choice of the counterion (Cl−, Br− or CF3SO3−) did not affect the library distributions (Fig. S15, S24, S26, S36, S43 and S52†), showing that the assembly was not templated by a specific counterion, as observed for other catenanes and more complex interlocked structures.18
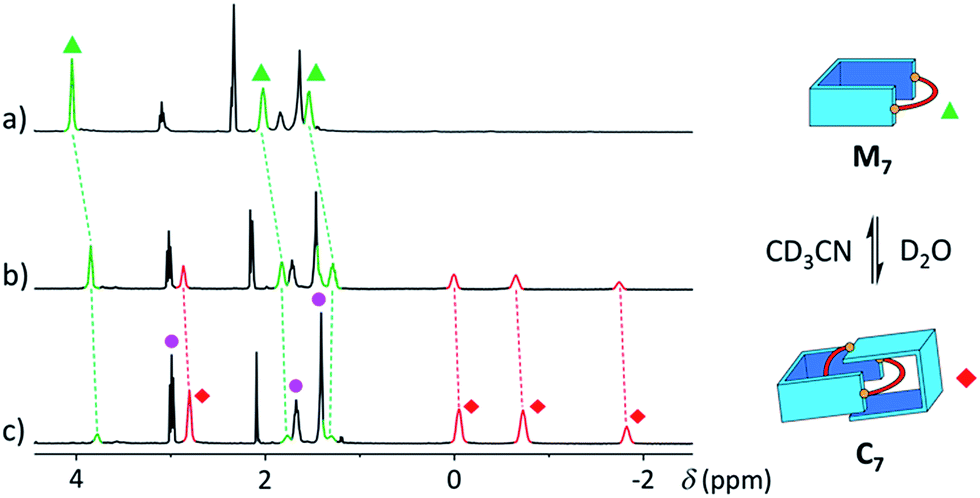 | ||
| Fig. 6 Partial 1H NMR spectra of the library formed from A and B7 (10 mM each) in (a) 30%, (b) 10%, and (c) 0% of CD3CN in D2O, showing the reversible polarity-dependent conversion of C7 into M7. | ||
Conclusions
The self-assembly of amphiphilic imine-based [2]catenanes in water is mostly driven by the need to minimize the hydrophobic surface area exposed to water. It does not require the complementary use of metal coordination6 or donor–acceptor π–π stacking.4,5,19 Even the charge repulsion between the positively charged motifs is overcome by the hydrophobic effect.The products of the libraries could not be isolated, but extensive NMR studies and tandem MS experiments, together with ion mobility spectrometry, allowed for the unambiguous identification and characterisation of the [2]catenanes within the libraries. More importantly, our system generated unexpected results regarding the complex role played by the length of the aliphatic chain. Indeed, the length of the aliphatic diamine controls both the yield and the conformation of the [2]catenanes. Evidently, odd-numbered aliphatic chains favour, and even-numbered chains disfavour, [2]catenane formation. Odd-even effects are often observed in solid phase,17 but are much less common in solution.4,20 Within the [2]catenanes, the aliphatic chains cannot adopt a fully relaxed zigzag conformation. Folding of odd-numbered chains (Fig. 1, insert), required to close the [2]catenanes, induces a chair-like turn that resembles most a relaxed zigzag conformation. On the other hand, folding of even-numbered chains generates unavoidable gauche interactions. In dynamic combinatorial libraries, the final equilibrium distribution can reflect clearly such small energetic differences8 (3.9 kJ mol−1 per gauche interaction).
The formation of the [2]catenanes is rather remarkable considering that imine condensation is not favoured in pure water, especially under dilute conditions.11 The presence of [2]catenanes systematically correlated with an increase in the overall yield of imine (Fig. 1), because the higher thermodynamic stability of the [2]catenanes shifts the equilibria in favour of imine condensation. The system presented here is still quite simple but allowed us to probe how solvophobic effects can direct the formation of interlocked molecules. The knowledge gained from this study will help constructing topologically more complex architectures, such as knots or interwoven materials, using the hydrophobic effect.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swiss National Science Foundation (PZ00P2_161270), the Deutsche Forschungsgemeinschaft (CRC 765 “Multivalency”) and the Department of Organic Chemistry at the University of Geneva are gratefully acknowledged for financial support. A. K. acknowledges financial support from the Alexander von Humboldt Foundation. We would like to thank Dr Naomi Sakai, who initially stimulated our interest on imine chemistry, and Prof. Stefan Matile for helpful comments on the manuscript. We also thank Jan Wollschläger and Ulrike Warzok for help with the MS measurements.Notes and references
- (a) R. S. Forgan, J. P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed; (b) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700–1712 RSC.
- For the DNA-based assembly of knots and catenanes, see: (a) J. Elbaz, Z.-G. Wang, F. Wang and I. Willner, Angew. Chem., Int. Ed., 2012, 51, 2349–2353 CrossRef CAS PubMed; (b) N. C. Seeman, Angew. Chem., Int. Ed., 1998, 37, 3220–3238 CrossRef CAS.
- (a) N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783–785 CrossRef CAS PubMed; (b) N. Ponnuswamy, F. B. L. Cougnon, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2014, 136, 8243–8251 CrossRef CAS PubMed.
- To the best of our knowledge, the only water-soluble [2]catenane whose formation was driven by the need to bury a purely aliphatic chain in its core was described in: F. B. L. Cougnon, N. Ponnuswamy, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 19129–19135 CrossRef CAS PubMed.
- H. Li, H. Zhang, A. D. Lammer, M. Wang, X. Li, V. M. Lynch and J. L. Sessler, Nat. Chem., 2015, 7, 1003–1008 CrossRef CAS PubMed.
- (a) M. Fujita, F. Ibukuro, H. Hagihara and K. Ogura, Nature, 1994, 367, 720–723 CrossRef CAS; (b) M. Fujita, F. Ibukuro and K. Ogura, J. Am. Chem. Soc., 1995, 117, 4175–4176 CrossRef CAS; (c) A. Hori, K. Kumazawa, T. Kusukawa, D. K. Chand, M. Fujita, S. Sakamoto and K. Yamaguchi, Chem.–Eur. J., 2001, 7, 4142–4149 CrossRef CAS PubMed.
- (a) O. Safarowsky, M. Nieger, R. Frölich and F. Vögtle, Angew. Chem., Int. Ed., 2000, 39, 1616–1618 CrossRef CAS; (b) M. Feigel, R. Ladberg, S. Engels, R. Herbst-Irmer and R. Frölich, Angew. Chem., Int. Ed., 2006, 45, 5698–5702 CrossRef CAS PubMed.
- (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (b) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (c) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- (a) M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2014 RSC; (b) K. S. Chichak, S. J. Cantrill, A. R. Pearse, S.-H. Chiu, G. W. V. Cave and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed; (c) D. A. Leigh, P. J. Lusby, S. J. Teat, A. J. Wilson and J. K. Y. Wong, Angew. Chem., Int. Ed., 2001, 113, 1586–1590 CrossRefG. Koshkakaryan, D. Cao, L. M. Klivansky, S. J. Teat, J. L. Tran and Y. Liu, Org. Lett., 2010, 12, 1528–1531 CrossRef CAS PubMed; (d) C. S. Wood, T. K. Ronson, A. M. Belenguer, J. J. Holstein and J. R. Nitschke, Nat. Chem., 2015, 7, 354–358 CrossRef CAS PubMed; (e) J. R. Price, J. K. Clegg, R. R. Fenton, L. F. Lindoy, J. C. McMurrie, G. V. Meehan, A. Parkin, D. Perkins and P. turner, Aust. J. Chem., 2009, 62, 1014–1019 CrossRef CAS.
- For selected examples of imine bonds particularly sensitive to hydrolysis, see: (a) W. Lu and T. H. Chang, J. Org. Chem., 2001, 66, 3467–3473 CrossRef CAS PubMed; (b) T. Hirashita, Y. Hayashi, K. Mitsui and S. Araki, J. Org. Chem., 2003, 68, 1309–1313 CrossRef CAS PubMed.
- (a) C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef; (b) I. Huc and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS PubMed; (c) V. Saggiomo and U. Lüning, Eur. J. Org. Chem., 2008, 4329–4333 CrossRef CAS; (d) V. Saggiomo and U. Lüning, Tetrahedron Lett., 2009, 50, 4663–4665 CrossRef CAS; (e) C. Givelet, J. Sun, D. Xu, T. J. Emge, A. Dhokte and R. Warmuth, Chem. Commun., 2011, 47, 4511–4513 RSC; (f) T. O. Omosun, M.-C. Hsieh, W. S. Childers, D. Das, A. K. Mehta, N. R. Anthony, T. Pan, M. A. Grover, K. M. Berland and D. G. Lynn, Nat. Chem., 2017, 49, 805–809 CrossRef PubMed.
- P. D. Kiser, M. Golczak and K. Palczewski, Chem. Rev., 2014, 114, 194–232 CrossRef CAS PubMed.
- (a) J. R. Nitschke, Angew. Chem., Int. Ed., 2004, 43, 3073–3075 CrossRef CAS PubMed; (b) M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem.–Eur. J., 2006, 12, 4069–4076 CrossRef CAS PubMed.
- (a) C. B. Minkenberg, L. Florusse, R. Eelkema, G. J. M. Koper and J. H. van Esch, J. Am. Chem. Soc., 2009, 131, 11274–11275 CrossRef CAS PubMed; (b) R. Nguyen, L. Allouche, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2009, 48, 1093–1096 CrossRef CAS PubMed.
- (a) P. M. S. D. Cal, J. B. Vicente, E. Pires, A. V. Coelho, L. F. Veiros, C. Cordeiro and P. M. P. Gois, J. Am. Chem. Soc., 2012, 134, 10299–10305 CrossRef CAS PubMed; (b) N. J. Gutiérrez-Moreno, F. Medrano and A. K. Yatsimirsky, Org. Biomol. Chem., 2012, 10, 6960–6972 RSC; (c) C. Arnal Hérault, A. Pasc, M. Michau, D. Cot, E. Petit and M. Barboiu, Angew. Chem., Int. Ed., 2007, 46, 8409–8413 CrossRef PubMed.
- (a) D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed; (b) D. Larsen, M. Pittelkow, S. Karmakar and E. T. Kool, Org. Lett., 2015, 17, 274–277 CrossRef CAS PubMed; (c) J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed; (d) E. Bartolami, Y. Bessin, V. Gervais, P. Dumy and S. Ulrich, Angew. Chem., Int. Ed., 2015, 54, 10183–10187 CrossRef CAS PubMed; (e) J. M. Priegue, D. N. Crisan, J. Martínez-Costas, J. R. Granja, F. Fernandez-Trillo and J. Montenegro, Angew. Chem., Int. Ed., 2016, 55, 7492–7495 CrossRef CAS PubMed.
- (a) F. Tao and S. L. Bernasek, Chem. Rev., 2007, 107, 1408–1453 CrossRef CAS PubMed; (b) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed; (c) K. Tomioka, T. Sumiyoshi, S. Narui, Y. Nagaoka, A. Lida, Y. Miwa, T. Taga, M. Nakano and T. Handa, J. Am. Chem. Soc., 2001, 123, 11817–11818 CrossRef CAS PubMed; (d) M. M. Thuo, W. F. Reus, C. A. Nijhuis, J. R. Barber, C. Kim, M. D. Schulz and G. M. Whitesides, J. Am. Chem. Soc., 2011, 133, 2962–2975 CrossRef CAS PubMed.
- (a) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed; (b) J.-F. Ayme, J. E. Beves, C. J. Campbell, G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, J. Am. Chem. Soc., 2015, 137, 9812–9815 CrossRef CAS PubMed; (c) R. A. Bilbeisi, T. Prakasam, M. Lusi, R. El-Khoury, C. Platas-Iglesias, L. J. Charbonnière, J.-C. Olsen, M. Elhabiri and A. Trabolsi, Chem. Sci., 2016, 7, 2524–2532 RSC; (d) A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS PubMed.
- (a) L. Fang, S. Basu, C.-H. Sue, A. C. Fahrenbach and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 396–399 CrossRef CAS PubMed; (b) L. Fang, C. Wang, A. C. Fahrenbach, A. Trabolsi, Y. Y. Botros and J. F. Stoddart, Angew. Chem., Int. Ed., 2011, 50, 1805–1809 CrossRef CAS PubMed.
- P. J. M. Stals, M. M. J. Smulders, R. Martín-Rapun, A. R. A. Palmans and E. W. A. Meijer, Chem.–Eur. J., 2009, 15, 2071–2080 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04901c |
| This journal is © The Royal Society of Chemistry 2018 |