DOI:
10.1039/C7NJ04200K
(Paper)
New J. Chem., 2018,
42, 458-464
Electrochemical fabrication of shape-controlled Cu2O with spheres, octahedrons and truncated octahedrons and their electrocatalysis for ORR†
Received
31st October 2017
, Accepted 20th November 2017
First published on 21st November 2017
Abstract
Transition metal oxide catalysts have been the focus of recent research for the oxygen reduction reaction (ORR). However, the relationship between morphology and electrocatalysis of metal oxide catalysts has not been well studied or understood hitherto. In the present work, cuprous oxides (Cu2O) with three different morphologies (sphere, octahedron, and truncated octahedron) have been synthesized employing a potentiostatic electrodeposition method. The as-prepared Cu2O has been characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), and transmission electron microscopy (TEM). Three different shapes of Cu2O crystals were used to study the shape-dependent electrocatalytic properties for ORR in alkaline media. It was found that the surface-specific activity of Cu2O with truncated octahedron shape toward ORR was higher than that of Cu2O with a sphere or octahedron morphology. The morphology–activity relationship of Cu2O as a cathode catalyst was further studied through periodic spin density functional theory (DFT) calculations. Comprehensive analysis of electrocatalysis exiperiments and DFT calculations reveals that Cu2O as a truncated octahedron preferentially exposes the (100) crystal planes, which are beneficial to stronger O2 adsorption and easier activation of adsorbed O2 on the surface of Cu2O. This finding furthers our understanding of Cu2O catalysis and provides new light on the relevant catalyst design criteria.
1 Introduction
Oxygen reduction reaction (ORR) contributes significantly to the energy efficiency of fuel cells and metal–air batteries because of the exceptionally high O–O bond energy (498 kJ mol−1) and the sluggish nature of ORR.1 Although Pt and its alloys are known as efficient catalysts for activation of the strong oxygen–oxygen double bond, their application is limited by high costs and scarce reserves.2 Therefore, efforts to explore non-noble metal cathode catalysts to replace the precious Pt in fuel cells and metal–air batteries have been going on to bring this efficient technology into real applications.3 Non-noble metal catalysts, including transition metal chalcogenides,4 nitrides,5,6 oxides,7 and heteroatom-doped carbonaceous materials (e.g. graphite, carbon nanotubes, graphene, and graphene oxides),8 have been recently identified as potential catalysts for ORR. Among these materials, non-noble-metal oxides are the most pursued alternative electrocatalysts due to their low cost, high yield, good stability, and high catalytic activity toward ORR.9–11 Cuprous oxide (Cu2O) is a p-type semiconductor with unique optical and electrical properties, which presents itself as a promising material in the fields of solar energy conversion, photocatalytic degradation, catalysis, and sensors.12,13 It can also easily react with oxygen in wet air to form CuO. This indicates that it can donate electrons to oxygen, which is in favor of ORR. So Cu2O has been gradually paid more and more attention for its potential utilization in electrocatalysis for ORR.14,15 Furthermore, Guo and his coworkers found that the low conductivity of Cu2O nanoparticles could be improved by combining them with high electrical conductive carbon materials such as graphene.14 Wang et al. reported that Cu2O nanocubes produced higher catalytic activity than both sphere-like and nanoporous Cu2O due to their large aspect ratio and preferentially exposed {100} crystal surface.15 We note that these reports did not consider the involved oxygen adsorption and activation of adsorbed O2 on the surfaces of Cu2O. We also infer that these different results may result from other differences in sample preparation.
It is generally observed that crystals with different types of exposed facets usually exhibit different physical and chemical properties.16 Different crystal phases, structure, and morphology will remarkably impact the physicochemical properties of these crystals.17–19 Therefore, the shape-controlled synthesis of nanocrystals presents an important direction for tuning the activity, stability, and selectivity of nanocrystal catalysts.20 Although cuprous oxide is expected to be a promising low-cost electrocatalyst for ORR,14,15 the morphology-related catalytic activity for ORR on Cu2O structures has not been fully realized. In addition, the main challenge for such studies comes from the synthetic routes for the controlled synthesis of Cu2O with different morphology. Some efforts have recently been devoted to the shape-controlled synthesis of Cu2O nanocrystals with various geometries.21–23 Depending on the reducing agent used and its strength, the reduction rate may be slow and the final product may be impure, and the amount of the reagent must be adequately controlled to avoid metallic copper formation. Siegfried and Choi demonstrated a methodological approach toward utilizing the preferential adsorption of surfactant during the galvanostatic process to obtain octahedral Cu2O crystals.24 Li et al. presented a facile electrodeposition of Cu2O onto activated carbon with polyvinylpyrrolidone (PVP) surfactant and used it as an air cathode to achieve high ORR activity.25 However, a great deal of impurities (surfactant) was introduced onto the surface of Cu2O microcrystals by the above synthetic method. Therefore, the electrodeposition method for the cuprous oxide preparation as well as the relationship between morphology and electrocatalysis still need to be further studied.
Herein, we wish to report a simple electrochemical route (potentiostatic) for the controlled synthesis of three Cu2O microcrystals (spheres, octahedrons, and truncated octahedrons) on the electrode surface in situ to further investigate the shape dependence of Cu2O toward ORR activity. SEM, TEM, XRD and XPS characterizations were carried out to compare crystal structure, surface area, and Cu oxidation states. The electrocatalysis performance for ORR was evaluated by CV and LSV in alkali media. The morphology–activity relationship of Cu2O as an ORR catalyst was also discussed through DFT calculations. The comprehensive analysis based on electrochemical experiments and density DFT calculations reveals that Cu2O as truncated octahedrons exhibits the best electrocatalytic properties toward ORR.
2 Experimental
2.1 Chemicals
Copper(II) acetate (Cu(OOCCH3)2), acetic acid (CH3CO2H), sodium hydroxide (NaOH), ethanol, isopropanol, and potassium hydroxide (KOH) were of analytical grade and from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Nafion solution was purchased from Sigma-Aldrich. Indium tin oxide (ITO) glass plates were purchased from Incole Union Technology Co., Ltd (Tianjin, China). Deionized water was used throughout the experiments.
2.2 Preparation of Cu2O crystals
Electrochemical deposition of Cu2O crystals onto a glass carbon disk electrode (GCDE, 5 mm in diameter, Pine Research Instruments) or an ITO plate (4 mm in width) was performed, respectively. A Ag/AgCl (3 M KNO3) electrode served as the reference electrode, and a platinum wire was used as the counter electrode. The electrolyte solution for deposition consisted of 0.02 M copper(II) acetate (Cu(OOCCH3)2) and 0.678 M acetic acid (CH3CO2H). The pH value of the solution was adjusted to 4.90 with sodium hydroxide (NaOH). Before each deposition, the solution was deaerated with nitrogen for 15 min until the solution was filled with nitrogen. The electrodeposition was conducted at 20, 50, and 70 °C, respectively. The electrodeposition potential was obtained according to the J–E curve: GCDE/0.02 MCu(OOCCH3)2/0.678 MCH3CO2H, which was registered from 0 to −1.0 V (vs. Ag/AgCl) (Fig. S1, ESI†). A cathodic current peak (Ic) placed at −0.45 V and the beginning of a second one (IIc) at −0.72 V were observed. The Ic and IIc processes were attributed to the electrochemical reduction of Cu(II) to Cu(I) with Cu2O formation and electrochemical reduction of Cu(I) to Cu(0) onto the working electrode, respectively (eqn (S1) and (S2), ESI†).26 From this analysis, a potential value of −0.45V was applied for electrodeposition. The j/t transients were recorded in a time range from 0 to 120 min (Fig. S2, ESI†). The prepared Cu2O samples were denoted by Cu2O-20, Cu2O-50, Cu2O-70, respectively.
2.3 Apparatus and characterization
XRD patterns were obtained on a Siemens D5005 X-ray diffractometer with Cu Kα radiation. XPS was determined on a Quantum-2000 Scanning ESCA Microprobe system using Al mono K radiation as the excitation source with a pass energy of 46.95 eV. The size and morphology of the composites were analyzed by scanning electron microscopy (SEM) on a JEOL 7500F scanning electron microscope and transmission electron microscopy (TEM) operating at 200 kV (Philips TECNAI G2). Cu2O loading was determined by Thermo 6300 inductively coupled plasma-atomic (ICP) emission spectrometry. The specific surface area was measured by a Coulter Omnisorp 100cx analyzer.
In order to obtain clear observation of the morphology of Cu2O crystals, ITO-coated glass plates were used to deposit Cu2O for surface characterization with the same procedures. In this study, there are no significant differences in the morphology when the Cu2O crystals are electrodeposited on GCDE or ITO (Fig. S3, ESI†). SEM imaging and XPS analysis of the as-deposited Cu2O were carried out on the ITO substrate itself. For XRD and specific surface area measurement, the Cu2O samples were scraped off from the ITO substrate. As for TEM measurement, the as-deposited Cu2O sample was removed carefully from the ITO electrode and then dispersed in isopropanol under ultrasonication and coated on the TEM grids.
2.4 Electrocatalytic measurements
The electrocatalysis properties of the Cu2O crystals were investigated by CV and LSV performed on a CHI 660C electrochemical workstation and Pine modulated speed rotator under ambient conditions. For identical loading of Cu2O crystals on the GCDE for electrocatalysis studies, the three Cu2O crystals were first scraped off from the deposited working electrodes. Then, catalyst suspensions were prepared by ultrasonically dispersing Cu2O crystals (10 mg) in a mixed solution (2 mL) of isopropanol and Nafion for 30 min. The ratio of Nafion to catalyst was kept at 0.2 during the preparations. Lastly, 5 μL of the suspension was dispersed onto the GCDE and dried at room temperature to obtain a catalyst loading of 128 μg cm−2. A Pt coil and double junction Ag/AgCl (3 M KNO3) acted as the counter and reference electrode, respectively. All potentials in this study refer to that of RHE. The potential difference between Ag/AgCl and RHE is 0.962 ± 0.002 V, based on the calibration measurement in H2 saturated 0.1 M KOH with two polished Pt wires as the working and counter electrodes, respectively, and the Ag/AgCl as the reference electrode. CVs were run at a scan rate of 1 mV s−1, and the average of the two potentials at which the current crossed zero was taken to be the thermodynamic potential for the hydrogen electrode reactions. So in 0.1 M KOH, E(RHE) = E(Ag/AgCl) + 0.962 V (Fig. S4, ESI†).
2.5 DFT Calculations
All calculations have been performed within Dmol327 density functional theory (DFT) using the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional. A (1 × 1) supercell with nine atomic layers, and with a vacuum of 15 Å, was employed for the calculation. The three outermost atomic layers were relaxed. In the computation, the inner electrons of the copper atoms were kept frozen and replaced by an effective core potential (ECP), the oxygen atoms were treated with an all-electron basis set. The valence electron functions were expanded into a set of numerical atomic orbitals by a double-numerical basis with polarization functions (DNP). Brillouin-zone integrations were performed using a 1 × 1 × 1 Monkhorst–Pack grid and a Methfessel–Paxton smearing of 0.005 Ha. The converge criteria judged by the energy, force, and displacement, respectively, were 2 × 10−5 Ha, 0.005 Å, and 0.004 Ha Å−1. The adsorption energy (Eads) of species X on the slab is defined as Eads = EA/slab − (Eslab + EX), where Eslab is the total energy of the slab and EX is the total energy of species X. A negative value corresponds to a stable adsorbate/slab system.
Cu2O has the cuprite (Pn3) structure, and each unit cell has two O and four Cu atoms, and each O atom is tetrahedrally coordinated with four Cu atoms and each Cu atom is linearly coordinated with two oxygen atoms (Fig. S5, ESI†). The structures of the (111) and (100) low index surfaces were studied (Fig. S1B and C, ESI†). The calculated bulk lattice constant for Cu2O is 4.32 Å and the dCu–O is 1.87 Å, agreeing reasonably with former theoretical results and the experimental value.28
3 Results and discussion
3.1 Morphology and composition characterization
Fig. 1 shows the SEM images of the three synthesized Cu2O morphologies: sphere-shaped (Cu2O-20), octahedral (Cu2O-50), and truncated octahedral (Cu2O-70). It can be seen that the bath temperature plays an important role in the formation of the deposit. Spherical crystals with rough surface could be observed on ITO at bath temperatures of 20 °C (Fig. 1a and b) and the diameter of the sphere is about 100 nm. Interestingly, when the bath temperature was increased to 50 °C, the collected brick-red samples are octahedrons with an average edge length of ∼110 nm (Fig. 1c and d). Fig. 1e and f show that the products exhibit a truncated octahedral shape (only the corners of the octahedra are truncated) with sizes of about 100 nm. The truncated corners exhibit {100} facets so Cu2O-70 has {100} facets exposed.29 The successful preparation of the above three Cu2O morphologies in this work suggests that the electrochemical approach can be applied to synthesize Cu2O with various morphologies, controlled size, and fascinating facets through controlling the bath temperature.
 |
| | Fig. 1 SEM images of the electrochemical synthesized Cu2O at a bath temperature of 20 °C (a and b), 50 °C (c and d), and 70 °C (e and f). Insets are the models of the corresponding samples. The samples were deposited on the ITO substrate. | |
The morphologies and structures of the Cu2O samples were further characterized by TEM and selected area electron diffraction (SAED). The TEM images (Fig. 2a–c) also confirm that spheres, octahedrons, and truncated octahedrons are formed. The HRTEM images (Fig. 2d–f) and the index of the spots in the SAED patterns (the right inset in Fig. 2d–f) indicate that the spherical product is polycrystalline, the octahedrons with eight {111} facets and the truncated octahedrons with crystals are polycrystalline. The surface region of Cu2O-50 (Fig. 2e) gives two sets of lattice fringes of 0.246 nm with an intersection angle of 60°. These lattice fringes respectively correspond to the lattice fringe of the (11![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (11) planes of the Cu2O structure (JCPDS card No. 34-1354), which further indicates that Cu2O-50 is composed of single crystals and these crystalline octahedrons mainly have their {111} crystal facets exposed. The lattice fringe of 0.214 nm of Cu2O-70 (Fig. 2f) can be assigned to the (200) plane of the Cu2O structure. The TEM and HRTEM images reveal that Cu2O-70 is composed of truncated octahedrons, which expose all eight {100} planes and the {111} facets of the single crystal. The TEM images show that, compared with Cu2O-20 (Fig. 2a), Cu2O-50 (Fig. 2b), and Cu2O-70 (Fig. 2c), the crystals possess much smoother surfaces and edges, which is consistent with the SEM results (Fig. 1c and e). The HRTEM image (Fig. 2d) also clearly reveals that the surface morphology of the Cu2O-20 crystal differs from that of Cu2O-50 and Cu2O-70. The lattice fringes of Cu2O are not distinct and continuous, which indicates spherical Cu2O-20 {111} and {100} crystal planes. The above results show that the growth rates along the different crystallographic planes can be changed when the temperature changed during the crystal growth process. The formation mechanism of Cu2O on the electrode accompanied with the morphology change may be explained in terms of the thermodynamics of the growth process.30–32 The increase in temperature makes the reduction of Cu(II) and the nucleation of Cu2O slower, which is helpful for the formation of thermodynamically stable single-crystalline seeds with {111} and {100} planes and the following growth of single-crystalline octahedron-like and truncated octahedron-like structures.
) and (11) planes of the Cu2O structure (JCPDS card No. 34-1354), which further indicates that Cu2O-50 is composed of single crystals and these crystalline octahedrons mainly have their {111} crystal facets exposed. The lattice fringe of 0.214 nm of Cu2O-70 (Fig. 2f) can be assigned to the (200) plane of the Cu2O structure. The TEM and HRTEM images reveal that Cu2O-70 is composed of truncated octahedrons, which expose all eight {100} planes and the {111} facets of the single crystal. The TEM images show that, compared with Cu2O-20 (Fig. 2a), Cu2O-50 (Fig. 2b), and Cu2O-70 (Fig. 2c), the crystals possess much smoother surfaces and edges, which is consistent with the SEM results (Fig. 1c and e). The HRTEM image (Fig. 2d) also clearly reveals that the surface morphology of the Cu2O-20 crystal differs from that of Cu2O-50 and Cu2O-70. The lattice fringes of Cu2O are not distinct and continuous, which indicates spherical Cu2O-20 {111} and {100} crystal planes. The above results show that the growth rates along the different crystallographic planes can be changed when the temperature changed during the crystal growth process. The formation mechanism of Cu2O on the electrode accompanied with the morphology change may be explained in terms of the thermodynamics of the growth process.30–32 The increase in temperature makes the reduction of Cu(II) and the nucleation of Cu2O slower, which is helpful for the formation of thermodynamically stable single-crystalline seeds with {111} and {100} planes and the following growth of single-crystalline octahedron-like and truncated octahedron-like structures.
 |
| | Fig. 2 TEM and HRTEM images of the electrochemical synthesized Cu2O at a bath temperature of 20 °C (a and d), 50 °C (b and e), and 70 °C (c and f). Insets in d, e, and f are SAED patterns of the corresponding samples. The samples were deposited on the ITO substrate. | |
As shown in Fig. 3a, the XRD patterns of the three as-prepared Cu2O samples are in good agreement with the characteristic diffractions peaks of cubic Cu2O (JCPDS No. 34-1354). The sharp and narrow diffraction peaks suggest that the products are well crystallized. The XRD curve of Cu2O-20 demonstrates that the compound consists of cubic Cu2O with a small amount of CuO. However, the peaks assigned to CuO are not found in the XRD patterns of Cu2O-50 and Cu2O-70. The (200) diffraction peak in the XRD pattern of truncated octahedral Cu2O crystals (Cu2O-70) is stronger than other diffraction peaks suggesting that truncated octahedral Cu2O crystals exclusively expose {100} crystal planes. Similar results are also found in the XRD pattern of octahedral Cu2O nanocrystals, which exclusively expose the {111} crystal plane. These observations confirm that octahedral Cu2O-50 is primarily dominated by the (111) facets. Thus, it can be futher concluded that each triangle facet of octahedral Cu2O-50 is a (111) facet. We have also found that the relative intensity ratio of the (111)/(200) reflection of Cu2O-70 is much lower than the values of Cu2O-20 and Cu2O-50 (Fig. 3b), further indicating preferential orientation growth in the (100) direction of Cu2O-70.
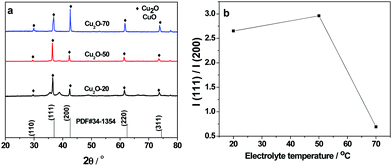 |
| | Fig. 3 (a) X-ray diffraction patterns of Cu2O-20, Cu2O-50 and Cu2O-70 and Cu2O JCPDS card (#34-1354); (b) plot line of relative intensity I(111)/I(200) as a function of electrolyte temperature. | |
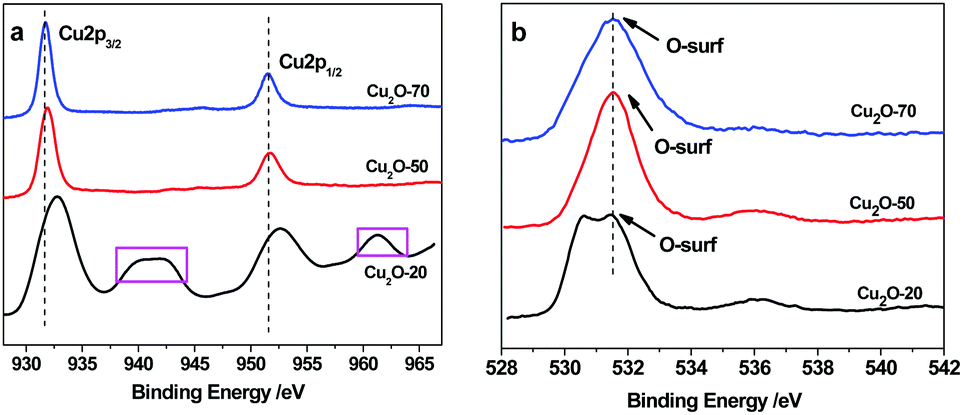
XPS analysis was conducted to confirm the chemical bonding properties and phase composition of the Cu2O deposits. As shown in Fig. 4a, the Cu2p peaks of Cu2O-70 at 931.7 and 951.8 eV corresponding to Cu2p3/2 and Cu2p1/2, respectively, reveal the +1 oxidation state.33 The occurrence of satellite feature peaks at ∼943.2 eV and ∼962.8 eV in the spectra of Cu2O-20 indicates the presence of CuO, because cupric oxide (Cu2+) has hole states in the Cu3d band (Cu 3d9 configuration). In contrast, the 3d band of cuprous oxide (Cu+) is filled (Cu 3d10), and the 4s band is unoccupied, thus no satellites are expected. The observed amount of CuO in Cu2O-20 is due to the slightly oxidized surface of Cu2O under exposure to the atmosphere at 20 °C. When the bath temperature increases up to 50 °C and 70 °C, the main peaks of Cu2p3/2 and 2p1/2 shift to the low-energy side; meanwhile, the satellites are not found. This result confirms the thermal reduction of CuO to Cu2O, which agrees with the findings of XRD. The above results demonstrate that the surface composition of the as-synthesized octahedral and truncated octahedral Cu2O nanocrystals remains as Cu2O. In addition, O1s spectral analysis allowed us to propose that the high initial reactivity was caused by the presence of non-lattice oxygen states on the surface of the different shaped Cu2O as shown in Fig. 4b. The binding energy of O1s in a crystal lattice is 528.5–529.7 eV and the binding energy of absorbed oxygen is 530.54–533.77 eV.34 The main peak at 531.4 eV was observed in the three spectra of the studied oxides, which suggests that most of the oxygen species exist in the form of “absorbed oxygen” (non-lattice oxygen) on the surfaces of the three kinds of Cu2O. However, a noticeable shoulder on the lower binding energy side (∼530.2 eV) is found in the O1s spectrum of the sphere oxide. This observation suggests the existence of additional oxygen species on the surface of this oxide. This should be attributed to small impurities of adsorbed water, carbonate groups, or hydroxyl groups.35 The “non-lattice” oxygen was found to be the highly active oxygen state and this type of oxygen is stabilized on the copper(I) oxide surface.36 So, we can propose that the obtained copper(I) oxides can be used for catalyzing ORR at low temperature. Furthermore, the XPS signals obtained after Ar2+ etching treatment show more defined signals. The composition analysis reveals that the Cu/O atomic ratio is about 0.73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.27:1, and 0.13:1 for Cu2O-20, Cu2O-50, and Cu2O-70, respectively. These values are much lower than the 2:1 atomic ratio of Cu2O, which indicates that O and O2 adsorb on the surface of the as-synthesized Cu2O is strong.34
:1, 0.27:1, and 0.13:1 for Cu2O-20, Cu2O-50, and Cu2O-70, respectively. These values are much lower than the 2:1 atomic ratio of Cu2O, which indicates that O and O2 adsorb on the surface of the as-synthesized Cu2O is strong.34
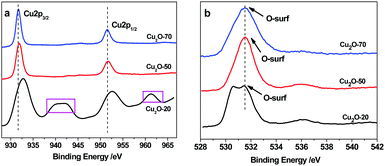 |
| | Fig. 4 XPS spectra of Cu2O-20, Cu2O-50, and Cu2O-70: (a) Cu2p3/2 and (b) O1s spectra. | |
3.2 Electrocatalysis toward ORR
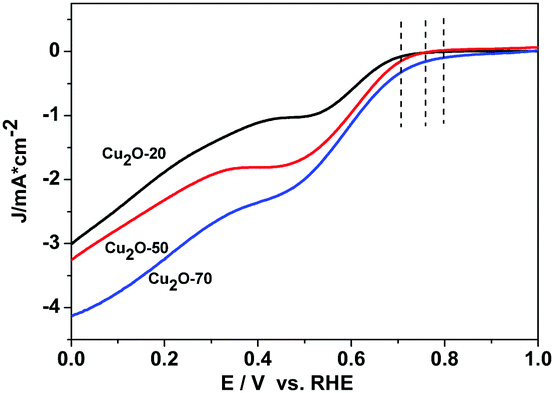
The catalytic activity of Cu2O-20, Cu2O-50, and Cu2O-70 used respectively as a catalyst for ORR was evaluated by cyclic voltammetry (Fig. S6, ESI†). In the CV curves of the three Cu2O modified electrodes in the N2-saturated 0.1 M KOH solution, a pair of redox peaks corresponding to the Cu(II)/Cu(I) redox couple can be observed.37 In the solution saturated with O2, the reduction and oxidation currents in the three Cu2O modified electrodes are enhanced remarkably, revealing that the three different Cu2O samples are all electrochemically active. At the potential of 0.65 V (vs. RHE), the current intensity of the ORR at the electrode with the Cu2O-70 catalyst is 4.20 times that of the electrode with the same catalyst in the N2-saturated solution. And this reduction potential (0.65 V) is more positive than that of the Cu2O-50 catalyst (0.63 V) and that of the Cu2O-20 catalyst (0.62 V), indicating that Cu2O-70 could catalyze ORR at a more positive potential. The rotating-disk voltammogram measurements were used to discuss the ORR kinetics of each sample. The ORR steady-state polarization plots for these Cu2O catalysts at a rotation rate of 1600 rpm are depicted in Fig. 5. As demonstrated by Fig. 5, the ORR activity is greatly morphology dependent. Compared with the spherical and octahedral Cu2O crystals (Cu2O-20 and Cu2O-50, dominated by {111} crystal planes), truncated octahedral Cu2O (Cu2O-70, dominated by {200} crystal planes) produces the highest ORR onset potential (Eonset) of 0.80 V and highest limiting current density of 4.12 mA cm−2 at 0 V of overpotential, followed by Cu2O-50 (0.76 V and 3.22 mA cm−2), then Cu2O-20 (0.71 V and 2.98 mA cm−2). Furthermore, the kinetic current density at 0.60 V (vs. RHE, Jk) of Cu2O-70 (1.85 mA cm−2) is also higher than that of Cu2O-20 (0.94 mA cm−2) and Cu2O-50 (1.67 mA cm−2), which indicates that an enhanced kinetic rate for the ORR can be achieved on truncated octahedral Cu2O as compared with the spherical and octahedral Cu2O crystals. The order of ORR activity based on these parameters increased as follows: Cu2O-70 > Cu2O-50 > Cu2O-20.
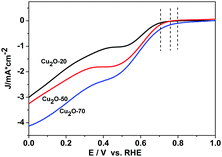 |
| | Fig. 5 ORR steady-state polarization plots for Cu2O-20, Cu2O-50 and Cu2O-70 catalysts at a rotation rate of 1600 rpm, scan rate: 5 mV s−1. | |
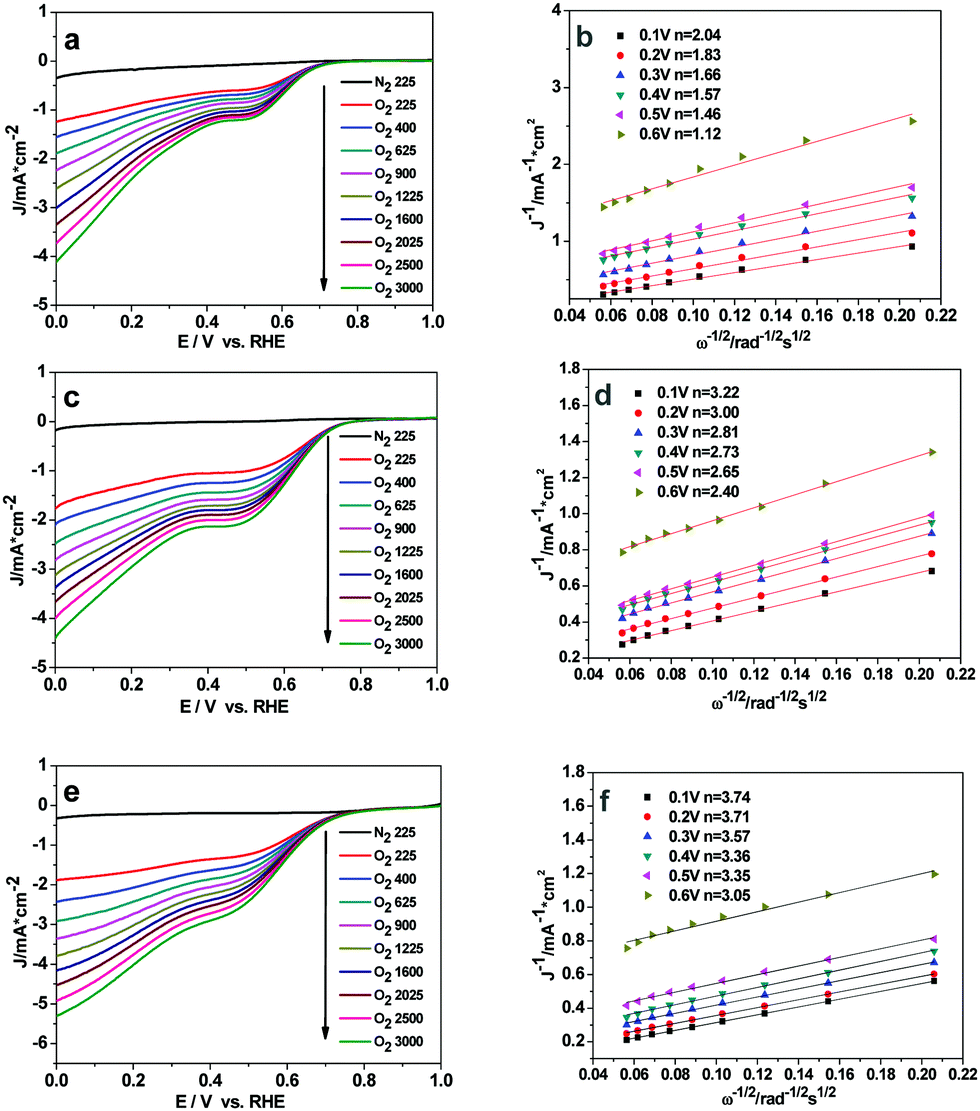
To quantitatively evaluate the ORR activity of the three Cu2O samples, the Koutecky–Levich plots (K–L, j−1vs. ω−1/2) based on the LSV results at various rotation speeds were used to calculate the corresponding number of transferred electrons in ORR (eqn (S3) and (S4), ESI†). The linear-sweep voltammetry (LSV) curves of the Cu2O-20, Cu2O-50, and Cu2O-70 catalysts at different rotation speeds are shown in Fig. 6a, c and e. The calculated Koutecky–Levich (K–L) plots for the three catalysts at a potential of 0.6 to 0.1 V (vs. RHE) are shown in Fig. 6b, d and f. The electron transfer number of Cu2O-70 estimated from the slope of the K–L plot is 3.74 at 0.1 V (vs. RHE), which is higher than that of the Cu2O-50 catalyst (n = 3.22) and that of the Cu2O-20 catalyst (n = 2.04). A diffusion control step at a potential of about 0.4 V (vs. RHE) can be observed from the LSV curves at a high rotation rate (Fig. 6a, c and e), which suggests that the ORR may undergo a 4-electron and 2-electron transfer combined pathway.38 The calculated number of electrons transferred per O2 molecule (n) further supports the higher ORR activity of truncated octahedral Cu2O-70 in agreement with LSV results.
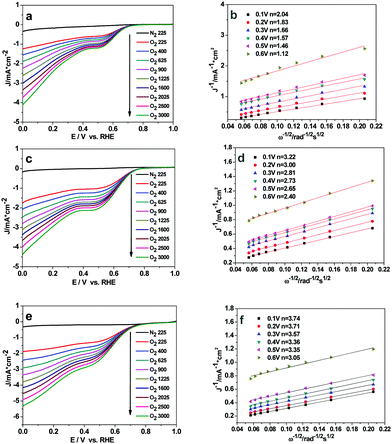 |
| | Fig. 6 Rotating-disk voltammograms recorded for (a) Cu2O-20, (c) Cu2O-50 and (e) Cu2O-70 electrodes in N2- or O2-saturated 0.1 M solution of KOH at different rotation rates; (b, d and f) are Koutecky–Levich plots of J−1versus ω−1/2 at different electrode potentials and the experimental data were obtained from (a, c and e). | |
3.3 Analysis of different activity by DFT
Where does this different ORR activity come from? Since the ORR is a surface reaction, specific surface area, average oxidation state of Cu, crystallinity and morphology may influence the catalyst performance in the ORR. However, in our case, there is not much difference in BET surface area among the samples (the BET surface area of Cu2O-20, Cu2O-50, and Cu2O-70 is 1.74, 0.93 and 0.74 m2 g−1, respectively), so morphology might play an important role in the catalyst performance. The previous experiments reported that octahedral Cu2O exposes (111) crystal planes and the truncated octahedra expose (100) and (111) crystal planes.29,39,40 To gain further insights into the morphology–activity relationship of Cu2O toward ORR, we have conducted a series of DFT calculations to address the adsorption mechanism of atomic O and molecular O2 on the Cu2O (111) and (100) surfaces. We systematically investigated the geometric and electronic structure of atomic O and molecular O2 adsorption on these Cu2O surfaces by analyzing the density of states (DOS) and differential charge density (Tables 1 and 2).
Table 1 Calculated adsorption energy (Eads, eV) and distances (R, Å) for oxygen adsorption on the Cu2O(100) surface
|
|
Conf. 1 |
Conf. 2 |
Conf. 3 |
|
E
ads/eV |
−1.71 |
−1.27 |
−1.83 |
|
R
Cu–O/Å |
1.880/1.883/2.126/2.192 |
1.932/1.931 |
1.852/1.853 |
|
R
O–O/Å |
1.51 |
1.36 |
1.43 |
| ΔRO–O/ΔÅ |
0.29 |
0.14 |
0.21 |
Table 2 Calculated adsorption energy (Eads, eV) and distances (R, Å) for oxygen adsorption on the Cu2O(111) surface
|
|
Conf. 1 |
Conf. 2 |
|
E
ads/eV |
−0.91 |
−0.77 |
|
R
Cu–O/Å |
2.232/2.237 |
2.063/1.877 |
|
R
O–O/Å |
1.37 |
1.37 |
| ΔRO–O/ΔÅ |
0.15 |
0.15 |
(1) O2 adsorption on Cu2O(100).
There are three configurations for O adsorption on Cu2O(100) (Fig. 7). The most stable configuration is that atomic O adsorbs on the two unsaturated surface Cu atoms with an adsorption energy of −1.71 eV, as shown in Fig. 7a. The distance between the O atom and the four unsaturated Cu atoms is 1.880 Å, 1.883 Å, 2.126 Å, and 2.192 Å, respectively. Furthermore, the bond length of O–O is lengthened to 1.51 Å and the elongation value of the O–O bond is 0.29 Å, compared to 1.22 Å of the free O2.41 The second most stable adsorption configuration is shown in Fig. 7b. The surface oxygen atom connects to two surface unsaturated Cu atoms with an adsorption energy of about −1.27 eV and the distance of O–Cu is 1.932 Å and 1.931 Å, respectively, and the distance of O–O is 1.36 Å. The last configuration is that the oxygen atom adsorbs on the bridge of Cu and O, the bond length of Cu–O (adsorbed O) is 1.852 Å and 1.853 Å, and the bond length of O–O is 1.43 Å, the adsorption energy is −1.83 eV as shown in Fig. 7c.
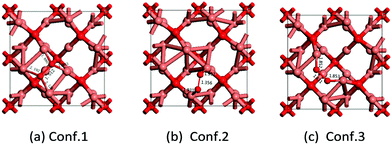 |
| | Fig. 7 Three configurations of O2 adsorption on Cu2O(100) (Cu atom in brown and O in red). | |
(2) O2 adsorption on Cu2O(111).
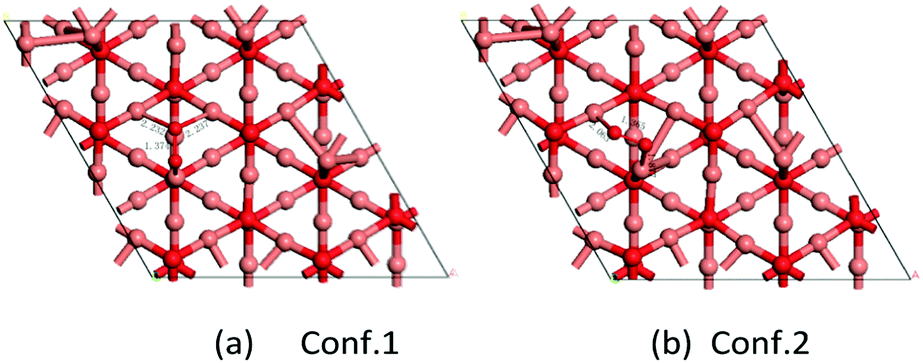
For molecular oxygen adsorption on the Cu-terminated surface of the Cu2O(111) lattice plane, there are two stable adsorption geometries, as shown in Fig. 8. Fig. 8a shows the most stable adsorption configuration with an adsorption energy of −0.91 eV. One O atom connects to one unsaturated Cu atom and the other O atom adsorbs on two saturated Cu atoms and the distances of O–Cusat are 2.232 and 2.237 Å, respectively. The bond length of O–O is 1.37 Å, and the elongation value of the O–O bond is 0.15 Å. Fig. 8b exhibits the other stable adsorption configuration with an adsorption energy of −0.77 eV. Molecular oxygen adsorbs on the surface of the two nearest Cu atoms (one is saturated and the other unsaturated). The distance of O–O is about 1.37 Å and the two bond lengths of Cu–O are 2.063 and 1.877 Å, respectively. According to our DFT calculations on the Cu2O-catalyzed ORR model, as well as the reports about oxygen adsorption on Cu2O surfaces by Wand and Feng,42 the E value of O adsorption (Eads) and the elongation value of the O–O bond (ΔRO–O) can serve as good descriptors for the catalytic activity of the corresponding surface toward ORR. The Eads is larger for O2 on Cu2O(100) surfaces than that on Cu2O(111) surfaces, indicating that O2 prefers to adsorb on Cu2O(100) surfaces. In addition, relatively larger elongation of the O–O bond was found on the Cu2O(100) surfaces, which means that the adsorbed O2 is more easily activated on the Cu2O(100) surfaces than on Cu2O(111) surfaces. Therefore, the higher activity of truncated octahedral Cu2O (Cu2O-70) toward ORR becomes understandable and most likely arises from the morphology peculiarity of the exposed Cu2O(100) facets at the side surface. Both our experimental results and theoretical calculations suggested that the ORR activities of Cu2O crystals should exhibit strong dependency on the exposure of surface facets.
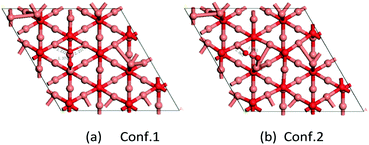 |
| | Fig. 8 Two configurations of O2 adsorption on Cu2O(111) (Cu atom in brown and O in red). | |
4 Conclusions
In this work, Cu2O crystals with three different morphologies (spheres, octahedron, and truncated octahedrons) were prepared by adjusting the reaction electrolyte temperature in the potentiostatic electrodeposition process. The truncated octahedral Cu2O exhibited the highest ORR activity in alkaline media among the three morphologies of Cu2O crystals. As illustrated in the results of HRTEM and XRD, the octahedral Cu2O exposes (111) facets while truncated octahedral Cu2O exposes the (100) and (111) facets. DFT calculations reveal that the adsorption of O2 on Cu2O(100) is found to be stronger and the activation of the adsorbed O2 on Cu2O(100) surfaces is much easier than that on Cu2O(111) surfaces. This might be the intrinsic reason for the superior activity of truncated octahedral Cu2O toward ORR. We have reason to believe that the present finding will deepen the understanding of non-noble metal oxide catalysis and guide the rational design of non-noble metallic oxide catalysts for the ORR.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (No. 21571034), the Natural Science Foundation of Fujian Province (No. 2014J01033), and a key item of the Education Department of Fujian Province (No. JA13085).
Notes and references
- C. Zhu and S. Dong, Nanoscale, 2013, 5, 1753 RSC.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. G. Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 1 Search PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371 CrossRef CAS PubMed.
- N. Mahmood, C. Zhang, J. Jiang, F. Liu and Y. Hou, Chem. – Eur. J., 2013, 19, 5183 CrossRef CAS PubMed.
- M. Liu, Y. Dong, Y. Wu, H. Feng and J. Li, Chem. – Eur. J., 2013, 19, 14781 CrossRef CAS PubMed.
- Y. Dong, Y. Wu, M. Liu and J. Li, ChemSusChem, 2013, 6, 2016 CrossRef CAS PubMed.
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2010, 22, 898 CrossRef CAS.
- Y. Liang, H. Wang, P. Diao, W. Chang, G. Hong, Y. Li, M. Gong, L. Xie, J. Zhou, J. Wang, T. Z. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2012, 134, 15849 CrossRef CAS PubMed.
- J. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333 CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612 CrossRef CAS PubMed.
- Y. Ye, L. Kuai and B. Geng, J. Mater. Chem., 2012, 22, 19132 RSC.
- M. Wang, J. Huang, Z. Tong, W. Li and J. Chen, J. Alloys Compd., 2013, 568, 26 CrossRef CAS.
- S. Deng, V. Tjoa, H. M. Fan, H. R. Tan, D. C. Sayle, M. Olivo, S. Mhaisalkar, J. Wei and C. H. Sow, J. Am. Chem. Soc., 2012, 134, 4905 CrossRef CAS PubMed.
- X. Y. Yan, X. L. Tong, Y. F. Zhang, X. D. Han, Y. Y. Wang, G. Q. Jin, Y. Qin and X. Y. Guo, Chem. Commun., 2012, 48, 1892 RSC.
- Q. Li, P. Xu, B. Zhang, H. Tsai, S. Zheng, G. Wu and H. L. Wang, J. Phys. Chem. C, 2013, 117, 13872 CAS.
- Q. Kuang, X. Wang, Z. Jiang, Z. Xie and L. Zheng, Acc. Chem. Res., 2014, 47, 308 CrossRef CAS PubMed.
- W. Xiao, D. Wang and X. W. Lou, J. Phys. Chem. C, 2010, 114, 1694 CAS.
- X. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638 CrossRef CAS PubMed.
- Y.-X. Chen, S.-P. Chen, Z.-Y. Zhou, N. Tian, Y.-X. Jiang, S.-G. Sun, Y. Ding and Z. L. Wang, J. Am. Chem. Soc., 2009, 131, 10860 CrossRef CAS PubMed.
- Y. Liang, L. Shang, T. Bian, C. Zhou, D. Zhang, H. Yu, H. Xu, Z. Shi, T. Zhang, L.-Z. Wu and C.-H. Tung, CrystEngComm, 2012, 14, 4431 RSC.
- W. Zhou, B. Yan, C. Cheng, C. Cong, H. Hu, H. Fan and T. Yu, CrystEngComm, 2009, 11, 2291 RSC.
- X. Wang, C. Liu, B. Zheng, Y. Jiang, L. Zhang, Z. Xie and L. Zheng, J. Mater. Chem. A, 2013, 1, 282 CAS.
- M. J. Siegfried and K.-S. Choi, Angew. Chem., Int. Ed., 2005, 44, 3218 CrossRef CAS PubMed.
- T. Yang, K. Li, Z. Liu, L. Pu and X. Zhang, J. Electrochem. Soc., 2017, 164, F270 CrossRef CAS.
- C. M. McShane and K. S. Choi, J. Am. Chem. Soc., 2009, 131, 2561 CrossRef CAS PubMed.
- B. Z. Sun, W. K. Chen, X. Wang and C. H. Lu, Appl. Surf. Sci., 2007, 253, 7501 CrossRef CAS.
- A. Werner and H. D. Hochheimer, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 25, 5929 CrossRef CAS.
- K. X. Yao, X. M. Yin, T. H. Wang and H. C. Zeng, J. Am. Chem. Soc., 2010, 132, 6131 CrossRef CAS PubMed.
- H. Li, R. Liu, R. Zhao, Y. Zheng, W. Chen and Z. Xu, Cryst. Growth Des., 2006, 6, 2795 CAS.
- H. Xu, W. Wang and W. Zhu, J. Phys. Chem. B, 2006, 110, 13829 CrossRef CAS PubMed.
- S. Guo, Y. Fang, S. Dong and E. Wang, Inorg. Chem., 2007, 46, 9537 CrossRef CAS PubMed.
- Y. K. Hsu, C. H. Yu, Y. C. Chen and Y. G. Lin, J. Power Sources, 2013, 242, 541 CrossRef CAS.
- J. Li, L. Liu, Y. Yu, Y. Tang, H. Li and F. Du, Electrochem. Commun., 2004, 6, 940 CrossRef CAS.
- D. A. Svintsitskiy, A. I. Stadnichenko, D. V. Demidov, S. V. Koscheev and A. I. Boronin, Appl. Surf. Sci., 2011, 257, 8542 CrossRef CAS.
- P. Grez, F. Herrera, G. Riveros, A. Ramírez, R. Henríquez, E. Dalchiele and R. Schrebler, Phys. Status Solidi A, 2012, 209, 2470 CrossRef CAS.
- L. Zhang, H. Li, Y. H. Ni, J. Li, K. M. Liao and G. C. Zhao, Electrochem. Commun., 2009, 11, 812 CrossRef CAS.
- X. Lv, W. Lv, W. Wei, X. Zheng, C. Zhang, L. Zhi and Q. H. Yang, Chem. Commun., 2015, 51, 3911 RSC.
- H. Bao, W. Zhang, D. Shang, Q. Hua, Y. Ma, Z. Jiang, J. Yang and W. Huang, J. Phys. Chem. C, 2010, 114, 6676 CAS.
- Z. Zheng, B. Huang, Z. Wang, M. Guo, X. Qin, X. Zhang, P. Wang and Y. Dai, J. Phys. Chem. C, 2009, 113, 14448 CAS.
- N. X. Lu, J. Q. Li, Y. J. Xu, W. K. Chen and Y. F. Zhang, J. Mol. Struct., 2004, 668, 51 CrossRef CAS.
- X. Yu, X. Zhang, X. Tian, S. Wang and G. Feng, Appl. Surf. Sci., 2015, 324, 53 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, equations, some tables and figures. See DOI: 10.1039/c7nj04200k |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |