
A phosphorescent fluoride probe based on Eu(III)-DO3A clicked with a 2,5-di(thien-2-yl)pyrrole scaffold†
Memduh
Bilmez‡
a,
Aysun
Degirmenci
a,
Melek Pamuk
Algi
ab and
Fatih
Algi
 *ac
*ac
aASUBTAM Memduh Bilmez BioNanoTech Lab., Aksaray University, TR-68100 Aksaray, Turkey. E-mail: falgi@aksaray.edu.tr
bHealth Services Vocational School, Aksaray University, TR-68100 Aksaray, Turkey
cDepartment of Biotechnology and Molecular Biology, Aksaray University, TR-68100 Aksaray, Turkey
First published on 27th November 2017
Abstract
The design, synthesis and physicochemical properties of a new phosphorescent fluoride probe 1 are described. The probe consists of a stable Eu(III) complex of a DO3A macrocycle on one side and a 2,5-di(thien-2-yl)pyrrole (SNS) scaffold on the other. They were linked together with a 1,2,3-triazole ring as a reliable connection unit, which was formed by Huisgen type copper-catalyzed [3+2] cycloaddition between the corresponding azide and alkyne. This novel material can be used as a turn-on phosphorescent fluoride probe. To the best of our knowledge, this is the first example of the combination of an SNS unit with a clicked Eu(III) complex of DO3A scaffold, which can induce a prominent luminogenic response to fluoride ions among other anions. Finally, its behavior as a two-input OR logic gate was also demonstrated using OH− and OAc− anions.
Introduction
Luminescent materials have attracted considerable attention, especially during the last three decades since they can lead to a variety of advanced technological applications in the field of sensors,1–7 imaging agents,8,9 molecular electronics and photonics,10 electrochromic materials,11–19 light-emitting diodes20 and so on. In particular, luminescent lanthanide complexes offer great promise in photonics due to the superior features of lanthanide ions most of which have characteristic luminescence with large Stokes shift, sharp emission peaks with high color purity and prolonged lifetimes (up to a few milliseconds).21–24 Owing to these unique features, lanthanide luminescence has been considered as one of the most powerful tools that can be used in a variety of physical, chemical and biological applications ranging from sensing to biomedical analyses, imaging, lighting and device applications.25–31 In spite of these advantages of lanthanide ions, they have very low absorption coefficients. For that reason, lanthanide ions require organic ligands to induce photoluminescence. The organic ligands should efficiently absorb and transfer energy to the lanthanide center upon excitation (antenna effect). Recently, lanthanide complexes with suitable organic ligands have been reported to be promising materials for sensing various analytes in terms of their superior optical features.32–36 In particular, intensity-based luminescent probes have been designed for use in (bio)analysis and imaging of specific target ions and/or molecules.37–43 Moreover, they have been widely used as labeling and imaging agents for biomolecules such as proteins, peptides, nucleic acids or antibodies.44–47Fluoride (F−) is a well-known anion that plays important roles in numerous health issues. Although fluoride has beneficial effects on teeth and bones, excess fluoride is responsible for serious disorders such as dental and skeletal fluorosis,48,49 cancer and neurotoxicity.50,51 Therefore, the design of novel molecular probes, which are selective and sensitive to fluoride ions (F−), has gained great attention. Fortunately, a number of examples concerning the sensing of fluoride ions have been reported to date in the literature.52–62
2,5-Dithienylpyrrole (SNS) is a highly versatile scaffold that can afford a variety of materials with decent properties.63–68 Recently, it has been reported that SNS with a pendant luminol arm could be used in the chemiluminogenic detection of metal cations (Cu2+, Fe2+), reactive oxygen species (superoxide radical anion) and/or blood stains (in forensic science).69 Furthermore, it has been revealed that SNS-based materials can be used to fabricate fluorescent sensors and/or probes for the detection of metal ions (Cu2+, Au3+ and Pb2+).70–72 The incorporation of fluorescent units on the SNS-based polymers provides both fluorogenic and electrochromic properties.73–79 Finally, it was found out that novel functions can be easily introduced to the SNS unit via rational design of the backbone structure.79
In this work, we present the design, synthesis and optical properties of a novel material, viz. europium(III) 2,2′,2′′-[10-({1-[2-(2,5-di-2-thienyl-1H-pyrrol-1-yl)ethyl]-1H-1,2,3-triazol-4-yl}methyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (1) (Fig. 1). This novel material 1 consists of a 2,5-di(thien-2-yl)pyrrole (SNS) unit and a stable Eu(III) complex of a DO3A macrocycle. They were linked together with a triazole ring that was formed by a Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition reaction. Therefore, we have envisaged that this unique design could provide not only the sensitization of Eu(III) luminescence by SNS as the antenna, but also a recognition site for anions on the saturated lanthanide metal center. To our delight, it was found that 1 can be utilized as a turn-on phosphorescent F− probe. To the best of our knowledge, this is the first example of an SNS derivative with a clicked Eu(III) complex of DO3A scaffold, which can induce a prominent luminogenic response to fluoride among other anions. Furthermore, it was shown that 1 could report the presence of AcO− and OH− anions in acetonitrile solution. Finally, the digital action of a two-input OR logic gate is also demonstrated using the OH− and OAc− anions.
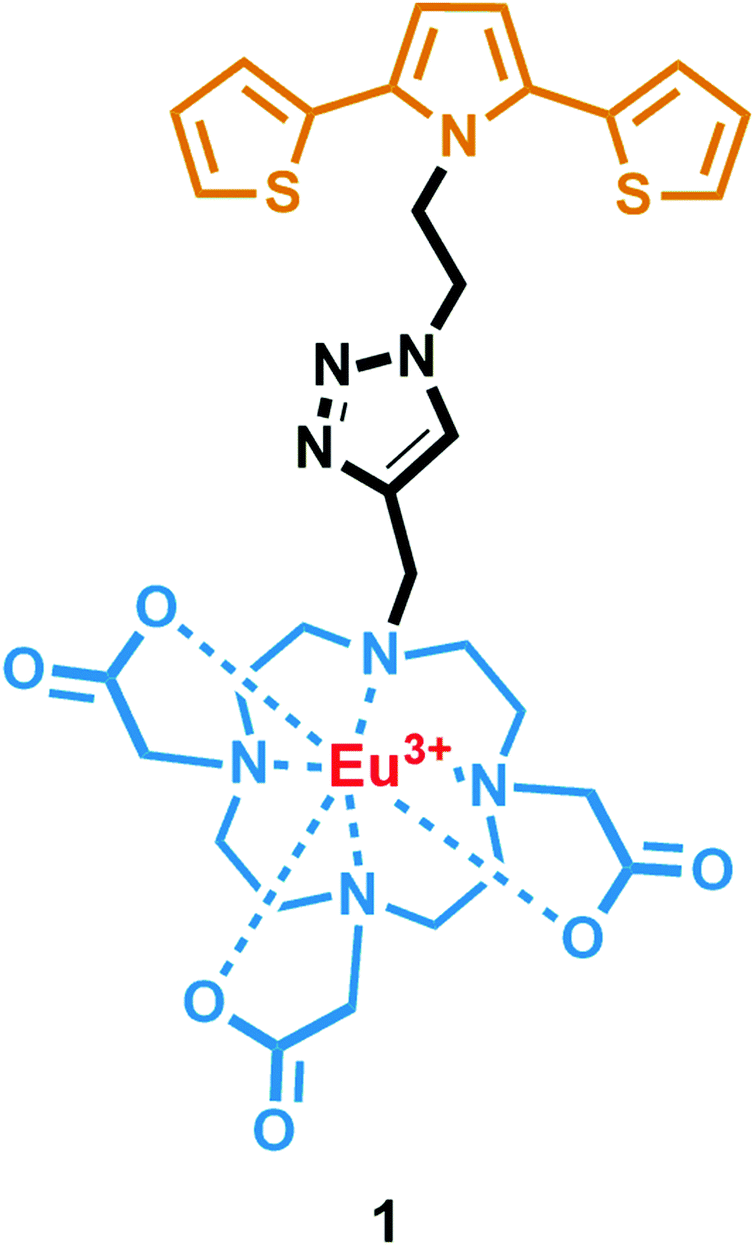 | ||
| Fig. 1 The structure of the target compound 1. | ||
Experimental section
General
All the chemicals were purchased from Sigma Aldrich Chemicals or Merck Company and used as received unless otherwise noted. The FTIR spectra were recorded on a Perkin Elmer Spectrum 100 model FTIR with attenuated total reflectance (ATR) and a Thermo Scientific Nicolet iS5 FT-IR Spectrometer with iD5-ATR. The 1H (300 MHz) and 13C (75 MHz) NMR spectra were recorded on a Bruker Ultrashield 300 NMR Spectrometer. Combustion analyses were carried out by using a LECO CHNS-932 analyzer. Matrix-assisted laser desorbtion/ionization (MALDI-TOF) mass spectra were recorded on a Bruker microflex LT MALDI-TOF MS system. The UV-Vis and fluorescence measurements were recorded on Varian Cary 50 and Varian Cary Eclipse spectrophotometers, respectively. Tetrabutylammonium salts were used in titrations and all the measurements were carried out immediately after the addition of ions. The melting points were determined on a Schorrp MPM-H2 model apparatus and are uncorrected. Column chromatography was performed on silica gel (60–200 mesh) from Merck Company. TLC was carried out on Merck 0.2 mm silica gel 60 F254 analytical aluminum plates. Compounds 4,8010,8212,83 and 1384 were synthesized according to a previously published procedure.Synthesis of 1,4-di(thiophen-2-yl)butane-1,4-dione (4)
To a suspension of AlCl3 (16 g, 0.12 mol) in dichloromethane (15 mL) a solution of thiophene 2 (9.61 mL, 0.12 mol) and succinyl chloride 3 (5.51 mL, 0.05 mol) in CH2Cl2 was added dropwise. The red mixture was stirred at room temperature for 18 h. This was then quenched with ice and concentrated HCl (5 mL). After stirring for 2 hours, the dark green organic phase was separated, and washed with 2 M HCl, H2O and NaHCO3 solution, respectively, and the organic layer was dried over MgSO4. After the evaporation of the solvent a blue-green solid remained, which was suspended in ethanol. The solid product was filtered and washed with ethanol and diethyl ether and this gave a green solid. The crude product was purified using column chromatography (CH2Cl2–hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)) and recrystallized from ethanol to give 41 as a white solid in 72% yield. M.p. 129–130 °C (lit. 126–127 °C); 1H NMR (400 MHz, CDCl3, δ): 7.75 (dd, 2H, J = 4.0–1.0 Hz), 7.57 (dd, 2H, J = 4.8–1.0 Hz), 7.08 (dd, 2H, J = 4.8–4.0 Hz), 3.30 (s, 4H); 13C NMR (100 MHz, CDCl3, δ): 191.5, 143.9, 133.8, 132.3, 128.3; FTIR (cm−1): 3101 (sp2 C–H), 2919 (sp3 C–H), 1649 (C
:1)) and recrystallized from ethanol to give 41 as a white solid in 72% yield. M.p. 129–130 °C (lit. 126–127 °C); 1H NMR (400 MHz, CDCl3, δ): 7.75 (dd, 2H, J = 4.0–1.0 Hz), 7.57 (dd, 2H, J = 4.8–1.0 Hz), 7.08 (dd, 2H, J = 4.8–4.0 Hz), 3.30 (s, 4H); 13C NMR (100 MHz, CDCl3, δ): 191.5, 143.9, 133.8, 132.3, 128.3; FTIR (cm−1): 3101 (sp2 C–H), 2919 (sp3 C–H), 1649 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
Synthesis of 1-(2-bromo-ethyl)-2,5-di(thiophen-2-yl)-1H-pyrrole (6)
2-Bromo ethylamine hydrobromide (0.74 g, 3.61 mmol) was heated to dissolve in ethanol (3 mL). After that, Et3N (0.5 mL) was added and the precipitate formed was separated by filtration in a 100 mL flask. The solvent of the liquid portion was removed under reduced pressure to give 2-bromo ethylamine (5). Next, 1,4-di(thiophen-2-yl)butane-1,4-dione (4) (805 mg, 3.22 mmol), 2-bromo ethylamine (5) (400 mg, 3.22 mmol) and p-toluenesulfonic acid (PTSA) (5 mg) were dissolved in 60 mL of anhydrous toluene and the reaction mixture was heated under reflux by using Dean–Stark apparatus until all the starting materials were consumed. After the reaction, the solvent was removed under reduced pressure. The residue was filtered through a short pad of silica gel by eluting with hexane to give 6 as a yellow solid in 30% yield, m.p. 70–74 °C; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.34 (dd, J = 1.35, J = 4.94, 2H), 7.11–7.06 (m, 4H), 6.33 (s, 2H), 4.50 (t, J = 7.71, 2H), 3.32 (t, J = 8.08, 2H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 134.0, 128.3, 127.4, 126.1, 125.9, 111.4, 46.1, 29.3; FTIR (cm−1): 3097 (sp2 C–H), 2953 (sp3 C–H), 1661 (CC), 696 (C–Br); anal calcd for C14H12BrNS2: C, 49.71; H, 3.58; N, 4.14; found: C, 49.60; H, 3.40; N, 4.25. MALDI-TOF MS calcd for C14H12BrNS2: 338.29 [M]+ Found: 339.35 [M + H]+.
Synthesis of 1-(2-azidoethyl)-2,5-di(thiophen-2-yl)-1H-pyrrole (7)
1-(2-Bromo-ethyl)-2,5-di(thiophen-2-yl)-1H-pyrrole (6) (86 mg, 0.25 mmol), potassium iodide (80 mg, 0.48 mmol) and sodium azide (50 mg, 0.76 mmol) were dissolved in anhydrous DMF (2 mL), and the mixture was heated to 50 °C. After the completion of the reaction, the mixture was allowed to cool to room temperature and poured into 100 mL of water and extracted with hexane (3 × 100 mL), and dried over MgSO4. The mixture was filtered and the solvent was removed under reduced pressure to give 7 as a brown viscose oil in 74% yield; 1H NMR (CDCl3, 400 MHz) δ/ppm: 7.34 (dd, J = 1.96, J = 4.37, 2H), 7.12–7.08 (m, 4H), 6.37 (s, 2H), 4.35 (t, J = 6.59, 2H), 3.24 (t, J = 6.60, 2H); 13C NMR (CDCl3, 100 MHz) δ/ppm: 133.1, 127.3, 126.5, 125.6, 124.9, 110.7, 49.5, 42.9; FTIR (cm−1): 3104 (sp2 C–H), 2959 (sp3 C–H), 2095 (–N3), 1659 (CC); anal calcd for C14H12N4S2: C, 55.97; H, 4.03; N, 18.65; found: C, 55.80; H, 3.90; N, 18.75. MALDI-TOF MS calcd for C14H12N4S2: 300.40 [M]+ found: 300.79 M+.
Synthesis of 1,4,7-tris(tert-butoxycarboxymethyl)-1,4,7,10-tetraazacyclododecane: (t-Bu)3DO3A (10)
Cyclen 8 (0.5 g, 2.91 mmol) and sodium bicarbonate (1.22 g, 14.52 mmol) were dissolved in acetonitrile (30 mL) and nitrogen gas was passed through the medium. After that, the mixture was cooled to 0 °C and tert-butylbromo acetate 9 (1.42 mL, 9.68 mmol) was added dropwise. After the addition of 9, the mixture was stirred at 0 °C for 1 h and stirred for 24 hours at room temperature. The reaction mixture was filtered, the solvent was removed under reduced pressure and the crude product was recrystallized from toluene. Cyclen-triester 1082 was obtained as a white solid in 55% yield; m.p. 208–210 °C; 1H NMR (400 MHz, CDCl3) δ/ppm: 3.38 (t, 4H), 3.30 (t, 2H), 3.11 (s, 4H), 2.94–2.89 (m, 12H), 1.47 (s, 27H); 13C NMR (100 MHz, CDCl3) δ/ppm: 170.5, 169.6, 81.8, 81.7, 58.2, 51.3, 51.1, 49.1, 48.6, 47.5, 28.2, 28.1; FTIR (cm−1): 2971 (sp3 C–H), 2851 (sp3 C–H), 1718 (CO), 1368 (C–N), 1146–1098 (C–O).
Synthesis of 1,4,7-tris(tert-butoxycarbonylmethyl)-10-(prop-2-yn)-1,4,7,10-tetraazacyclododecane (12)
(t-Bu)3DO3A 10 (100 mg, 0.2 mmol) was dissolved in acetonitrile (4 mL) and placed in a 100 mL round-bottomed flask under argon atmosphere. Potassium carbonate (60 mg, 0.43 mmol) and propargyl bromide 11 (27 μL) were added to the solution. The reaction mixture was stirred at room temperature for 4 h. After that, the mixture was filtered to get rid of potassium carbonate and the solvent was removed under reduced pressure. The crude product was re-dissolved in dichloromethane (100 mL) and extracted with water (3 × 20 mL) and brine (3 × 20 mL) and the organic layer was dried over MgSO4. After solvent evaporation under reduced pressure, a yellowish oil of 1283 remained in 86% yield; 1H NMR (CDCl3, 400 MHz) δ/ppm: 3.25 (s, 2H), 3.11–3.01 (m, 6H), 2.92–2.77 (m, 12H), 2.42 (m, 4H), 2.08 (s, 1H), 1.42–1.37 (m, 27H); 13C NMR (CDCl3, 100 MHz) δ/ppm: 173.5, 173.0, 82.7, 82.4, 79.1, 72.87, 56.7, 55.8, 51.5, 50.9, 49.9, 49.3 (–NCH2–), 42.9, 28.0; FTIR (cm−1): 3297 (sp C–H), 2975 (sp3 C–H), 2830 (sp3 C–H), 2130 (C–C triple bond), 1721 (CO), 1368 (C–N), 1103–1156 (C–O).
Synthesis of 1,4,7-tris(carboxymethyl)-10-(prop-2-ynyl)-1,4,7,10-tetraazacyclododecane (13)
1,4,7-Tris(tert-butoxycarbonylmethyl)-10-(prop-2-ynyl)-1,4,7,10-tetraazacyclododecane (12) (182 mg, 0.33 mmol) was dissolved in dichloromethane (2.5 mL) and trifluoroacetic acid (TFA) (2.5 mL) was added and the solution was stirred at room temperature for 24 h. The solvent was removed in vacuo. Then dichloromethane (2 × 5 mL) was added and removed under vacuum, followed by diethyl ether (2 × 5 mL) and removed under vacuum. The cyclen-triacid bis-trifluoroacetate salt 1384 was obtained as a hygroscopic white solid in 85% yield; 1H NMR (D2O, 300 MHz) δ/ppm: 3.82–3.77 (m, 6H), 3.60 (bs, 2H), 3.26–3.15 (m, 18H); 13C NMR (D2O, 75 MHz) δ/ppm: 174.3, 171.9, 77.6, 72.6, 56.8, 54.4, 53.2, 53.1, 52.8, 51.7, 49.7, 48.8, 45.4, 43.6, 36.9, 31.3; FTIR (cm−1): 3400 (O–H) 3221 (sp C–H), 2986 (sp3 C–H), 2860 (sp3 C–H), 2131 (C–C triple bond), 1734 (CO), 1367 (C–N), 1103–1156 (C–O); LC MS (m/z): M+ calculated: 384.20, measured [M + H]+: 385.50.
Synthesis of Eu(III) complex 14
A solution of 1,4,7-tris(carboxymethyl)-10-(prop-2-ynyl)-1,4,7,10-tetraazacyclododecane (13) (76 mg, 0.2 mmol) and EuCl3·6H2O (87 mg, 0.24 mmol) was prepared in water (5 mL) and adjusted to pH 7 by the addition of NaOH. On mixing the pH fell to ∼5, due to the release of protons on complexation. The solution was adjusted to pH 6 using NaOH and was stirred at room temperature for 72 h. The solution was filtered and lyophilized giving a white hydroscopic solid 1381 in 60% yield. M.p. 298 °C (decomposed); LC MS (m/z) M+ calculated: 534.10, measured [M + Na + H2O]+: 575.48, [M + H2O + H]+: 553.58 and [M + H]+: 535.25. FTIR (cm−1): 3221 (sp C–H), 2868 (sp3 C–H), 2130 (C–C triple bond), 1684 (CO), 1389 (C–N), 1204–1081 (C–O).
Synthesis of SNS–cyclen Eu(III) complex 1
CuI (15 mg, 20% mmol) was added to a solution of azide 7 (30 mg, 0.1 mmol) and the alkyne 14 (43 mg, 0.08 mmol) in anhydrous acetonitrile (50 mL), and the mixture was heated under reflux. After the completion of the reaction, the mixture was allowed to cool to room temperature and filtered through a short pad of Celite 545, and the solvent was removed under reduced pressure to give a pure khaki colored powder solid 1 in a yield of 60%. M.p. 194 °C (decomposed); UV-Vis (CH3CN, λmax): 243, 306 nm; Luminescence (CH3CN, λmax): 412, 590, 615, 695 nm; FTIR (cm−1): 3107 (sp2 C–H), 2950–2867 (sp3 C–H), 1681 (CO), 1600 (CC aromatic and CC triazole overlapped), 1437 (NN, triazole) 1398 (C–N), 1202–1078 (C–O). Anal calcd for C31H37EuN8O6S2: C, 44.66; H, 4.47; N, 13.44; found: C, 44.40; H, 4.30; N, 13.55. LC MS (m/z) M+ calculated: 834.15, measured [M + Na]+: 857.10, [M + H]+: 835.13.
Results and discussion
Synthesis
In order to obtain the target molecule 1, initial efforts were directed towards the construction of a clickable SNS unit and Eu(III)-DO3A macrocycle. The synthesis of the target compound 1 started with the acylation of thiophene 2 with succinyl chloride 3 to give 1,4-di(thien-2-yl)butane-1,4-dione 4 in 72% yield.80 Next, the bromo-SNS unit was synthesized in 30% yield by a Paal–Knorr reaction of the dione 4 and 2-bromoethylamine 5 in the presence of a catalytic amount of p-toluenesulfonic acid (PTSA) by using a Dean–Stark trap. The nucleophilic substitution reaction of bromo-SNS 6 with NaN3 in the presence of a catalytic amount of KI in DMF afforded azide 7 in 74% yield (Scheme 1).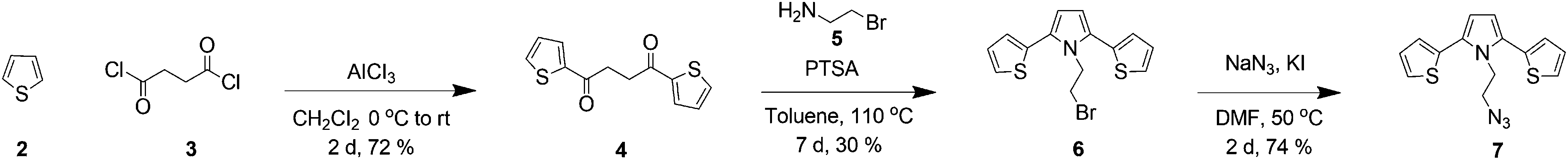 | ||
| Scheme 1 The strategic route for the synthesis of SNS-azide 7. | ||
On the other hand, the synthesis of the Eu(III) complex of the DO3A macrocycle 14 was carried out in four steps according to a slightly modified procedure.81,82 For this purpose, cyclen8 was chosen as the starting compound. Cyclen-triester 10 and bis-alkynyl-cyclen-triester 12 were synthesized according to a previously reported procedure.82,83 The deprotection of bis-alkynyl-cyclentriester with trifluoroacetic acid (TFA) gave triacid 13.84 The complexation of triacid 13 was carried out with EuCl3.6H2O salt with careful adjustment of the pH (around 6.0) in water to afford Eu(III)-DO3A 14 in 80% yield (Scheme 2).81 All of the compounds obtained from these reactions were characterized by the traditional spectroscopic methods (1H and 13C NMR, mass spectroscopy, FTIR and elemental analysis etc.) in order to prove the structures explicitly (see ESI† for details, Fig. S1–S24).
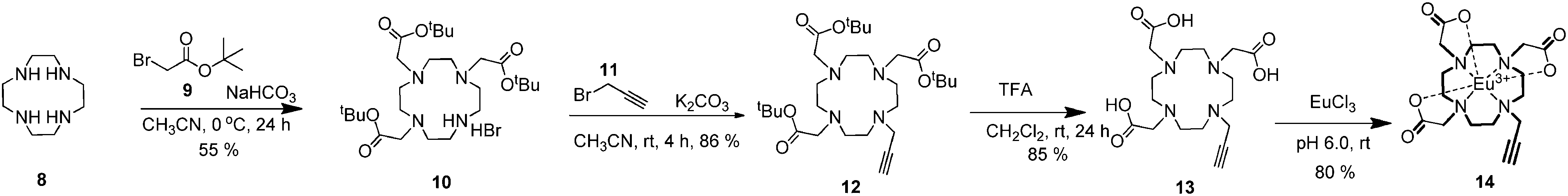 | ||
| Scheme 2 The synthesis of Eu(III)-DO3A 14. | ||
After the synthesis of SNS–azide 7 and Eu(III)-DO3A 14, we investigated the click reaction between 7 and 14 in order to obtain the target compound 1via the regioselective Cu(I)-catalyzed alkyne-azide 1,3-dipolar cycloaddition (CuAAC) reaction. Interestingly, standard click reaction conditions, where CuSO4·5H2O and Na-ascorbate were used as the catalyst system under a variety of conditions (solvents and/or solvent mixtures such as H2O, alcohol, THF etc., or variation of temperature: rt and/or 40 °C), did not furnish compound 1 at all.81 Furthermore, all of our attempts to obtain 1 by using an outstanding CuAAc catalyst85 reported by Ozcubukcu et al. under different conditions unfortunately failed. These results encouraged us to use relatively harsh conditions to accomplish the click reaction. In this context, we have previously reported the synthesis of 1,4-benzoquinone-containing 1,2,3-triazoles by using CuI as a catalyst at 80 °C in an organic solvent under inert atmosphere.86 Fortunately, it was found that the treatment of 7 and 14 with a catalytic amount of CuI at 80 °C in anhydrous acetonitrile under argon atmosphere provided the desired compound 1 in 60% yield (Scheme 3).
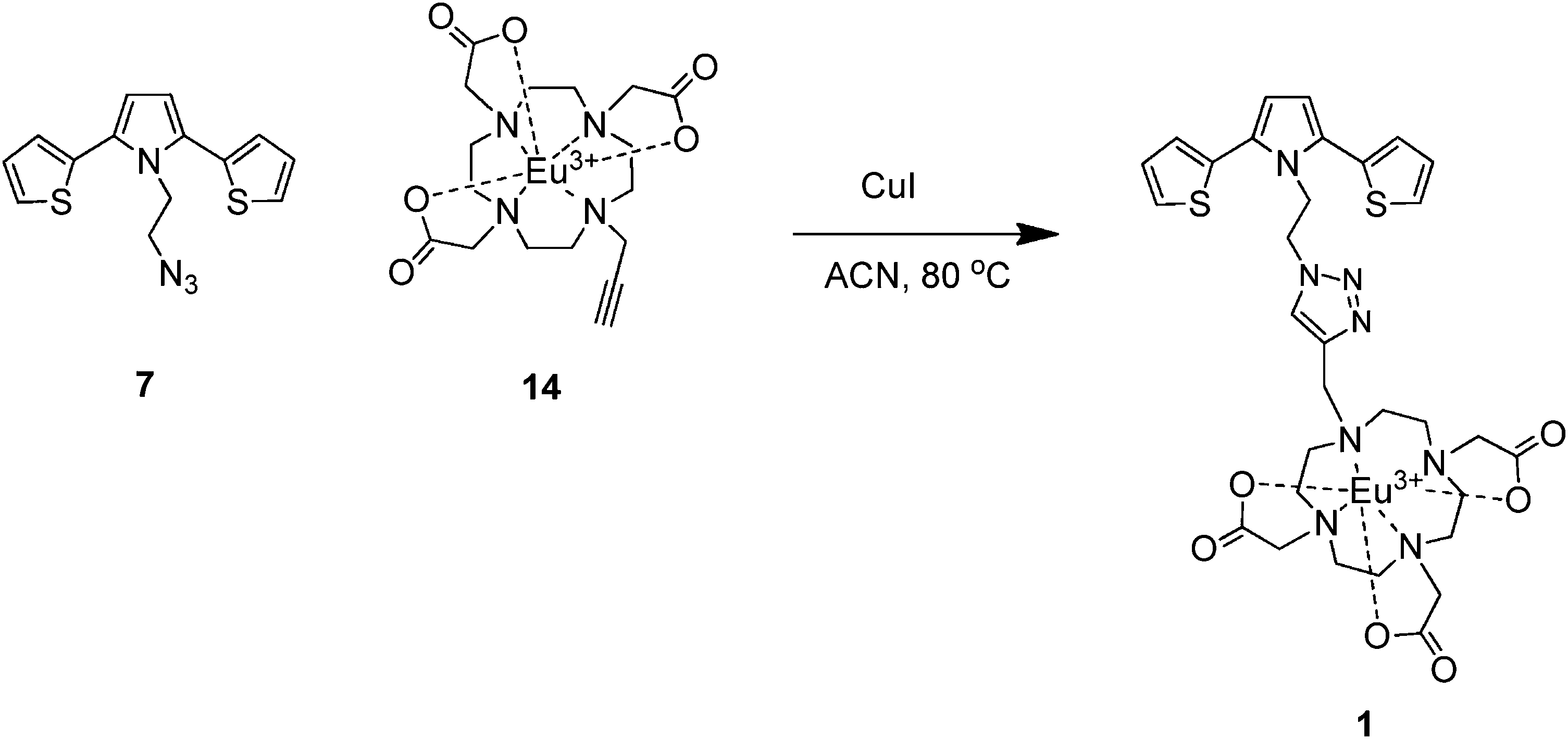 | ||
| Scheme 3 The synthesis of 1via Huisgen 1,3-dipolar cycloaddition. | ||
Photophysical properties
First of all, the photophysical properties of the target compound 1 were investigated in CH3CN. The UV-Vis absorption spectrum of 1 (5 × 10−5 M) exhibits two absorption bands centered at 243 and 306 nm with extinction coefficients (ε) of 16515 and 12367 M−1 cm−1, respectively (Fig. S25, ESI†). On the other hand, the fluorescence emission spectrum of 1 was characterized by a broad emission peak with a λmax(em) at 411 nm when excited (λexc) at 305 nm (Fig. S26, ESI†). The anion responsiveness of 1 was investigated in the same solution by spectrophotometric titrations with different anions. Compound 1 was weakly luminescent (Φ87 = 0.029). It was observed that the phosphorescence spectrum of 1 showed insignificant changes upon the addition of BF4−, PF6−, ClO4−, H2PO4−, N3−, CN−, Cl−, Br− and I− anions. Fortunately, however, the emission intensity of 1 increased dramatically (ca. 6-fold) when F− ions were added to the solution of 1 (Fig. 2). To our serendipitous advantage, compound 1 responds to fluoride anions very quickly due to the fast interaction process. It was shown that the emission intensity increases immediately after the addition of the anion, thus providing a rapid (zero-wait)88 detection method (Fig. S27, ESI†). It was also found that the emission intensity increased in the presence of OH− and AcO− anions, but to a smaller extent when compared to fluoride anions. It should be noted that fluoride and hydroxide anions are isoelectronic, both of which have the same charge and similar ionic radius (1.285 A for F−vs. 1.32 A for OH−).89Fig. 3 illustrates the relative phosphorescence emission intensity of 1 (0.8 × 10−5 M) in the presence of various anions (2 equiv.) in CH3CN. | ||
| Fig. 2 Phosphorescence spectra of 1 (0.8 × 10−5 M) in CH3CN in the presence of various anions (2 equiv.) (λexc = 305 nm). | ||
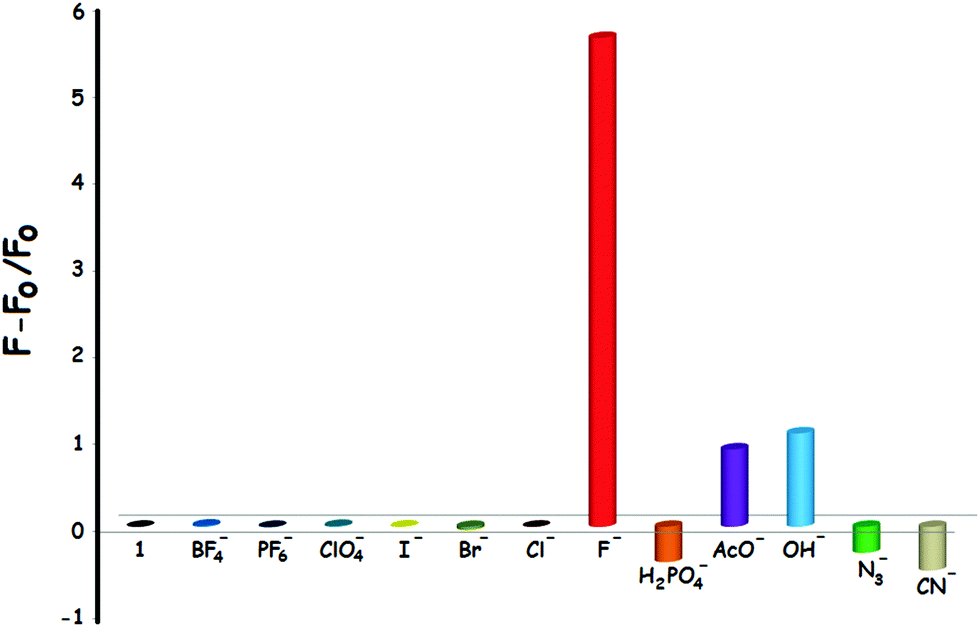 | ||
| Fig. 3 Relative phosphorescence emission intensity of 1 (0.8 × 10−5 M) in the presence of various anions (2 equiv.) in CH3CN. The emissions data of 1 were recorded at 615 nm; excitation was at 305 nm (λexc = 305 nm). | ||
The effect of F− concentration on the emission of 1 was investigated in detail. Fig. 4 depicts the phosphorescence spectral changes of 1 (0.8 × 10−5 M) as a function of F− concentration (0.0–2.0 equiv.) in CH3CN at room temperature. It is important to note that a progressive increase in the phosphorescence emission intensity (Φ = 0.17) was observed as the concentration of the ion increased. The changes in the phosphorescence emission profile of 1 upon the addition of F− anions indicated the formation of a complex between the Eu(III) center in 1 and F−. The stoichiometry of the complex between 1 and F− was determined on the basis of the Job plot analysis, which pointed out a 1:2 ratio (see ESI,† Fig. S28). It has recently been demonstrated that the europium complexes of DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) derivatives having a water molecule at the ninth coordination site could be very selective for F− sensors. Therefore, the fluoride ion displaces the coordinated water molecule during complexation and this enhances europium-based luminescence. Moreover, it has been shown that a fluoride anion sandwiched between the two Eu complexes.90,91 In this context, our result for the stoichiometric ratio between 1 and F− is consistent with literature examples about lanthanide-based fluoride probes. It has been indicated that upon binding of F− anions to 1, the phosphorescence emission of the lanthanide ion is turned on.
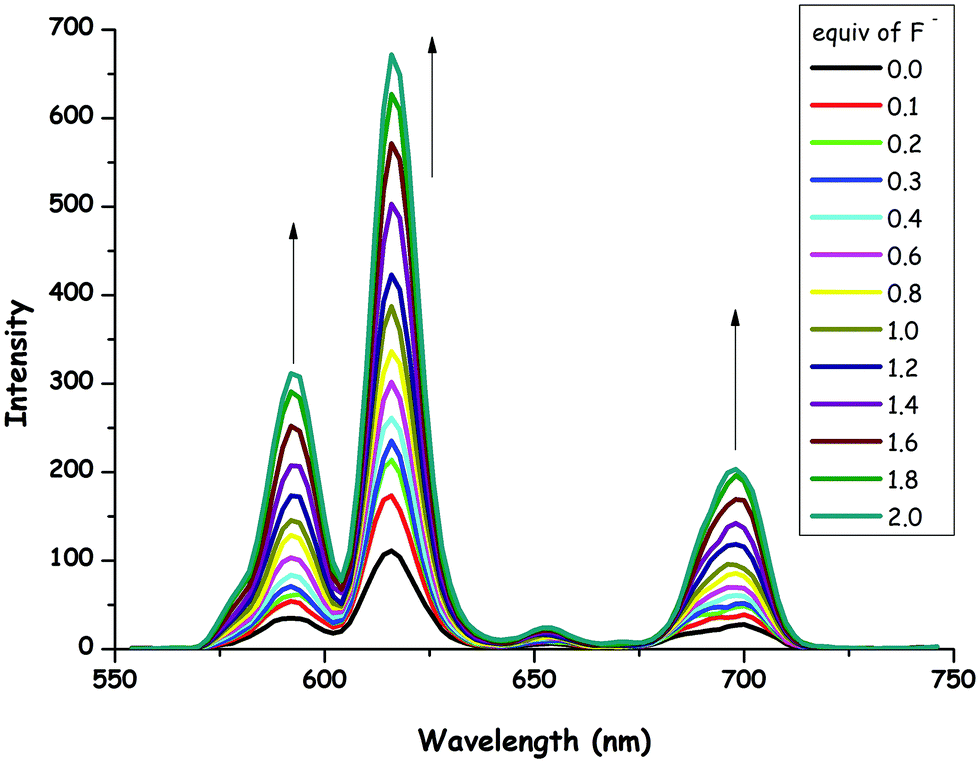 | ||
| Fig. 4 Phosphorescence emission spectra of 1 (0.8 × 10−5 M) as a function of various amounts (0.0, 0.1, 0.2, 0.3, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8 and 2.0 equiv.) of F− anions in CH3CN at room temperature (λexc = 305 nm). | ||
On the basis of the above spectrophotometric titrations, the binding constant (Ka) of 1 with F− was determined from the emission intensity data following the steady-state fluorometric method,2,4,70,71 in which I0 refers to the phosphorescence intensities of solutions of 1. When I0/(I − I0) is plotted against [F−]−1, Ka was calculated to be 2.5 × 105 from the ratio of the intercept/slope with a good correlation coefficient (R = −0.99297) (see ESI,† Fig. S29). Also, the detection limit2,4,70,71 of 1 for F− was calculated to be 0.5 × 10−6 M, which is below the levels defined by the United States Environmental Protection Agency (EPA)92 (see ESI,† Fig. S30).
As was previously mentioned, the emission intensity of 1 increased to a smaller extent in the presence of OH− (Fig. 5) and OAc− (Fig. 6) anions when compared to F− anion. These anions could also bind to the Eu(III) center as in the case of fluoride, thus resulting in an increase in phosphorescence intensity as shown in Fig. 5 and 6. The binding constants (Ka) of 1 with OH− and OAc− were calculated to be 0.56 × 104 and 0.95 × 106, respectively (see ESI,† Fig. S31 and S32). Also, the detection limits of 1 for OH− and OAc− were found to be 0.70 × 10−6 M and 0.87 × 10−6 M, respectively (see ESI,† Fig. S33 and S34).
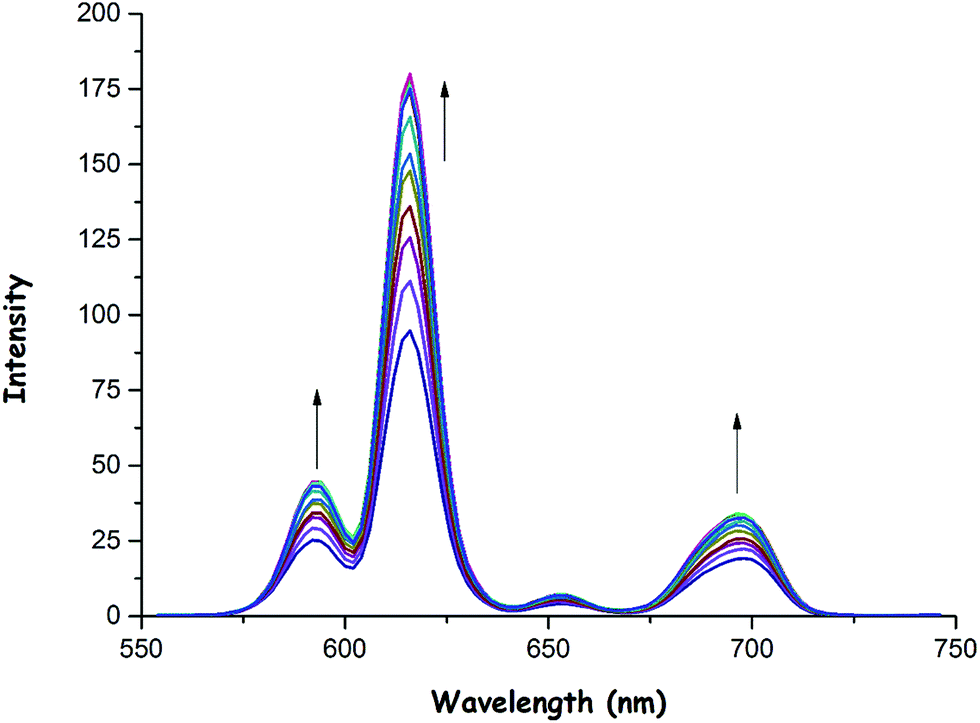 | ||
| Fig. 5 Phosphorescence emission spectra of 1 (0.8 × 10−5 M) as a function of various amounts (0.0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.2, 1.4, 1.6, 1.8 and 2.0 equiv.) of OH− anions in CH3CN at room temperature (λexc = 305 nm). | ||
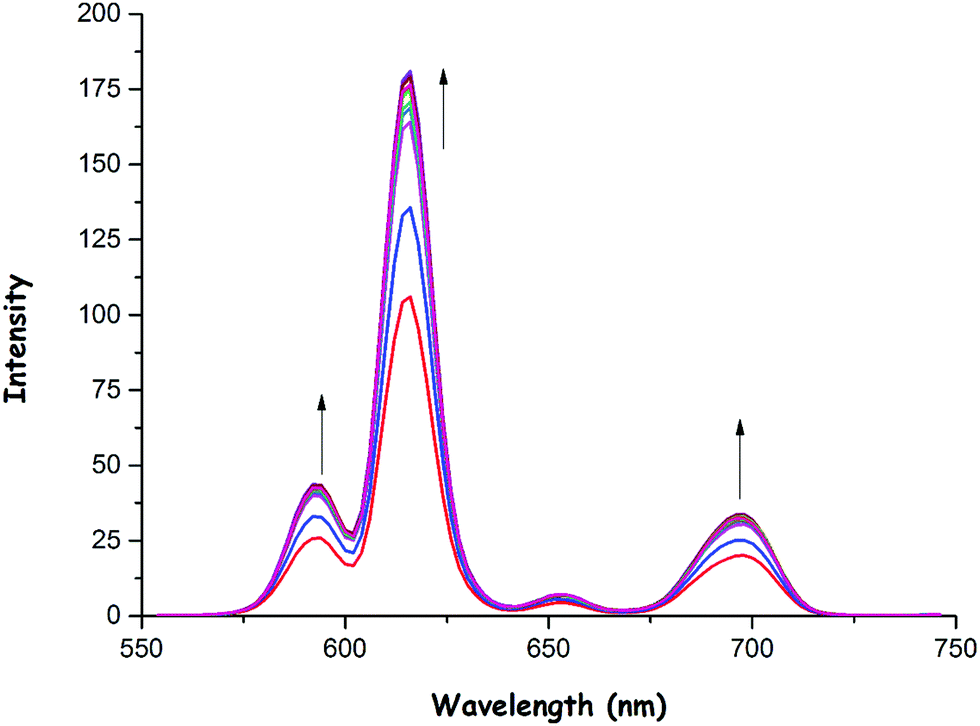 | ||
| Fig. 6 Phosphorescence emission spectra of 1 (0.8 × 10−5 M) as a function of various amounts (0.0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0 and 2.0 equiv.) of OAc− anions in CH3CN at room temperature (λexc = 305 nm). | ||
These observations led us to explore the possibility of OR7,93,94 logic gate95 behavior of probe 1 on the basis of the fact that 1 was responsive to both OH− and AcO− anions as two different chemical inputs. Fortunately, the addition of both inputs produced a well-defined positive signal (Table 1). It was found that the emission intensities of 1 in the presence of OH− (Fig. 5) and OAc− (Fig. 6) anions were approximately at the same level, which indicated the turn-on state of the probe 1. Importantly, these results show that probe 1 allows the construction of a two-input OR logic gate with the OH− and AcO− anions as depicted in Table 1. Remarkably, the output is in good agreement with the truth table of an OR logic gate.
| Input 1 | Input 2 | Output |
|---|---|---|
| 0 | 0 | 0 |
| 0.8 × 10−5 M OH− | 0 | 1 |
| 0 | 0.8 × 10−5 M OAc− | 1 |
| 0.8 × 10−5 M OH− | 0.8 × 10−5 M OAc− | 1 |
Conclusion
In summary, the design and synthesis of a new phosphorescent fluoride probe 1 were described. The probe consists of a stable Eu(III) complex of a DO3A macrocycle and a 2,5-di(thien-2-yl)pyrrole (SNS) scaffold. They were linked together with a 1,2,3-triazole ring as a reliable connection unit, which was formed by the Cu(I) catalyzed 1,3-dipolar cycloaddition reaction between the corresponding azide and alkyne. This novel material can be used as a turn-on phosphorescent fluoride (F−) probe. To the best of our knowledge, this is the first example of an SNS derivative with a clicked Eu(III) complex of DO3A scaffold, which can induce a prominent luminogenic response to fluoride among other anions. Its behavior as a two-input OR logic gate was also demonstrated. Considering the multitude of choices in lanthanide metal selection, we are confident that this design can be easily extended to a variety of novel supramolecular architectures, which can easily find some practical applications in many fields. Further work to show the versatility of this type of material (as ion sensors, MRI agents and/or electrochromic materials etc.) is currently underway in our laboratories and the results will be reported in due time.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors are grateful to the Scientific and Technological Research Council of Turkey (TUBITAK, Grant No. 112T668) and European Cooperation in Science and Technology (CMST COST Action CM 1006 European F-Element Network (EUFEN)). F. A. is indebted to the Turkish Academy of Sciences (TUBA) for an Outstanding Young Investigator Award (GEBIP). M. B. and A. D. are indebted to TUBITAK for graduate fellowships. F. A. thanks S. Ozcubukcu for providing a small amount of their click catalyst as a gift.References
- H. Weng and B. Yan, Inorg. Chem. Commun., 2016, 63, 11–15 CrossRef CAS.
- M. Pamuk and F. Algi, Tetrahedron Lett., 2012, 53, 7010–7012 CrossRef CAS.
- M. Pamuk and F. Algi, Tetrahedron Lett., 2012, 53, 7117–7120 CrossRef CAS.
- S. Karakaya and F. Algi, Tetrahedron Lett., 2014, 55, 5555–5559 CrossRef CAS.
- A. Yoldas and F. Algi, RSC Adv., 2015, 5, 7868–7873 RSC.
- M. Pamuk-Algi, Tetrahedron, 2016, 72, 1558–1565 CrossRef.
- M. Pamuk-Algi, J. Fluoresc., 2016, 26, 487–496 CrossRef PubMed.
- L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS.
- Y. Song, E. K. Kohlmeir and T. J. Meade, J. Am. Chem. Soc., 2008, 130, 6662–6663 CrossRef CAS PubMed.
- B. J. Coe and N. R. M. Curati, Comments Inorg. Chem., 2004, 25, 147–184 CrossRef CAS.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- A. Cihaner and F. Algi, Adv. Funct. Mater., 2008, 18, 3583–3589 CrossRef CAS.
- M. Icli, M. Pamuk, F. Algi, A. M. Onal and A. Cihaner, Chem. Mater., 2010, 22, 4034–4044 CrossRef CAS.
- M. Icli, M. Pamuk, F. Algi, A. M. Onal and A. Cihaner, Org. Electron., 2010, 11, 1255–1260 CrossRef CAS.
- M. Pamuk, S. Tirkes, A. Cihaner and F. Algi, Polymer, 2010, 51, 62–68 CrossRef CAS.
- M. Ozkut-Icli, Z. Oztas, F. Algi and A. Cihaner, Org. Electron., 2011, 12, 1505–1511 CrossRef.
- M. Ozkut-Icli, M. Pamuk-Algi, Z. Oztas, F. Algi, A. M. Onal and A. Cihaner, Macromolecules, 2012, 45, 729–734 CrossRef.
- O. Celikbilek, M. Ozkut-Icli, F. Algi, A. M. Onal and A. Cihaner, Org. Electron., 2012, 13, 206–213 CrossRef CAS.
- M. Pamuk-Algi, S. Tirkes, S. Ertan, E. G. C. Ergun, A. Cihaner and F. Algi, Electrochim. Acta, 2013, 109, 766–774 CrossRef.
- J. H. Burroughes, D. D. C. Bradley, A. A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- J. C. G. Bunzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- J. C. G. Bunzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- J. P. Leonard and T. Gunnlaugsson, J. Fluoresc., 2005, 15, 585–595 CrossRef CAS PubMed.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- J. C. G. Bunzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- J. C. G. Bunzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC.
- M. Tropiano and S. Faulkner, Chem. Commun., 2014, 50, 4696–4698 RSC.
- A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4742 RSC.
- F. Lux, S. Roux, P. Perriat and O. Tillement, Curr. Inorg. Chem., 2011, 1, 117–129 CrossRef CAS.
- T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef CAS.
- S. Shinoda and H. Tsukube, Analyst, 2011, 136, 431–435 RSC.
- L. M. P. Lima and R. Tripier, Curr. Inorg. Chem., 2011, 1, 36–60 CrossRef CAS.
- G. Angelovski and I. Mamedov, Curr. Inorg. Chem., 2011, 1, 76–90 CrossRef CAS.
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652–1666 RSC.
- T. Gunnlaugsson and J. P. Leonard, Chem. Commun., 2005, 3114–3131 RSC.
- A. Thibon and V. C. Pierre, Anal. Bioanal. Chem., 2009, 394, 107–120 CrossRef CAS PubMed.
- E. J. New, D. Parker, D. G. Smith and J. W. Walton, Curr. Opin. Chem. Biol., 2010, 14, 238–246 CrossRef CAS PubMed.
- X. Wang, H. Chang, J. Xie, B. Zhao, B. Liu, S. Xu, W. Pei, N. Ren, L. Huang and W. Huang, Coord. Chem. Rev., 2014, 273-274, 201–212 CrossRef CAS.
- L.-J. Xu, G.-T. Xu and Z.-N. Chen, Coord. Chem. Rev., 2014, 273-274, 47–62 CrossRef CAS.
- E. Pershagen and K. E. Borbas, Coord. Chem. Rev., 2014, 273-274, 30–46 CrossRef CAS.
- Q. Li, C. Wang, H. Tan, G. Tang, J. Gao and C.-H. Chen, RSC Adv., 2016, 6, 17811–17817 RSC.
- J. P. Montgomery, B. J. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef PubMed.
- M. Rajendran, E. Yapici and L. W. Miller, Inorg. Chem., 2014, 53, 1839–1853 CrossRef CAS PubMed.
- D. Geißler, S. Linden, K. Liermann, K. D. Wegner, L. J. Charbonnière and N. Hildebrandt, Inorg. Chem., 2014, 53, 1824–1838 CrossRef PubMed.
- L. Ancel, A. Niedzwiecka, C. Lebrun, C. Gateau and P. Delange, C. R. Chim., 2013, 6, 515–523 CrossRef.
- J. J. Clarkson and J. McLoughlin, Int. Dent. J., 2000, 50, 119–128 CrossRef CAS PubMed.
- M. Shanthi, B. V. Reddy, V. Venkataramana, S. Gowrisankar, B. V. Thimma Reddy and S. Chennupati, J. Int. Oral Health, 2014, 6, 106–110 CAS.
- S. Ayoob and A. K. Gupta, Crit. Rev. Environ. Sci. Technol., 2006, 36, 433–487 CrossRef CAS.
- E. Bassin, D. Wypij, R. Davis and M. Mittleman, Cancer Causes Control, 2006, 17, 421–428 CrossRef PubMed.
- P. Ashokkumar, H. Weibhoff, W. Kraus and K. Rurack, Angew. Chem., Int. Ed., 2014, 53, 2225–2229 CrossRef CAS PubMed.
- C. Zou, Q. Qiao, M. Zhao, D. Mao, D. Wang, L. Feng, J. Cui and Z. Xu, RSC Adv., 2014, 4, 43746–43751 RSC.
- T. Nishimura, S.-Y. Xu, Y.-B. Jiang, J. S. Fossey, K. Sakurai, S. D. Bull and T. D. James, Chem. Commun., 2013, 49, 478–480 RSC.
- J.-M. Zhou, W. Shi, N. Xu and P. Cheng, Inorg. Chem., 2013, 52, 8082–8090 CrossRef CAS PubMed.
- P. Hou, S. Chen, H. Wang, J. Wang, K. Voitchovsky and X. Song, Chem. Commun., 2014, 50, 320–322 RSC.
- B. Sui, B. Kim, Y. Zhang, A. Frazer and K. D. Belfield, ACS Appl. Mater. Interfaces, 2013, 5, 2920–2923 CAS.
- X. Chen, T. Leng, C. Wang, Y. Shen and W. Zhu, Dyes Pigm., 2017, 141, 299–305 CrossRef CAS.
- C. G. Gulgas and T. M. Reineke, Inorg. Chem., 2008, 47, 1548–1559 CrossRef CAS PubMed.
- Q. M. Wang and H. Tamiaki, J. Photochem. Photobiol., A, 2009, 206, 124–128 CrossRef CAS.
- S. K. Kim and J. Yoon, Chem. Commun., 2002, 770–771 RSC.
- E. J. Cho, B. J. Ryu, Y. J. Lee and K. C. Nam, Org. Lett., 2005, 7, 2607–2609 CrossRef CAS PubMed.
- F. Algi and A. Cihaner, Tetrahedron Lett., 2008, 49, 3530–3533 CrossRef CAS.
- A. Yavuz, B. Bezgin, L. Aras and A. M. Onal, J. Electroanal. Chem., 2010, 639, 116–122 CrossRef CAS.
- P. Camurlu, Z. Bicil, C. Gultekin and N. Karagoren, Electrochim. Acta, 2012, 63, 245–250 CrossRef CAS.
- A. Cihaner and F. Algi, Electrochim. Acta, 2008, 54, 786–792 CrossRef CAS.
- G. G. Abashev, A. Y. Bushueva and E. V. Shklyaeva, Chem. Heterocycl. Compd., 2011, 47, 130–154 CrossRef CAS.
- F. Ekiz, F. Oguzkaya, M. Akin, S. Timur, C. Tanyeli and L. Toppare, J. Mater. Chem., 2011, 21, 12337–12342 RSC.
- D. Asil, A. Cihaner, F. Algi and A. M. Onal, Electroanalysis, 2010, 22, 2254–2260 CrossRef CAS.
- M. Pamuk-Algi, Z. Oztas and F. Algi, Chem. Commun., 2012, 48, 10219–10221 RSC.
- Z. Oztas, M. Pamuk and F. Algi, Tetrahedron, 2013, 69, 2048–2051 CrossRef CAS.
- A. Degirmenci, D. Iskenderkaptanoglu and F. Algi, Tetrahedron Lett., 2015, 56, 602–607 CrossRef CAS.
- A. Cihaner and F. Algi, J. Electroanal. Chem., 2008, 614, 101–106 CrossRef CAS.
- A. Cihaner and F. Algi, Electrochim. Acta, 2008, 53, 2574–2578 CrossRef CAS.
- A. Cihaner and F. Algi, Electrochim. Acta, 2008, 54, 665–670 CrossRef CAS.
- S. Tirkes, J. Mersini, Z. Oztas, M. Pamuk-Algi, F. Algi and A. Cihaner, Electrochim. Acta, 2013, 90, 295–301 CrossRef CAS.
- M. Pamuk-Algi, Z. Oztas, S. Tirkes, A. Cihaner and F. Algi, Org. Electron., 2013, 14, 1094–1102 CrossRef.
- D. Asil, F. Algi, A. Cihaner and A. M. Onal, J. Electroanal. Chem., 2008, 618, 87–93 CrossRef CAS.
- M. Pamuk-Algi, A. Cihaner and F. Algi, Tetrahedron, 2014, 70, 5064–5072 CrossRef.
- L. F. Schweiger, K. S. Ryder, D. G. Morris, A. Glidlec and J. M. Cooperc, J. Mater. Chem., 2000, 10, 107–114 RSC.
- G. J. Stasiuk and M. P. Lowe, Dalton Trans., 2009, 9725–9727 RSC.
- A. Beeby, L. M. Bushby, D. Maffeo and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 2002, 1, 48–54 RSC.
- C. Vanasschen, N. Bouslimani, D. Thono and J. F. Desreux, Inorg. Chem., 2011, 50, 8946–8958 CrossRef CAS PubMed.
- M. Jauregui, W. S. Perry, C. Allain, L. R. Vidler, M. C. Willis, A. M. Kenwright, J. S. Snaith, G. J. Stasiuk, M. P. Lowe and S. Faulkner, Dalton Trans., 2009, 6283–6285 RSC.
- S. Ozcubukcu, E. Ozkal, C. Jimeno and M. A. Pericas, Org. Lett., 2009, 11, 4680–4683 CrossRef CAS PubMed.
- F. Algi and M. Balci, Synthesis, 2009, 1341–1347 CrossRef CAS.
- For the determination of quantum yields see: M. Montalti, L. Prodi, M. Zaccheroni, L. Charbonniere, L. Douce and R. Ziessel, J. Am. Chem. Soc., 2001, 123, 12694–12695 CrossRef CAS PubMed.
- W. Xu, Y. Zhou, D. Huang, M. Su, K. Wang, M. Xiang and M. Hong, J. Mater. Chem. C, 2015, 3, 2003–2015 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- T. Liu, A. Nonat, M. Beyler, M. Regueiro-Figueroa, K. N. Nono, O. Jeannin, F. Camerel, F. Debaene, S. Cianferani-Sanglier, R. Tripier, C. Platas-Iglesias and L. J. Charbonniere, Angew. Chem., 2014, 126, 7387–7391 CrossRef.
- L. M. P. Lima, A. Lecointre, J.-F. Morfin, A. de Blas, D. Visvikis, L. J. Charbonniere, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2011, 50, 12508–12521 CrossRef CAS PubMed.
- http://www.who.int/water_sanitation_health/dwq/chemicals/fluoride.pdf, accessed 2004.
- A. P. De Silva, I. M. Dixon, H. Q. N. Gunaratne, T. Gunnlaugsson, P. R. S. Maxwell and T. E. Rice, J. Am. Chem. Soc., 1999, 121, 1393–1394 CrossRef CAS.
- B. Turfan and E. U. Akkaya, Org. Lett., 2002, 4, 2857–2859 CrossRef CAS PubMed.
- B. Feringa, For molecular logic systems, see: Molecular Switches, Wiley VCH, Weinheim, Germany, 2001, pp. 339–361 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR, FTIR, and MALDI-TOF MS spectra. See DOI: 10.1039/c7nj03569a |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |