
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-step synthesis of Pt based electrocatalysts encapsulated by polyoxometalate for methanol oxidation†
Shan
Gao
a,
Xiaoxuan
Yang
a,
Mei-Jie
Wei
a,
Song
Liang
b,
Hong-Ying
Zang
 *a,
Hua-Qiao
Tan
a,
Yong-Hui
Wang
a and
Yang-Guang
Li
*a
*a,
Hua-Qiao
Tan
a,
Yong-Hui
Wang
a and
Yang-Guang
Li
*a
aInstitute of Functional Material Chemistry, Key Lab of Polyoxometalate, Science of Ministry of Education, Key Laboratory of Nanobiosensing and Nanobioanalysis at Universities of Jilin Province, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, Jilin, P. R. China. E-mail: zanghy100@nenu.edu.cn; liyg658@nenu.edu.cn
bKey Laboratory of Bionic Engineering Ministry of Education, Jilin University, No. 5988 Renmin Street, Changchun, China
First published on 14th November 2017
Abstract
Methanol oxidation is a very important reaction in diret methanol fuel cells. Developing stable and efficient Pt-based catalysts with a convenient method has been on the stage recently. Herein, we employed a convenient one-pot method to synthesize Pt nanoparticles encapsulated by polyoxometalates (POMs) spreading on a few layers of graphene oxide with a nickel foam as a conductive substrate. Four kinds of samples with different Pt loadings were finally obtained by adjusting the H2PtCl6 concentration, named as PPGN-n (n = 1, 2, 3, 4). The microscopic structure of the sample was characterized using scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy. It was proved that small-sized Pt nanoparticles with a diameter of ca. 2.47 nm spreaded on graphene oxide (GO) with the nickle foam support were successfully synthesized. The synthesized electrocatalysts exhibited higher electrocatalytic activities than commercial Pt/C for methanol oxidation in electrocatalytic tests. The mass activity of PPGN-1 of 250.6 mA mg−1 can be achieved. During the synthesis, POM was used as a bifunctional reagent; as a reductant for reducing chloroplatinic acid and a stabilizer for the regulation of the Pt nanoparticle size simultaneously. POM cooperated rapid synthesis of Pt nanoparticles can offer a new route for designing and synthesising electrocatalysts.
1. Introduction
Nowadays, fossil fuels are declining, and research as well as invention to replace fossil fuels with renewable clean energy has become important. Development of new clean energy is a necessary choice against the energy crisis all over the world.1,2 Consequently, the discovery of fuel cells which can directly convert chemical energy into electrical energy has attracted great interest from scientists. Mobile phones, laptops and cars with fuel cells as power generators are already available. Among them, direct methanol fuel cells (DMFCs) are very promising energy storage and conversion devices due to their high efficiency, low operation temperature, cleanness and safety.3,4 However, low kinetic constants for the methanol oxidation reaction (MOR), scarcity of noble metals and noble catalysts being poisoned by intermediate products have hampered the large-scale commercial application of DMFCs.5,6 The invention of a new electrocatalyst is an effective strategy to solve this issue. Besides, Pt-based catalysts are widely used as MOR electrocatalysts. In the design of catalysts, more and more nanoscale carbon materials are introduced into the catalyst system for immobilization of Pt nanoparticles due to their excellent physical and chemical properties, especially graphene. The development of a convenient and green preparation methodology for the synthesis of Pt-modified carbon-based nanomaterials is the current focus.Carbonaceous species such as Vulcan XC-72,7 carbon nanotubes,8 graphene9 and carbon cloth10 as the catalyst carriers have become the focus in recent years, because of their excellent stability in acidic or alkaline solutions, low-cost, and good electronic conductivity. Graphene is a new type of two-dimensional material discovered by Geim et al. in 2004,11 and carbon atoms are interconnected by an sp2 hybrid orbital. Graphene oxide (GO) is a precursor of graphene prepared by chemical oxidation with a great quantity of oxygen-containing groups attached to the surface and the edge as well as some defects.12,13 Moreover, the oxygenous functional groups on the GO surface provide modified active sites that can anchor metal particles. Although GO is inferior to graphene in electrical conductivity, GO has unique characteristics,14 such as hydrophilicity, more reactive sites, high chemical controllability and easy functionalization, which are more suitable for preparing functional nanomaterials. The choice of an appropriate carrier can improve the dispersion of noble metal nanoparticles, promote the transmission of electrons, and improve the electrochemical performance. Accordingly, GO having a high specific surface area is selected as a catalyst carrier. The surface oxy-group contributes to the dispersion of noble metal nanoparticles and improves the electrochemical activity of the catalyst.
At present, many groups have successfully synthesized platinum-loaded graphene nanocatalysts. Duan et al. used sodium borohydride to synthesize platinum-loaded graphene aerogels (Pt/GOA).15 Wang et al. prepared three dimensional network graphene using the chemical vapor deposition (CVD) method, followed by loading Pt with graphene (Pt/G3DN).16 Shi et al. used an ionic liquid to functionalize the graphene and then used it as a carrier to support Pt (Pt/IL1–IL2/GN).17 However, the preparation of the above electrocatalysts often requires cumbersome experimental steps and the introduction of reducing agents. Moreover, experimental equipment is also complex and expensive. As a consequence, a convenient synthetic way is urgently required to prepare a highly efficient MOR electrocatalyst.
Polyoxometalates (POMs)18,19 are nanoscale metal oxygen anionic clusters with high electronegativity, an oxygen-rich surface, a special redox potential and exhibit electron transfer capability, and are widely used in electrical devices, electrodes and electrochemical capacitors, and other fields.20–23 According to the previous literature, POMs can be used as reducing agents to restore noble metal nanoparticles and can be coated on the surface of the nanoparticles to form a wrapping structure.24 At the same time, POM can be used as a connecting agent, through hydrogen bonding, to connect the carbon-based carrier and noble metal nanoparticles.25
In general, the preparation of electrodes by the drop coating method needs the use of an adhesive to immobilize the catalyst on the surface, but only the surface layer participates in the reaction. The nickel foam has 3D porous structures which can improve the utilization of platinum and the catalytic performance of MOR directly as a working electrode. Herein, we used the Keggin type H3PMo12O40 as a reducing agent and a stabilizing agent, selecting the 3D nickel foam as the conductive substrate, preparing Pt/POM/GO/NF multiple hybrid nanomaterials at room temperature in one step. In this work, POM acts as a reducing reagent to reduce Pt nanoparticles, also as a stabilizer to limit the agglomeration of nanoparticles and control the particle size. Electrochemical tests showed that the nanocatalysts all showed good electrocatalytic activity and durability.
2. Experimental part
2.1 Reagents and materials
All reagents were of analytical grade and were not further purified. H2PtCl6·6H2O was purchased from Third Party Pharmaceutical Technology Co. Ltd. H3PMo12O40, natural flake graphite and commercial Pt/C catalysts were purchased from Johnson Matthey. Ultrapure water was used throughout the experiment.2.2 Preparation of graphene oxide
Graphene oxide (GO) is obtained by the oxidation of natural flake graphite with an improved method.26 The preparation process is briefly described below: first, natural flake graphite (3 g) was added to a mixture of sulfuric acid (360 mL) and phosphoric acid (40 mL) and then KMnO4 (18 g) was added. The temperature was raised to 50 °C for 12 h. After that ice water (400 mL) was added, followed by dropping an appropriate amount of hydrogen peroxide into the solution and the color of the solution became golden yellow. The product was washed with hydrochloric acid and ultrapure water. Finally the sample was freeze-dried to collect the solids.2.3 Preparation of a Pt/POM/GO/NF hybrid catalyst
In order to remove impurities such as oil on the surface of the nickel foam, the untreated nickel foam was placed in 4 M hydrochloric acid, ethanol and ultrapure water successively for ultrasonic cleaning. A certain concentration of chloroplatinic acid solution was prepared (2.5, 5, 7.5 and 10 mM) using water and ethylene glycol with a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as a solvent. 4 mg mL−1 GO solution (3 mL), 5 mM H3PMo12O40 solution (3 mL) and the as-prepared chloroplatinic acid solution (3 mL) were mixed to form a homogeneous solution. The washed nickel foam was soaked in 3 mL mixed uniform solution for 2 min. The reaction system was rotated for 5 minutes using a vortex mixer (Qilinbeier Vortex-5), and then the nickle foam was washed with ultrapure water several times, finally drying at 50 °C. Pt/POM/GO/NF hybrid catalyst was successfully synthesized. The Pt/POM/GO/NF-n (n = 1, 2, 3, 4) composites were obtained by changing the concentration of chloroplatinic acid from low to high, referred to as PPGN-n (n = 1, 2, 3, 4). In order to further study the effect of POM and GO on the morphology and properties of the composites in the synthesis, the comparative samples were synthesized without adding POM or GO or both, while the other synthetic conditions were kept the same. These three comparative catalysts are referred to as PGN, PPN and PN, respectively. To obtain more information about the composite material, nickel foam was etched using HCl to facilitate subsequent characterization.
:1 as a solvent. 4 mg mL−1 GO solution (3 mL), 5 mM H3PMo12O40 solution (3 mL) and the as-prepared chloroplatinic acid solution (3 mL) were mixed to form a homogeneous solution. The washed nickel foam was soaked in 3 mL mixed uniform solution for 2 min. The reaction system was rotated for 5 minutes using a vortex mixer (Qilinbeier Vortex-5), and then the nickle foam was washed with ultrapure water several times, finally drying at 50 °C. Pt/POM/GO/NF hybrid catalyst was successfully synthesized. The Pt/POM/GO/NF-n (n = 1, 2, 3, 4) composites were obtained by changing the concentration of chloroplatinic acid from low to high, referred to as PPGN-n (n = 1, 2, 3, 4). In order to further study the effect of POM and GO on the morphology and properties of the composites in the synthesis, the comparative samples were synthesized without adding POM or GO or both, while the other synthetic conditions were kept the same. These three comparative catalysts are referred to as PGN, PPN and PN, respectively. To obtain more information about the composite material, nickel foam was etched using HCl to facilitate subsequent characterization.
2.4 Electrochemical measurements
The Cyclic voltammetry (CV) or Chronoamperometry of the catalyst was tested using a Princeton Electrochemical Workstation (PMC CHS08A) at 25 °C. The as-prepared nickel foam has been used as the working electrode, Pt plate and saturated calomel electrode (SCE) as the counter electrode and reference electrode, respectively. Commercial Pt/C was used as a comparative working electrode, 2 mg of the Pt/C catalyst was dispersed in Nafion aqueous solution, 3 μL of the above Nafion solution was accurately added to the surface of the treated nickel foam to prepare a comparative electrode. Before electrochemical measurement, high purity nitrogen was bubbled through the electrolyte for 30 min to remove the dissolved oxygen in the solution. Since the nickel foam was unstable under acidic conditions, all electrochemical tests were carried out under alkaline conditions. Furthermore, Pt has higher resistance to CO poisoning in alkaline media than acidic media. For the methanol oxidation test, the performance of different catalysts was tested in 1 mol L−1 CH3OH + 0.1 mol L−1 KOH solution and the electrochemical surface area (ECSA) of the catalyst was tested in 0.1 mol L−1 KOH solution. Electrochemical tests were all carried out at 50 mV s−1. The properties of commercial Pt/C or other contrast catalysts were also tested under the same conditions.2.5 Characterization of catalysts
The samples were characterized using field emission scanning electron microscopy (FESEM; Hitachi SU-8010) and Transmission electron microscopy (TEM; JEOL-2100F). XPS was performed on a VG ESCALAB MKII spectrometer using a Mg Kα (1253.6 eV) achromatic X-ray source. The Raman spectra were recorded using the HR800 spectrometer.3. Results and discussion
The typical synthesis of the nano-electrocatalyst PPGN is shown in Fig. 1. It can be proved that the experimental operation is convenient and the catalyst is obtained in one step. Moreover, the as-prepared catalyst has the structure that Pt nanoparticles encapsulated by POM are anchored to the GO layer. Fig. 2a shows the scanning electron microscope (SEM) image of PPGN-1 and it can be seen that a large number of particles are uniformly dispersed on the surface of the nickel foam. Fig. S1 (ESI†) is the SEM image of the nickel foam before (a) and after (b) the reaction. Observing these two graphs, we can find that bare nickel foam has a smooth surface,27,28 an irregular polygonal area covering the entire skeleton. Some of the surfaces also have a pit that may be caused by the production of nickel foam. After the reaction, the surface of the nickel foam becomes rough evidently, but the polygonal lines are still clearly visible, indicating that the hybrid material cladding the surface is thin. Fig. S2a, d, g (ESI†) are the SEM of the PPGN-2, 3, 4 catalysts. It is observed that nanoparticles are uniformly distributed on the nickel foam skeleton, which is similar to the morphology of PPGN-1. Furthermore, as the concentration of Pt in the reaction solution increases, the loading of Pt nanoparticles is increased and meanwhile, the boundaries between the particles become blurred and tend to form larger particles, which indicates the occurrence of agglomeration. The transmission electron microscopy (TEM) images of PPGN-1, 2, 3 and 4 samples are shown in Fig. 2b and Fig. S2b, e, h (ESI†), and their high-revolution transmission electron microscopy (HRTEM) image is shown in Fig. 2c and Fig. S2c, f, i (ESI†). The same conclusion can be obtained from these pictures. It can be seen in Fig. 2c and d that the average particle size of Pt nanoparticles in PPGN-1 is 2.47 nm and its lattice spacing is 0.23 nm, which is attributed to the face centered cubic (fcc) lattice structure of Pt, corresponding to the (111) plane.29 The layered stripe is clearly visible in Fig. S2c, f, i (ESI†), demonstrating that the platinum-loaded GO is of few layers, which can preserve the structural stability of the catalyst. The EDX spectra of PPGN-1 capture the obvious C, O, P, Mo, Pt and Ni peaks as shown in Fig. S3 (ESI†), where the intensity of Ni elements is much larger than other elements, indicating that the content of the GO complex supported on nickel foam is low. Fig. 2e–h shows the elemental mapping of the PPGN-1 catalyst.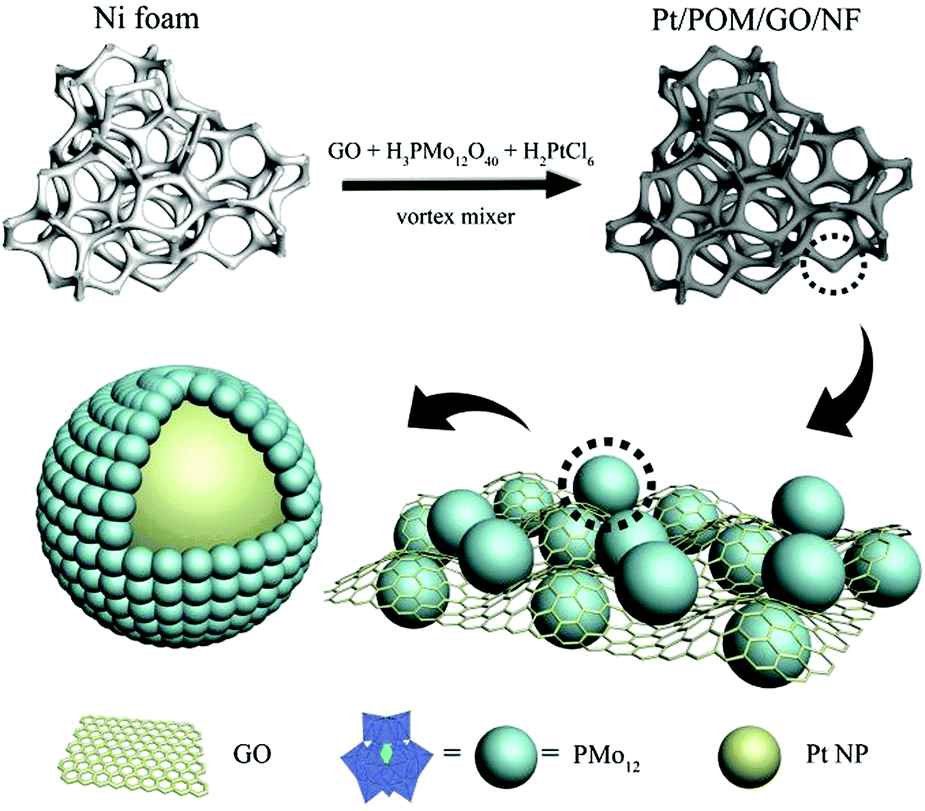 | ||
| Fig. 1 Schematic illustration of PPGN. | ||
 | ||
| Fig. 2 The SEM (a), TEM (b) and HRTEM (c and d) of PPGN-1 and the corresponding elemental mapping of P, Mo and Pt (e–h). | ||
The micrographs of the three comparative samples (PGN, PPN, PN) are shown in Fig. S4 (ESI†). When H3PMo12O40 is absent in PGN, the exposed Pt nanoparticles are formed on the surface and the slice structure is attributed to GO in Fig. S4a and b (ESI†). We did not find the microstructure of the noble metal nanoparticles loaded on GO. PPN was obtained without adding GO, and the surface of the nickel foam used on the substrate became uneven by analyzing its SEM in Fig. S4c and d (ESI†), due to the fact that POM was strongly acidic and corrosive. When GO and POM are not present in the reaction system, PN is obtained and its SEM is shown in Fig. S4e and f (ESI†). The exposed Pt nanoparticles with a larger diameter are formed on the surface of the nickel foam, while the surface becomes rough. From these three experiments, it can be confirmed that POM has the role of regulating the growth of Pt particles and effectively control the size of the particle. Simultaneously, the lack of GO for the samples makes nanoparticles larger in size and smaller in specific surface area. Therefore, the introduction of POM and GO into the reaction system is necessary.
The PPGN sample is characterized using XPS, which is a spectrum that can be used to obtain the composition of the sample surface, element valence, functional group and other relevant information of the surface composition of the sample. Fig. 3a is the survey XPS spectrum of PPGN-1, the emergence of the C 1s, O 1s, Pt 4f and Mo 3d peaks can prove the existence of C, O, Pt and Mo. This conclusion is in accordance with EDX. Compared with raw material GO in Fig. S5 (ESI†), there are also four different types of carbon present in PPGN-1 in Fig. 3b, which are C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O, CO and O–CO, corresponding to 284.6 eV, 286.6 eV, 288.0 eV and 288.9 eV.30,31 In Table S1 (ESI†), the electron binding energy and percentage of the four types of chemical bonds are listed, and the proportion of graphite-like carbon is increased after the reaction, which indicates that POM can reduce oxygen-containing groups on GO and reconstruct the sp2 hybrid of the carbon structure. The increase in the C–O ratio is due to the participation of POM absorbed on graphene oxide in the sample. Fig. 3c and Fig. S6 (ESI†) show the four catalysts all having Pt 4f7/2 and Pt 4f5/2 peaks, exhibiting the presence of Pt(0) and Pt(II) species. In Fig. 3c, the two main peaks appearing at 70.8 eV and 74.1 eV belong to Pt(0).32 The two weak peaks appearing in 71.5 eV and 74.8 eV belong to Pt(II), probably due to Pt oxides on the sample surface.33Fig. 3d shows two peaks at 232.5 eV and 235.7 eV, which are attributed to Mo 3d5/2 and Mo 3d3/2 from Mo, respectively.34 This also demonstrates the presence of POM and the successful modification of POM into the catalyst system.
C, C–O, CO and O–CO, corresponding to 284.6 eV, 286.6 eV, 288.0 eV and 288.9 eV.30,31 In Table S1 (ESI†), the electron binding energy and percentage of the four types of chemical bonds are listed, and the proportion of graphite-like carbon is increased after the reaction, which indicates that POM can reduce oxygen-containing groups on GO and reconstruct the sp2 hybrid of the carbon structure. The increase in the C–O ratio is due to the participation of POM absorbed on graphene oxide in the sample. Fig. 3c and Fig. S6 (ESI†) show the four catalysts all having Pt 4f7/2 and Pt 4f5/2 peaks, exhibiting the presence of Pt(0) and Pt(II) species. In Fig. 3c, the two main peaks appearing at 70.8 eV and 74.1 eV belong to Pt(0).32 The two weak peaks appearing in 71.5 eV and 74.8 eV belong to Pt(II), probably due to Pt oxides on the sample surface.33Fig. 3d shows two peaks at 232.5 eV and 235.7 eV, which are attributed to Mo 3d5/2 and Mo 3d3/2 from Mo, respectively.34 This also demonstrates the presence of POM and the successful modification of POM into the catalyst system.
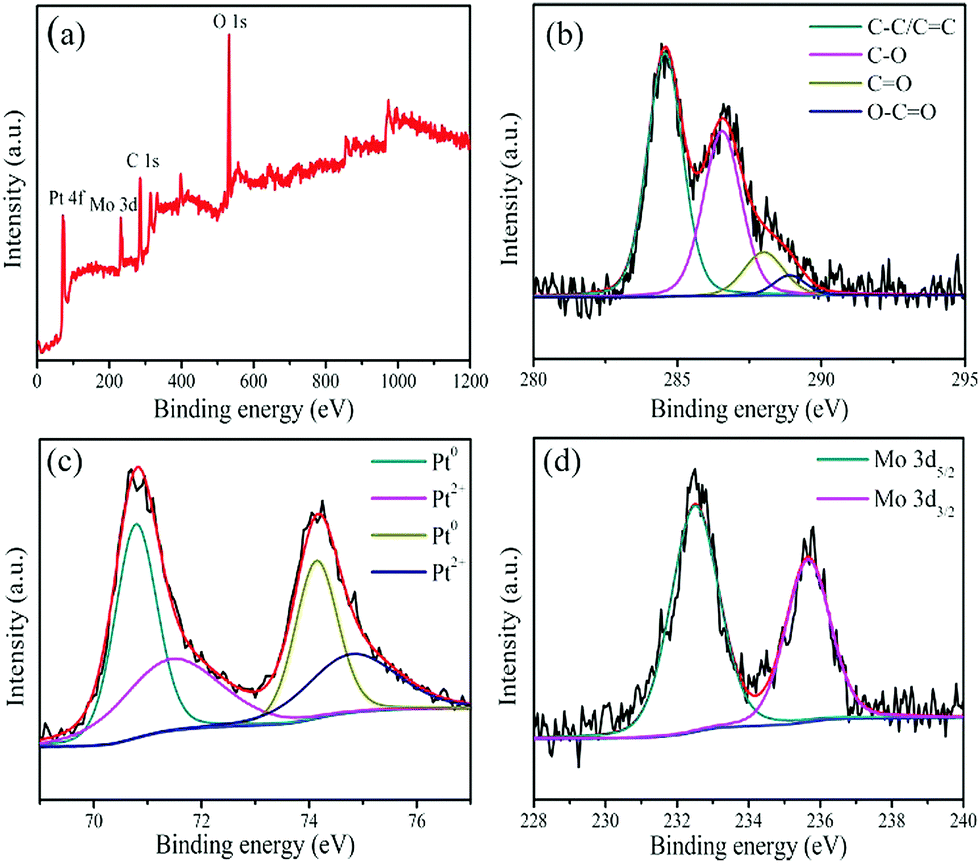 | ||
| Fig. 3 XPS of the PPGN-1 survey spectrum (a), C 1s (b), Pt 4f (c) and Mo 3d (d). | ||
Raman spectroscopy is an effective method to analyze the structure of carbon materials. The D peak at ∼1358 cm−1 and the G peak at ∼1600 cm−1 in Fig. 4 are the two characteristic peaks of the carbon element. The D-band represents a disordered structure in graphene, characterizing defects or edges in graphene.35 The G peak represents a graphitized carbon atom and is an ordered graphite structure with sp2 bonds. The peak intensity ratio of the D peak to the G peak stands for the degree of graphitization of the sample. The calculated values are GO (0.89) and PPGN-1 (1.10). The increase in ID/IG values indicates the reduction of partial O-containing groups, forming a small conjugated structure, which is consistent with the previous literature.36
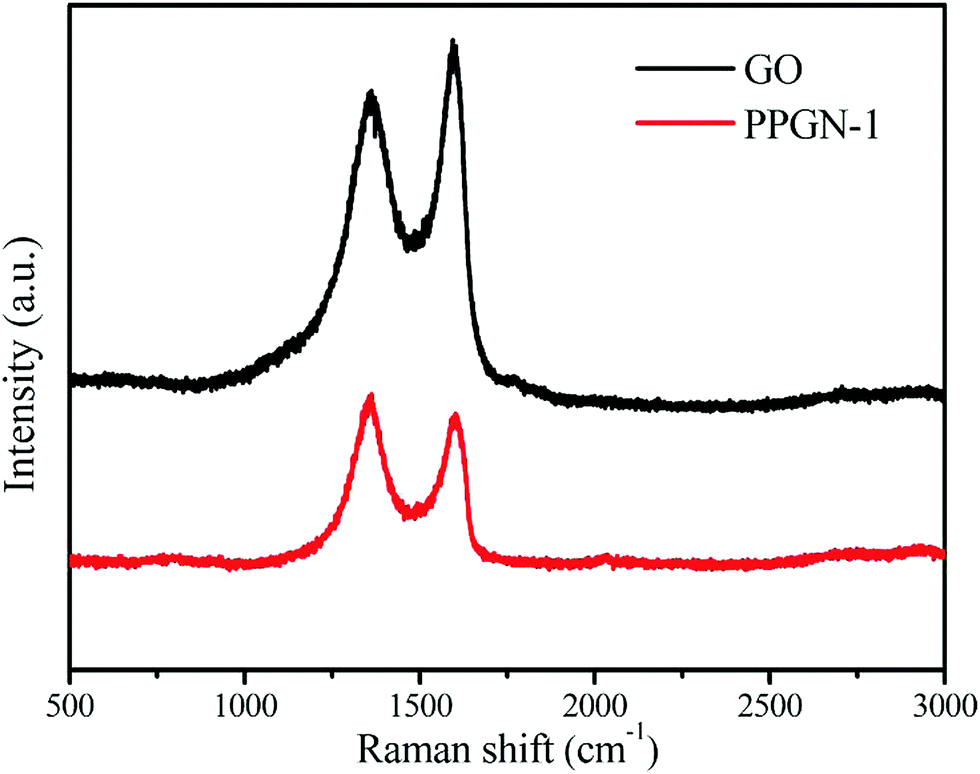 | ||
| Fig. 4 The Raman spectrum of GO and PPGN-1. | ||
The electrochemical MOR performance of the PPGN-n sample is demonstrated by the MOR activity test and the durability test, as shown in Fig. 5. The performance data of the electrocatalysts are summarized in Table 1. The electrochemical surface area (ECSA) was estimated by calculating the area of H desorption (QH), on the basis of the following formula:37
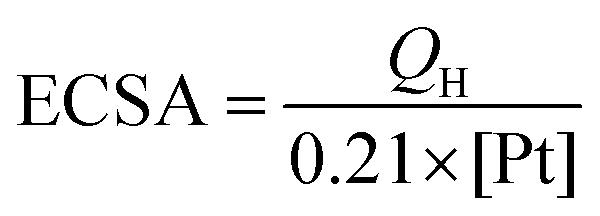
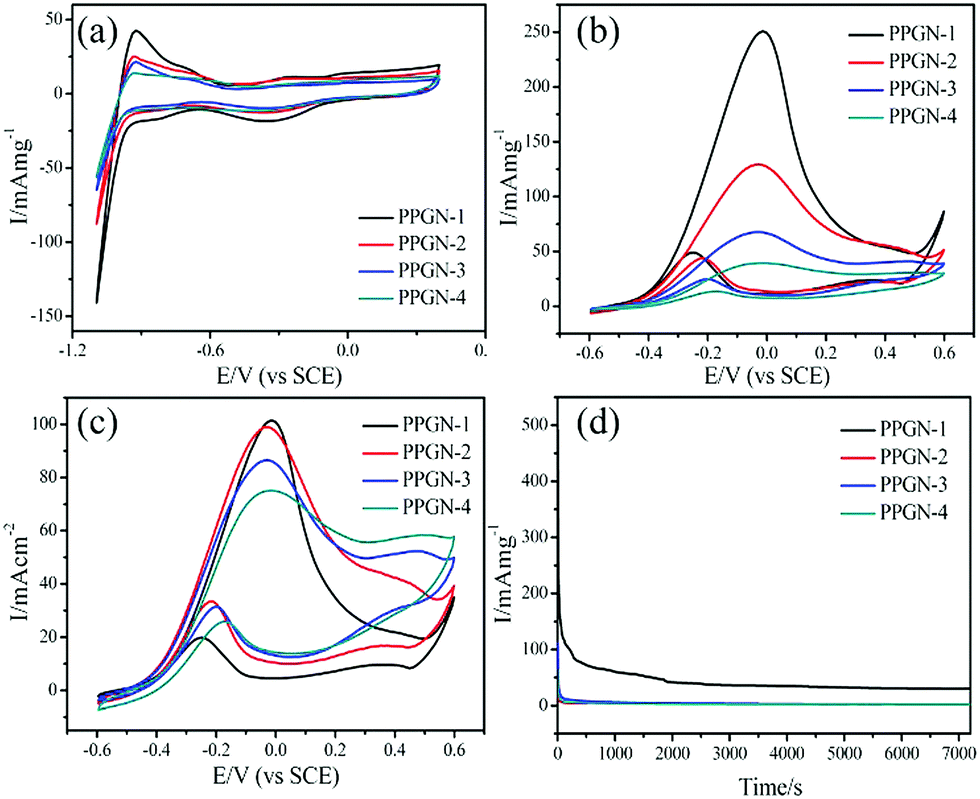 | ||
| Fig. 5 CV curves of PPGN-n in 0.1 mol L−1 KOH solution (a) and in 1 mol L−1 CH3OH + 0.1 mol L−1 KOH solution (b and c). The Chronoamperometry of PPGN-n in 1 mol L−1 CH3OH + 0.1 mol L−1 KOH solution (d). | ||
| Sample | ECSA (m2 g−1) | I f (mA mg−1) | I b (mA mg−1) | I f (mA cm−2) |
|---|---|---|---|---|
| PPGN-1 | 69.3 | 250.6 | 48.9 | 101.4 |
| PPGN-2 | 40.1 | 129.4 | 43.7 | 98.9 |
| PPGN-3 | 36.1 | 67.6 | 24.6 | 86.5 |
| PPGN-4 | 26.8 | 39.3 | 13.5 | 75.1 |
0.21 mC cm−2 represents the monolayer adsorption charge of hydrogen on the Pt surface.38 [Pt] is the amount of Pt supported in the sample. From Fig. 5a, we can observe that PPGN-1 has a maximum ECSA value of 69.3 m2 g−1. The peak appearing at −0.9 V in Fig. 5a refers to the desorption peak of Pt nanoparticles on the (111) plane. Fig. 5b shows the mass activity of PPGN samples with different Pt loadings for the electrocatalytic oxidation of methanol.
These peaks are observed in Fig. 5b due to the backward scan and forward scan, respectively, corresponding to the carbon–oxygen intermediate product peak and the methanol oxidation peak. Mass activity is a vital indicator of the catalyst capability. The mass activity of the forward scan is 250.6, 129.4, 67.6 and 39.3mA mg−1 for the samples of PPGN-1, 2, 3 and 4. PPGN-1 with the highest value exhibiting the best Pt utilization. Because the size of Pt nanoparticles loaded in PPGN-1 is smaller and can effectively improve the catalytic activity of MOR. The MOR specific activity of the PPGN-n multicomponent catalyst is shown in Fig. 5c. Likewise, the catalytic performance of PPGN-1 is the best. The poisoning resistance of the hybrid catalyst material is determined by the ratio of If/Ib. It can be seen that PPGN-1 has the strongest anti-intoxication ability, the If/Ib value is 5.10. Similarly, PGN, PPN, PN, commercially available Pt/C and Ni foam were tested for methanol oxidation and the results are shown in Fig. S7 (ESI†) and the current densities were 61.9, 58.3, 44.8, 31.2 and 19.3 mA cm−2, respectively. So the following conclusions can be drawn: firstly, POM and GO in the reaction system have played a crucial role, the lack of one of them will lead to a lower performance of the sample. Secondly, a series of samples prepared by one-step synthesis showed excellent methanol oxidation activity, even higher than that of the commercial Pt/C catalyst. At last, since POM is not stable under alkaline conditions and may decompose into molybdenum oxide, the process still needs further study.39 However, the main role of POM is to control the particle size in the formation of nanoparticles, preventing excessive growth of Pt nanoparticles. Table S2 (ESI†) lists the MOR performance of the different electrocatalysts in the previous literature under alkaline conditions.
The electrochemical durability test of the PPGN-n electrocatalyst is shown in Fig. 5d. The current intensity of all the catalysts decreased rapidly at the beginning of the test and eventually reached a platform. The intermediate product of methanol oxidation continues to occupy the active site causing the rapid decline in current. The stability of the electrocatalyst can be determined by the current density of the platform region.40,41 It can be concluded that from Fig. 5d that PPGN-1 held the highest mass activity until 7200 s. Therefore the PPGN-1 catalyst has excellent durability for the electrocatalytic oxidation of methanol.
4. Conclusions
In this work, we report a convenient and rapid method for one-step synthesis of Pt/POM/GO/NF electrocatalysts. Through a series of characterization, it was found that POM played a significant role in controlling the particle size of Pt nanoparticles. Moreover, the defects in graphene oxide provide more active sites for supporting noble metal particles. The synthesized catalyst has a good effect on the direct electrocatalysis of methanol oxidation under alkaline conditions, and even higher than the commercial Pt/C catalyst, where the maximum current density can reach 250.6 mA mg−1. In particular, the value of If/Ib of PPGN-1 reached 5.10, showing excellent resistance to CO poisoning. We hope that our synthetic strategy will provide inspiration for the future design of DMFC anode catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely thank the financial support from the Natural Science Foundation of China (No. 21471028 and 21673098), National Key Basic Research Program of China (No. 2013CB834802), Changbai Mountain Scholarship, Natural Science Foundation of Jilin Province (No. 20150101064JC).Notes and references
- J. Potocnik, Science, 2007, 315, 810–811 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- Y. J. Hu, P. Wu, Y. J. Yin, H. Zhang and C. X. Cai, Appl. Catal., B, 2012, 111-112, 208–217 CrossRef CAS.
- Y. C. Xin, J. G. Liu, Y. Zhou, W. M. Liu, J. Gao, Y. Xie, Y. Yin and Z. G. Zou, J. Power Sources, 2011, 196, 1012–1018 CrossRef CAS.
- X. H. Yan, R. Z. Wu, J. B. Xu, Z. T. Luo and T. S. Zhao, J. Power Sources, 2016, 311, 188–194 CrossRef CAS.
- R. Baronia, J. Goel, S. Tiwari, P. Singh, D. Singh, S. P. Singh and S. K. Singhal, Int. J. Hydrogen Energy, 2017, 42, 10238–10247 CrossRef CAS.
- A. A. Siller-Ceniceros, M. E. Sánchez-Castro, D. Morales-Acosta, J. R. Torres-Lubian, E. Martínez G. and F. J. Rodríguez-Varela, Appl. Catal., B, 2017, 209, 455–467 CrossRef CAS.
- L. M. Zhang, X. L. Sui, L. Zhao, J. J. Zhang, D. M. Gu and Z. B. Wang, Carbon, 2016, 108, 561–567 CrossRef CAS.
- H. J. Huang, J. X. Zhu, D. B. Li, C. Shen, M. M. Li, X. Zhang, Q. G. Jiang, J. F. Zhang and Y. P. Wu, J. Mater. Chem. A, 2017, 5, 4560–4567 CAS.
- A. L. Wang, X. J. He, X. F. Lu, H. Xu, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2015, 54, 3669–3673 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Z. Q. Jiang, X. S. Zhao and A. Manthiram, Int. J. Hydrogen Energy, 2013, 38, 5875–5884 CrossRef CAS.
- C. X. Xu, Y. C. Cao, R. Kumar, X. Wu, X. Wang and K. Scott, J. Mater. Chem., 2011, 21, 11359–11364 RSC.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed.
- J. L. Duan, X. L. Zhang, W. J. Yuan, H. L. Chen, S. Jiang, X. W. Liu, Y. F. Zhang, L. M. Chang, Z. Y. Sun and J. Du, J. Power Sources, 2015, 285, 76–79 CrossRef CAS.
- M. J. Wang, X. F. Song, Q. Yang, H. Hua, S. G. Dai, C. G. Hu and D. P. Wei, J. Power Sources, 2015, 273, 624–630 CrossRef CAS.
- G. Y. Shi, Z. H. Wang, J. F. Xia, S. Bi, Y. Li, F. F. Zhang, L. Xia, Y. H. Li, Y. Z. Xia and L. H. Xia, Electrochim. Acta, 2014, 142, 167–172 CrossRef CAS.
- V. Goovaerts, K. Stroobants, G. Absillis and T. N. Parac-Vogt, J. Inorg. Biochem., 2015, 150, 72–80 CrossRef CAS PubMed.
- D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- D. Ma, L. Y. Liang, W. Chen, H. M. Liu and Y. F. Song, Adv. Funct. Mater., 2013, 23, 6100–6105 CrossRef CAS.
- K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356–5360 CrossRef CAS PubMed.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Arino, Chem. Soc. Rev., 2012, 41, 7464–7478 RSC.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- S. W. Li, X. L. Yu, G. J. Zhang, Y. Ma, J. N. Yao, B. Keita, N. Louis and H. Zhao, J. Mater. Chem., 2011, 21, 2282–2287 RSC.
- S. W. Li, X. L. Yu, G. J. Zhang, Y. Ma, J. N. Yao and P. de Oliveira, Carbon, 2011, 49, 1906–1911 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- M. S. Guo, Y. Cheng, Y. N. Yu and J. B. Hu, Appl. Surf. Sci., 2017, 416, 439–445 CrossRef CAS.
- Y. F. Wu, L. Gan, S. P. Zhang, B. C. Jiang, H. O. Song, W. T. Li, Y. Pan and A. M. Li, Chem. Eng. J., 2017, 316, 146–153 CrossRef CAS.
- L. Y. Ruan, E. B. Zhu, Y. Chen, Z. Y. Lin, X. Q. Huang, X. F. Duan and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 12577–12581 CrossRef CAS PubMed.
- Y. J. Li, W. Gao, L. J. Ci, C. M. Wang and P. M. Ajayan, Carbon, 2010, 48, 1124–1130 CrossRef CAS.
- J. H. Guo, Y. T. Shi, H. W. Zhou, X. C. Wang and T. L. Ma, RSC Adv., 2017, 7, 2051–2057 RSC.
- P. R. kasturi, R. K. Selvan and Y. S. Lee, RSC Adv., 2016, 6, 62680–62694 Search PubMed.
- H. J. You, F. L. Zhang, Z. Liu and J. X. Fang, ACS Catal., 2014, 4, 2829–2835 CrossRef CAS.
- Y. Q. Duan, Y. Sun, L. Wang, Y. Dai, B. B. Chen, S. Y. Pan and J. L. Zou, J. Mater. Chem. A, 2016, 4, 7674–7682 CAS.
- X. L. Wang, C. Li and G. Q. Shi, Phys. Chem. Chem. Phys., 2014, 16, 10142–10148 RSC.
- Y. X. Xu, K. X. Sheng, C. Li and G. Q. Shi, J. Mater. Chem., 2011, 21, 7376–7380 RSC.
- Y. Y. Zhang, F. P. Li, X. Q. Liu, J. X. Lu and G. R. Zhang, Electrochim. Acta, 2017, 242, 165–172 CrossRef CAS.
- A. Pozio, M. D. Francesco, A. Cemmi, F. Cardellini and L. Giorgi, J. Power Sources, 2002, 105, 13–19 CrossRef CAS.
- S. Khadempir, A. Ahmadpour, M. T. H. Mosavian, N. Ashraf, F. F. Bamoharram, R. Fernández-Pacheco, J. M. de la Fuente and S. G. Mitchell, RSC Adv., 2016, 6, 5359–5366 RSC.
- Z. H. Wang, C. C. Lu, W. Kong, Y. H. Zhang and J. J. Li, J. Alloys Compd., 2017, 690, 95–100 CrossRef CAS.
- Y. Sun, Y. J. Zhou, C. Zhu, L. L. Hu, M. M. Han, A. Q. Wang, H. Huang, Y. Liu and Z. H. Kang, Nanoscale, 2017, 9, 5467–5474 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj03593d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |