
Microwave-assisted MgO NP catalyzed one-pot multicomponent synthesis of polysubstituted steroidal pyridines†
Anam
Ansari
a,
Abad
Ali
ab,
Mohd
Asif
a and
Shamsuzzaman
 *a
*a
aSteroid Research Laboratory, Department of Chemistry, Aligarh Muslim University, Aligarh 202 002, India. E-mail: shamsuzzaman9@gmail.com; Tel: +91941100346
bOrganometallic Synthesis and Catalysis Group, Chemical Engineering Division, CSIR-National Chemical Laboratory (CSIR-NCL), Dr Homi Bhabha Road, Pune 411 008, Maharashtra, India
First published on 20th November 2017
Abstract
The present study reports a highly efficient and green synthetic route for the synthesis of steroidal pyridines. The synthetic methodology involves a microwave-assisted one-pot multicomponent reaction using MgO NPs as a heterogeneous, mild and reusable catalyst. The synthesized MgO NPs were characterized by FTIR, TGA/DTA and XRD analyses. The remarkable features of this protocol include simple operational procedure, shorter reaction profiles, mild reaction conditions, minimal chemical waste and economic viability. The recyclability of the catalyst and high yield of products make the proposed method a sustainable alternative.
1. Introduction
Steroids are a group of important polycyclic compounds with multiple chiral centers and a rigid skeleton, exhibiting diverse and profound biological activities in living organisms.1,2 They include great variation in structure and encompass compounds of vital importance to life, such as cholesterol, bile acids, sex hormones, vitamin D, corticoid hormones, cardiac aglycones, antibiotics and insect molting hormones.3 Steroids regulate a wide variety of physiological and developmental processes and several steroidal compounds with suitable substitution modifying their molecular properties are widely used as anti-inflammatory, contraceptive, anti-androgenic, progestational, diuretic, anabolic, and anticancer agents as well as in other applications.4,5 The active constituents of pharmaceuticals are structurally complex organic molecules that have a precise arrangement of chemical groups and introduction of heteroatomic substituents into organic molecules is one of the most powerful ways of adding value and function to them.6 The replacement of one or more atoms in the structure of the maternal steroids and incorporation of a heteroatom or heterocyclic ring in a steroidal moiety not only affects the chemical properties of these molecules but also results in the alteration of its biological properties and a marked enhancement in pharmacological activities.7–9 Pyridines represent an important class of bioactive molecules and are particularly useful synthetic intermediates in the preparation of complex nitrogenous natural products and pharmaceutical targets.10,11 Pyridines have a widespread natural occurrence and are used to treat a broad spectrum of medical conditions, such as asthma, epilepsy, cancer and kidney diseases.12Catalysis is the key to sustainable synthetic chemistry and over the past few decades, heterogeneous catalysts have dominated organic catalytic chemistry and enabled the expeditious synthesis and discovery of plentiful valuable organic compounds, including natural products, pharmaceuticals and advanced materials. Several catalysts used in heterogenous catalytic processes consist of porous solid materials with a high surface area that offer the active sites on which chemical reactions are facilitated.13,14 Basic oxides play a major role in catalytic chemistry and MgO is one of the most privileged oxides in the field of catalysis. Its catalytic interest lies in its essentially basic surface character, which makes it an effective catalyst and catalyst support.15 MgO is an attractive metal oxide because of its purely ionic nature and simple cubic rock-salt structure. With high surface area and nanocrystalline structure, MgO has promising applications in the domain of catalysis and is exploited in various organic reactions and also allows the high yield synthesis of significant molecules.16 In recent years, extensive research has been conducted employing MgO as a catalyst for several base catalyzed organic transformations,17 in toxic waste remediation and as an additive in refractory, paint and super-conductor products,18,19 with simple MgO,20–22 high surface area MgO,23 nanosized MgO24 and MgO supported metal catalysts being used.25 MgO has been known to promote Cannizaro reactions and hydrogen transfer reactions as well as Michael, Wittig and Knoevenagel condensations.26
Microwave-assisted heating under controlled conditions is an invaluable technology for synthetic and medicinal chemists because assistance of microwaves in chemical reactions dramatically reduces the reaction time, typically from days or hours to minutes or even seconds and is capable of improving product yield as well as being a safe and convenient method.27 In addition, microwave-assisted organic synthesis technology often facilitates the discovery of innovative reaction pathways, because the extreme reaction conditions attainable by microwave heating sometimes lead to unusual reactivity that cannot be always duplicated by conventional heating.28 The catalytic synthetic protocol coupled with microwave-assisted multicomponent reactions has been successfully employed to generate molecular complexity and selective functionalization, and to discover new chemical entities of widespread application.29 In continuation of our previous lab work,30–35 here we describe a microwave-assisted one-pot multicomponent synthesis of steroidal pyridines in the presence of MgO NPs as a heterogeneous catalyst.
2. Experimental
2.1. Materials and methods
All glass apparatus was oven-dried prior to use. The chemicals and solvents used in this study were of ACS grade and used directly without additional steps of purification. The microwave synthesis was performed in an Anton Paar Monowave 300 microwave synthesizer. The melting points were determined on a Biogen digital auto melting point apparatus. The IR spectra were recorded on KBr pellets with a Perkin Elmer FT-IR Spectrometer spectrum Two and the values are given in cm−1. The 1H NMR and 13C NMR spectra were run in CDCl3 on a Bruker Avance II 400 NMR Spectrometer (operating at 400 MHz for 1H NMR and at 100 MHz for 13C NMR) with TMS as an internal standard and the values are given in parts per million (ppm) (δ). Elemental analysis was recorded on a Perkin Elmer 2400 CHN Elemental Analyzer. X-ray diffraction (XRD) patterns of the samples were obtained at room temperature with a step of 0.02°, using a Rigaku SmartLab diffractometer configured with a rotating anode CuKα source in the range of 10° < 2θ < 80° at 40 kV. The TGA/DTA analysis was performed on Shimadzu DTG-60H (simultaneous DTA-TG) apparatus. Thin-layer chromatography (TLC) plates were coated with silica gel G and exposed to iodine vapors to check the homogeneity as well as the progress of the reaction. Sodium sulfate (anhydrous) was used as a drying agent.2.2. General method for the preparation of MgO nanoparticles
MgO NPs were prepared by the precipitation method using magnesium nitrate and sodium hydroxide as precursors. As a precipitating agent a 0.2 M solution of NaOH was slowly added dropwise into a 0.1 M solution of magnesium nitrate in 50 mL of water with vigorous stirring. The pH was maintained at 10.5 by controlled addition of NaOH solution. After the complete precipitation procedure, the mixture was stirred at room temperature for 1–2 h. A white precipitate of Mg(OH)2 is obtained, which was washed thoroughly with distilled water and centrifuged at 4000 rpm for 15 min. The procedure was repeated several times until the precipitate was free from any trace of impurities. The precipitate was dried at 100 °C followed by calcination at 400 °C resulting in the formation of MgO NPs.2.3. General method for the synthesis of steroidal pyridines (4–9, 12a–d, 13a–d)
To a mixture of steroidal ketones (1–3 and 10), substituted aromatic aldehyde, malononitrile/methyl cyanoacetate and ammonium acetate in EtOH, MgO NPs were added as catalyst. The reaction mixture was irradiated in a microwave synthesis reactor (Anton Paar, Monowave 300) at 70 °C. After the completion of the reaction (monitored by TLC), the catalyst was separated by filteration from the reaction mixture and then the reaction mixture was poured into ice cold water. The precipitate thus obtained was filtered, washed with water, air dried and recrystallized from ethanol to afford the respective products.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1620 (CC). 1H NMR (400 MHz, CDCl3, ppm): δ 7.9–7.2 (m, 5H, aromatic), 7.4 (s, 2H, NH2, exchangeable with D2O), 3.66 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 2.11 (s, 3H, OCOCH3), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 170.3, 164.5, 159.9, 156.0, 135.1, 129.2, 128.5, 128.5, 127.2, 122.4, 120.2, 115.7, 85.4, 73.0, 56.4, 52.7, 48.2, 45.1, 43.2, 39.5, 39.5, 37.9, 36.4, 35.8, 33.9, 32.3, 28.1, 27.9, 27.5, 27.4, 25.9, 24.4, 23.7, 23.7, 22.5, 21.5, 19.0, 15.0, 12.6. Anal. calcd for C39H53N3O2; C, 78.61; H, 8.97; N, 7.05 found; C, 78.59; H, 8.99; N, 7.07; MS (EI): m/z 595 [M+˙].
N), 1625 (CC), 765 (C–Cl). 1H NMR (400 MHz, CDCl3, ppm): δ 7.8–7.9 (m, 5H, aromatic), 7.3 (s, 2H, NH2, exchangeable with D2O), 3.42 (m, 1H, C3-αH, W1/2 = 16 Hz, axial), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 165.7, 160.0, 155.0, 137.1, 129.1, 128.2, 128.2, 127.6, 127.6, 126.2, 114.7, 85.4, 60.8, 56.0, 52.9, 47.9, 43.1, 42.2, 39.5, 39.5, 38.8, 37.0, 36.4, 35.8, 35.0, 33.5, 28.9, 27.8, 27.0, 25.1, 24.6, 23.1, 23.1, 22.0, 19.2, 15.0, 12.6. Anal. calcd for C37H50ClN3; C, 77.66; H, 8.81; N, 7.34 found; C, 77.63; H, 8.84; N, 7.30; MS (EI): m/z 571/573 [M+˙].
N), 1620 (CC). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.9 (m, 5H, aromatic), 7.4 (s, 2H, NH2, exchangeable with D2O), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 164.7, 159.9, 155.0, 136.1, 129.1, 129.1, 128.-, 127.1, 127.1, 126.2, 114.7, 85.4, 56.5, 52.9, 48.9, 45.9, 43.1, 41.1, 40.0, 39.5, 39.2, 36.3, 35.4, 30.0, 28.2, 27.6, 27.0, 26.5, 25.6, 24.2, 23.0, 23.0, 22.3, 22.1, 19.0, 16.0, 13.6. Anal. calcd for C37H51N3; C, 82.63; H, 9.56; N, 7.81 found; C, 82.60; H, 9.52; N, 7.84; MS (EI): m/z 537 [M+˙].
O, ester), 1735 (OCOCH3), 1640 (CN), 1630 (CC), 1150 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.8 (m, 5H, aromatic), 7.7 (s, 2H, NH2, exchangeable with D2O), 3.91 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 2.21 (s, 3H, OCOCH3), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 170.3, 164.2, 160.0, 156.5, 151.9, 138.2, 129.1, 129.1, 129.0, 127.1, 127.1, 123.9, 102.6, 73.0, 56.6, 52.9, 51.8, 48.0, 45.1, 43.2, 39.5, 39.2, 37.0, 36.9, 35.8, 33.9, 32.3, 28.3, 28.0, 27.0, 27.0, 25.2, 24.6, 23.0, 23.0, 22.0, 21.4, 19.4, 16.0, 13.6. Anal. calcd for C40H56N2O4; C, 76.39; H, 8.98; N, 4.45 found; C, 76.43; 8.95; N, 4.59; MS (EI): m/z 628 [M+˙].
O, ester), 1630 (CN), 1620 (CC), 1145 (C–O), 768 (C–Cl). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.9 (m, 5H, aromatic), 7.3 (s, 2H, NH2, exchangeable with D2O), 3.89 (s, 3H, COOCH3), 3.72 (m, 1H, C3-αH, W1/2 = 16 Hz, axial), 2.21 (s, 3H, OCOCH3), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 164.9, 161.0, 156.2, 151.9, 138.0, 129.0, 129.0, 128.9, 127.2, 127.2, 123.9, 102.6, 60.8, 55.2, 52.9, 51.9, 48.0, 43.1, 43.2, 39.5, 39.3, 38.5, 37.0, 36.5, 35.0, 35.0, 33.8, 28.1, 28.5, 27.8, 25.9, 24.0, 23.0, 23.0, 22.7, 19.0, 15.0, 12.6. Anal. calcd for C38H53ClN2O2; C, 75.40; H, 8.83; N, 4.63 found; C, 75.38; H, 8.80; N, 4.67; MS (EI): m/z 604/606 [M+˙].
O, ester), 1636 (CN), 1625 (CC), 1148 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.9–7.4 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 3.89 (s, 3H, COOCH3), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 164.9, 160.3, 156.2, 151.9, 138.0, 129.0, 129.0, 128.8, 127.1, 127.1, 123.9, 101.6, 56.5, 52.8, 51.9, 48.9, 45.9, 43.1, 40.2, 40.1, 39.7, 39.5, 36.5, 35.3, 28.9, 28.3, 28.0, 27.0, 26.2, 25.9, 24.1, 23.5, 23.5, 22.5, 22.1, 19.0, 16.0, 13.6. Anal. calcd for C38H54N2O2; C, 79.95; H, 9.53; N, 4.91 found; C, 79.91; H, 9.56; N, 4.95; MS (EI): m/z 570 [M+˙].
N), 1620 (CC). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.2 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 169.2, 165.6, 163.9, 156.5, 142.8, 140.0, 130.0, 129.9, 129.5, 127.9, 127.9, 123.8, 115.7, 98.7, 77.5, 73.9, 55.9, 55.3, 51.5, 50.9, 39.5, 38.8, 37.6, 36.4, 35.0, 31.8, 28.2, 28.2, 23.2, 22.1, 21.1, 19.3, 11.3. Anal. calcd for C33H39N3O2; C, 77.76; H, 7.71; N, 8.24 found; C, 77.80; H, 7.67; N, 8.20; MS (EI): m/z 509 [M+˙].
N), 1625 (CC), 1440 (NO). 1H NMR (400 MHz, CDCl3, ppm): δ 8.6–7.7 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.31 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 169.2, 165.6, 162.9, 156.0, 148.9, 140.5, 138.9, 132.5, 131.1, 126.4, 125.7, 121.8, 115.7, 98.9, 77.5, 73.6, 55.9, 54.5, 53.5, 51.0, 38.3, 38.3, 37.0, 37.0, 32.0, 31.8, 28.5, 28.2, 24.2, 21.9, 21.0, 19.5, 11.5. Anal. calcd for C33H38N4O4; C, 71.46; H, 6.91; N, 10.10 found; C, 71.43; H, 6.95; N, 10.07; MS (EI): m/z 554 [M+˙].
N), 1620 (CC), 765 (C–Cl). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.5 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.31 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 167.2, 164.6, 162.9, 156.9, 151.8, 146.1, 140.8, 139.3, 132.3, 130.8, 128.2, 122.0, 110.7, 78.7, 74.5, 58.8, 56.8, 56.0, 51.0, 50.1, 38.9, 38.5, 37.0, 37.0, 32.0, 31.8, 28.3, 28.2, 25.0, 21.8, 21.2, 19.3, 11.6. Anal. calcd for C33H38ClN3O2; C, 72.84; H, 7.04; N, 7.72 found; C, 72.88; H, 7.00; N, 7.69; MS (EI): m/z 543/545 [M+˙].
N), 1625 (CC), 1120 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.2–6.9 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.83 (s, 3H, OCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 167.2, 165.6, 162.9, 156.8, 150.6, 148.3, 140.8, 136.3, 131.9, 129.8, 125.7, 122.5, 121.0, 99.7, 77.5, 73.8, 56.8, 56.5, 55.9, 52.0, 51.0, 39.8, 38.5, 38.2, 37.0, 32.4, 32.0, 31.8, 28.3, 28.2, 25.0, 22.1, 21.0, 19.3, 11.5. Anal. calcd for C35H43N3O4; C, 73.78; H, 7.61; N, 7.38 found; C, 73.75; H, 7.64; N, 7.35; MS (EI): m/z 569 [M+˙].
O, ester), 1735 (OCOCH3), 1640 (CN), 1635 (CC), 1145 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.5–7.2 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 169.2, 165.9, 164.5, 157.4, 151.2, 148.9, 140.8, 139.2, 139.2, 130.2, 127.8, 127.4, 121.8, 106.7, 78.4, 73.8, 56.8, 55.0, 50.5, 50.5, 50.0, 38.1, 38.0, 37.4, 37.2, 32.0, 31.8, 28.3, 28.2, 25.0, 21.8, 21.0, 19.3, 11.5. Anal. calcd for C34H42N2O4; C, 75.25; H, 7.80; N, 5.16 found; C, 75.21; H, 7.83; N, 5.14; MS (EI): m/z 542 [M+˙].
O, ester), 1733 (OCOCH3), 1630 (CN), 1620 (CC), 1145 (C–O), 1430 (NO). 1H NMR (400 MHz, CDCl3, ppm): δ 8.2–7.2 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 166.0, 165.9, 164.2, 157.6, 151.2, 148.4, 140.7, 139.6, 132.7, 130.7, 128.25, 122.7, 121.7, 106.7, 77.3, 74.5, 56.7, 56.6, 56.1, 50.09, 50.01, 39.5, 38.2, 37.2, 37.01, 31.9, 31.8, 28.2, 28.01, 24.28, 22.56, 21.04, 19.38, 11.85. Anal. calcd for C34H41N3O6; C, 69.48; H, 7.03; N, 7.15 found; C, 69.51; H, 7.00; N, 7.19; MS (EI): m/z 587 [M+˙].
O, ester), 1733 (OCOCH3), 1640 (CN), 1625 (CC), 1150 (C–O), 748 (C–Cl). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.5 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.31 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 168.2, 165.9, 164.2, 157.5, 151.0, 148.8, 139.0, 138.8, 130.3, 129.3, 128.5, 128.5, 121.8, 106.5, 76.4, 73.8, 58.8, 56.0, 55.5, 51.0, 50.1, 38.9, 38.3, 37.3, 37.0, 32.0, 31.0, 28.2, 28.2, 25.0, 22.1, 21.0, 19.3, 11.8. Anal. calcd for C34H41ClN2O4; C, 70.76; H, 7.16; N, 4.85 found; C, 70.73; H, 7.18; 4.89; MS (EI): m/z 576/578 [M+˙].
O, ester), 1735 (OCOCH3), 1630 (CN), 1630 (CC), 1146 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.2–6.9 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 3.83 (s, 3H, OCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 169.2, 165.9, 164.2, 157.6, 151.2, 149.3, 140.8, 140.4, 138.9, 132.9, 121.8, 113.5, 111.0, 106.7, 77.4, 73.8, 56.6, 56.5, 53.8, 52.0, 51.5, 51.5, 50.8, 38.5, 38.1, 37.4, 37.4, 32.0, 31.8, 28.4, 28.2, 25.2, 22.1, 21.0, 19.3, 11.5. Anal. calcd for C36H46N2O6; C, 71.73; H, 7.69; N, 4.65 found; C, 71.70; H, 7.72; N, 4.62; MS (EI): m/z 602 [M+˙].
N), 1620 (CC). 1H NMR (400 MHz, CDCl3, ppm): δ 7.9–7.2 (m, 5H, aromatic), 7.4 (s, 2H, NH2, exchangeable with D2O), 3.66 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 2.11 (s, 3H, OCOCH3), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 170.3, 164.5, 159.9, 156.0, 135.1, 129.2, 128.5, 128.5, 127.2, 122.4, 120.2, 115.7, 85.4, 73.0, 56.4, 52.7, 48.2, 45.1, 43.2, 39.5, 39.5, 37.9, 36.4, 35.8, 33.9, 32.3, 28.1, 27.9, 27.5, 27.4, 25.9, 24.4, 23.7, 23.7, 22.5, 21.5, 19.0, 15.0, 12.6. Anal. calcd for C39H53N3O2; C, 78.61; H, 8.97; N, 7.05 found; C, 78.59; H, 8.99; N, 7.07; MS (EI): m/z 595 [M+˙].
N), 1625 (CC), 765 (C–Cl). 1H NMR (400 MHz, CDCl3, ppm): δ 7.8–7.9 (m, 5H, aromatic), 7.3 (s, 2H, NH2, exchangeable with D2O), 3.42 (m, 1H, C3-αH, W1/2 = 16 Hz, axial), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 165.7, 160.0, 155.0, 137.1, 129.1, 128.2, 128.2, 127.6, 127.6, 126.2, 114.7, 85.4, 60.8, 56.0, 52.9, 47.9, 43.1, 42.2, 39.5, 39.5, 38.8, 37.0, 36.4, 35.8, 35.0, 33.5, 28.9, 27.8, 27.0, 25.1, 24.6, 23.1, 23.1, 22.0, 19.2, 15.0, 12.6. Anal. calcd for C37H50ClN3; C, 77.66; H, 8.81; N, 7.34 found; C, 77.63; H, 8.84; N, 7.30; MS (EI): m/z 571/573 [M+˙].
N), 1620 (CC). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.9 (m, 5H, aromatic), 7.4 (s, 2H, NH2, exchangeable with D2O), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 164.7, 159.9, 155.0, 136.1, 129.1, 129.1, 128.-, 127.1, 127.1, 126.2, 114.7, 85.4, 56.5, 52.9, 48.9, 45.9, 43.1, 41.1, 40.0, 39.5, 39.2, 36.3, 35.4, 30.0, 28.2, 27.6, 27.0, 26.5, 25.6, 24.2, 23.0, 23.0, 22.3, 22.1, 19.0, 16.0, 13.6. Anal. calcd for C37H51N3; C, 82.63; H, 9.56; N, 7.81 found; C, 82.60; H, 9.52; N, 7.84; MS (EI): m/z 537 [M+˙].
O, ester), 1735 (OCOCH3), 1640 (CN), 1630 (CC), 1150 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.8 (m, 5H, aromatic), 7.7 (s, 2H, NH2, exchangeable with D2O), 3.91 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 2.21 (s, 3H, OCOCH3), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 170.3, 164.2, 160.0, 156.5, 151.9, 138.2, 129.1, 129.1, 129.0, 127.1, 127.1, 123.9, 102.6, 73.0, 56.6, 52.9, 51.8, 48.0, 45.1, 43.2, 39.5, 39.2, 37.0, 36.9, 35.8, 33.9, 32.3, 28.3, 28.0, 27.0, 27.0, 25.2, 24.6, 23.0, 23.0, 22.0, 21.4, 19.4, 16.0, 13.6. Anal. calcd for C40H56N2O4; C, 76.39; H, 8.98; N, 4.45 found; C, 76.43; 8.95; N, 4.59; MS (EI): m/z 628 [M+˙].
O, ester), 1630 (CN), 1620 (CC), 1145 (C–O), 768 (C–Cl). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.9 (m, 5H, aromatic), 7.3 (s, 2H, NH2, exchangeable with D2O), 3.89 (s, 3H, COOCH3), 3.72 (m, 1H, C3-αH, W1/2 = 16 Hz, axial), 2.21 (s, 3H, OCOCH3), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 164.9, 161.0, 156.2, 151.9, 138.0, 129.0, 129.0, 128.9, 127.2, 127.2, 123.9, 102.6, 60.8, 55.2, 52.9, 51.9, 48.0, 43.1, 43.2, 39.5, 39.3, 38.5, 37.0, 36.5, 35.0, 35.0, 33.8, 28.1, 28.5, 27.8, 25.9, 24.0, 23.0, 23.0, 22.7, 19.0, 15.0, 12.6. Anal. calcd for C38H53ClN2O2; C, 75.40; H, 8.83; N, 4.63 found; C, 75.38; H, 8.80; N, 4.67; MS (EI): m/z 604/606 [M+˙].
O, ester), 1636 (CN), 1625 (CC), 1148 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.9–7.4 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 3.89 (s, 3H, COOCH3), 1.04 (C10-CH3), 1.04 (C13-CH3), 0.91 and 0.96 (other methyl proton). 13C NMR (100 MHz, CDCl3, ppm): δ 164.9, 160.3, 156.2, 151.9, 138.0, 129.0, 129.0, 128.8, 127.1, 127.1, 123.9, 101.6, 56.5, 52.8, 51.9, 48.9, 45.9, 43.1, 40.2, 40.1, 39.7, 39.5, 36.5, 35.3, 28.9, 28.3, 28.0, 27.0, 26.2, 25.9, 24.1, 23.5, 23.5, 22.5, 22.1, 19.0, 16.0, 13.6. Anal. calcd for C38H54N2O2; C, 79.95; H, 9.53; N, 4.91 found; C, 79.91; H, 9.56; N, 4.95; MS (EI): m/z 570 [M+˙].
N), 1620 (CC). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.2 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 169.2, 165.6, 163.9, 156.5, 142.8, 140.0, 130.0, 129.9, 129.5, 127.9, 127.9, 123.8, 115.7, 98.7, 77.5, 73.9, 55.9, 55.3, 51.5, 50.9, 39.5, 38.8, 37.6, 36.4, 35.0, 31.8, 28.2, 28.2, 23.2, 22.1, 21.1, 19.3, 11.3. Anal. calcd for C33H39N3O2; C, 77.76; H, 7.71; N, 8.24 found; C, 77.80; H, 7.67; N, 8.20; MS (EI): m/z 509 [M+˙].
N), 1625 (CC), 1440 (NO). 1H NMR (400 MHz, CDCl3, ppm): δ 8.6–7.7 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.31 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 169.2, 165.6, 162.9, 156.0, 148.9, 140.5, 138.9, 132.5, 131.1, 126.4, 125.7, 121.8, 115.7, 98.9, 77.5, 73.6, 55.9, 54.5, 53.5, 51.0, 38.3, 38.3, 37.0, 37.0, 32.0, 31.8, 28.5, 28.2, 24.2, 21.9, 21.0, 19.5, 11.5. Anal. calcd for C33H38N4O4; C, 71.46; H, 6.91; N, 10.10 found; C, 71.43; H, 6.95; N, 10.07; MS (EI): m/z 554 [M+˙].
N), 1620 (CC), 765 (C–Cl). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.5 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.31 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 167.2, 164.6, 162.9, 156.9, 151.8, 146.1, 140.8, 139.3, 132.3, 130.8, 128.2, 122.0, 110.7, 78.7, 74.5, 58.8, 56.8, 56.0, 51.0, 50.1, 38.9, 38.5, 37.0, 37.0, 32.0, 31.8, 28.3, 28.2, 25.0, 21.8, 21.2, 19.3, 11.6. Anal. calcd for C33H38ClN3O2; C, 72.84; H, 7.04; N, 7.72 found; C, 72.88; H, 7.00; N, 7.69; MS (EI): m/z 543/545 [M+˙].
N), 1625 (CC), 1120 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.2–6.9 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.83 (s, 3H, OCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 167.2, 165.6, 162.9, 156.8, 150.6, 148.3, 140.8, 136.3, 131.9, 129.8, 125.7, 122.5, 121.0, 99.7, 77.5, 73.8, 56.8, 56.5, 55.9, 52.0, 51.0, 39.8, 38.5, 38.2, 37.0, 32.4, 32.0, 31.8, 28.3, 28.2, 25.0, 22.1, 21.0, 19.3, 11.5. Anal. calcd for C35H43N3O4; C, 73.78; H, 7.61; N, 7.38 found; C, 73.75; H, 7.64; N, 7.35; MS (EI): m/z 569 [M+˙].
O, ester), 1735 (OCOCH3), 1640 (CN), 1635 (CC), 1145 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.5–7.2 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 169.2, 165.9, 164.5, 157.4, 151.2, 148.9, 140.8, 139.2, 139.2, 130.2, 127.8, 127.4, 121.8, 106.7, 78.4, 73.8, 56.8, 55.0, 50.5, 50.5, 50.0, 38.1, 38.0, 37.4, 37.2, 32.0, 31.8, 28.3, 28.2, 25.0, 21.8, 21.0, 19.3, 11.5. Anal. calcd for C34H42N2O4; C, 75.25; H, 7.80; N, 5.16 found; C, 75.21; H, 7.83; N, 5.14; MS (EI): m/z 542 [M+˙].
O, ester), 1733 (OCOCH3), 1630 (CN), 1620 (CC), 1145 (C–O), 1430 (NO). 1H NMR (400 MHz, CDCl3, ppm): δ 8.2–7.2 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 166.0, 165.9, 164.2, 157.6, 151.2, 148.4, 140.7, 139.6, 132.7, 130.7, 128.25, 122.7, 121.7, 106.7, 77.3, 74.5, 56.7, 56.6, 56.1, 50.09, 50.01, 39.5, 38.2, 37.2, 37.01, 31.9, 31.8, 28.2, 28.01, 24.28, 22.56, 21.04, 19.38, 11.85. Anal. calcd for C34H41N3O6; C, 69.48; H, 7.03; N, 7.15 found; C, 69.51; H, 7.00; N, 7.19; MS (EI): m/z 587 [M+˙].
O, ester), 1733 (OCOCH3), 1640 (CN), 1625 (CC), 1150 (C–O), 748 (C–Cl). 1H NMR (400 MHz, CDCl3, ppm): δ 7.7–7.5 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.31 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 168.2, 165.9, 164.2, 157.5, 151.0, 148.8, 139.0, 138.8, 130.3, 129.3, 128.5, 128.5, 121.8, 106.5, 76.4, 73.8, 58.8, 56.0, 55.5, 51.0, 50.1, 38.9, 38.3, 37.3, 37.0, 32.0, 31.0, 28.2, 28.2, 25.0, 22.1, 21.0, 19.3, 11.8. Anal. calcd for C34H41ClN2O4; C, 70.76; H, 7.16; N, 4.85 found; C, 70.73; H, 7.18; 4.89; MS (EI): m/z 576/578 [M+˙].
O, ester), 1735 (OCOCH3), 1630 (CN), 1630 (CC), 1146 (C–O). 1H NMR (400 MHz, CDCl3, ppm): δ 7.2–6.9 (m, 5H, aromatic), 7.74 (s, 2H, NH2, exchangeable with D2O), 4.32 (m, 1H, C3-αH, W1/2 = 17 Hz, axial), 3.89 (s, 3H, COOCH3), 3.83 (s, 3H, OCH3), 2.21 (s, 3H, OCOCH3), 1.30 (C10-CH3), 1.04 (C13-CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 169.2, 165.9, 164.2, 157.6, 151.2, 149.3, 140.8, 140.4, 138.9, 132.9, 121.8, 113.5, 111.0, 106.7, 77.4, 73.8, 56.6, 56.5, 53.8, 52.0, 51.5, 51.5, 50.8, 38.5, 38.1, 37.4, 37.4, 32.0, 31.8, 28.4, 28.2, 25.2, 22.1, 21.0, 19.3, 11.5. Anal. calcd for C36H46N2O6; C, 71.73; H, 7.69; N, 4.65 found; C, 71.70; H, 7.72; N, 4.62; MS (EI): m/z 602 [M+˙].
3. Results and discussion
MgO NPs were synthesized employing a simple precipitation method using Mg(NO3)2 as a precursor.36 The FT-IR spectrum for the synthesized Mg(OH)2 and calcined MgO NPs is depicted in Fig. 1. In the FT-IR spectrum of Mg(OH)2 (Fig. 1a) the sharp and intense peak at 3700 cm−1 is due to the –OH group in Mg(OH)2. It is well known that H2O and CO2 molecules are easily chemisorbed onto the MgO surface when exposed to the atmosphere. In the spectrum of the calcined sample shown in Fig. 1b, the broad band at 3450 cm−1 is associated with the OH stretching vibrations of surface-adsorbed water molecules, while those at 1012 and 1634 cm−1 are associated with their bending vibrations.37 In addition, a very weak band corresponding to the adsorption of gas phase CO2 is visible around 2425 cm−1 and CO3−2 is visible at around 1385 cm−1.38 The bands that appeared at low frequencies of 836 and 652 cm−1 correspond to stretching vibrations of Mg–O–Mg bonding.39 The band at 3699 cm−1 in the FTIR spectrum of the dried sample completely disappears after calcination, which is due to the decomposition of Mg(OH)2 to MgO NPs. | ||
| Fig. 1 FTIR spectrum of (a) Mg(OH)2 and (b) MgO NPs. | ||
In order to examine the thermal stability of the Mg(OH)2, TGA/DTA was carried out between 0 °C and 800 °C. Thermal analysis (Fig. 2) illustrates the temperature at which dehydration of the hydroxide occurs as the material transitions to the oxide form. The TGA curve (Fig. 2a) shows one small major weight loss in the temperature range of 250–600 °C, which is related to the decomposition of Mg(OH)2 and crystallization of MgO. Fig. 2b shows the DTA curve of the as-prepared Mg(OH)2. As can be seen from the curve, the strong endothermic peak observed at about 398 °C in the DTA curve could be attributed to the decomposition of Mg(OH)2.
| Mg(OH)2 ↔ MgO + H2O |
 | ||
| Fig. 2 (a) TGA and (b) DTA profiles of the as-prepared Mg(OH)2. | ||
The structural phase of the MgO NPs was determined by the X-ray diffraction pattern. Fig. 3 shows the XRD pattern of the MgO NPs prepared by the precipitation method. As shown in the figure, the observed peaks at 2θ = 36.63°, 42.45°, 61.84°, 74.11° and 78.04° are associated with the [111], [200], [220], [311] and [222] planes, respectively, which are in good accordance with the reported literature.40 Furthermore, no characteristic peaks of other impurities were observed, which indicated that the product had a high purity. In addition, the strong and sharp diffraction peaks indicate that the MgO NPs are well crystallized. The grain sizes corresponding to the [111], [200], [220], [311] and [222] lattice planes of the MgO NPs were calculated using the Debye–Scherrer formula, and are listed in Table 1.

 | ||
| Fig. 3 XRD pattern of the MgO NPs. | ||
| Entry | 2θ | FWHM | Miller index (hkl) | Grain size (nm) |
|---|---|---|---|---|
| 1 | 36.63 | 0.6940 | 111 | 12.05 |
| 2 | 42.45 | 0.6078 | 200 | 14.02 |
| 3 | 61.84 | 0.7604 | 220 | 12.17 |
| 4 | 74.11 | 0.4584 | 311 | 21.70 |
| 5 | 78.04 | 0.9056 | 222 | 11.29 |
Polysubstituted pyridines are of particular interest and are known to be synthesized by different synthetic strategies including multicomponent reaction profiles. Many of the existing literature show synthesis of pyridine derivatives through a multicomponent reaction involving aromatic aldehydes, and malononitrile/active methylene compounds, in the presence of different catalysts such as Au/MgO, SnCl2·H2O, Zn–VCO3 hydrotalcite and MgO.41–45 In continuation of our work on the synthesis of steroidal heterocyclic compounds,46–49 we challenge ourselves with a one-pot multicomponent approach to prepare steroidal polysubstituted pyridines utilizing MgO NPs as a catalyst.
Steroidal ketones (1–3) employed for the present investigation were conveniently synthesized by the known literature method,50–54 and synthesis of compounds 12a–d and 13a–d was achieved from commercially available pregnenolone acetate (10). The product was obtained by reacting steroidal ketones (1–3 and 10) with malononitrile/methylcyanoacetate, substituted aromatic aldehyde and ammonium acetate in EtOH using MgO NPs as a catalyst (Scheme 1). MgO NPs were found to efficiently catalyze the reaction giving steroidal pyridines with an easy workup procedure.
 | ||
| Scheme 1 Synthesis of steroidal pyridine derivatives. | ||
Encouraged by the outcome, our interest was focused on the optimization of the reaction conditions in terms of effect of different catalysts and solvents by means of a model reaction following the convenient procedure starting from steroidal ketone 1, benzaldehyde (11a), malononitrile and ammonium acetate in ethanol. As depicted in Table 2, many metal oxides were evaluated as a potential catalyst for the synthesis of compound 4 in the presence of ethanol as a solvent. The probe commenced with the synthesis of model compound 4 without any catalyst in ethanol as a solvent and it was observed that a very low yield (25%) of product is obtained in 24 h (Table 2, entry 1). Then we test the reaction with different catalysts and the examination of the outcomes (Table 2) shows that the presence of a catalyst increases the product yield and reduces the reaction time. This indicates that the basic catalysts such as ZnO, CaO, K2CO3, and Al2O3 all have capacity to catalyze the proposed synthetic protocol, although the yields of the product were typically low. Among all the catalysts screened, MgO NPs proved to be the most effective heterogeneous catalyst giving 80% product yield in 6 h. Structurally, MgO is an oxide with a rock-salt structure, which means that on the surface, each Mg2+ is surrounded by five O2− ions. MgO was reported to exhibit surface defects such as edges, corners and kinks, and these surface defects were believed to play a role in splitting of the chemical bonds of the adsorbed molecules and thereby, may influence the catalytic performance of the MgO.55 Chemically, an interesting feature related to MgO is that it is an irreducible oxide, with a very electropositive cation (Mg2+), and oxygen vacancies, which are anion vacancies with trapped electrons.56 The nature of the MgO catalyst, particularly its acidic/basic properties and binding of the substrate to the active sites, seems to play an important role in enhancing the efficiency of the synthesis of the target compounds.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Timec (h) | Yieldd (%) |
| a Reaction conditions: steroidal ketone 1 (1 mmol), malononitrile (1 mmol), benzaldehyde 11a (1 mmol), ammonium acetate (1 mmol), MgO NPs (0.16 g), ethanol (15 mL), 70 °C. b Reaction conditions: steroidal ketone 1 (1 mmol), malononitrile (1 mmol), benzaldehyde 11a (1 mmol), ammonium acetate (1 mmol), MgO NPs (0.16 g), ethanol (15 mL), 70 °C-MW. c Reaction progress monitored by TLC. d Isolated yield of products. | |||||
| 1 | Nil | Ethanol | 70 | 24 | 25 |
| 2 | Al2O3 | Ethanol | 70 | 10 | 65 |
| 3 | K2CO3 | Ethanol | 70 | 6 | 72 |
| 4 | CaO | Ethanol | 70 | 8 | 70 |
| 5 | ZnO | Ethanol | 70 | 8 | 75 |
| 6 | MgO NPs | Methanol | 70 | 6 | 77 |
| 7 | MgO NPs | DMSO | 70 | 7 | 68 |
| 8 | MgO NPs | DMF | 70 | 7 | 70 |
| 9 | MgO NPs | Acetonitrile | 70 | 6.5 | 72 |
| 10 | MgO NPs | Chloroform | 70 | 8 | 67 |
| 11 | MgO NPs | Toluene | 70 | 8 | 62 |
| 12 | MgO NPs | Dichloromethane | 70 | 8 | 65 |
| 13 | MgO NPs | Ethanol | 70 | 6 | 79 |
| 14b | MgO Nps | Ethanol | 70, MW | 20 (min) | 89 |

With the best catalytic system chosen, we subsequently investigated the effect of solvent and it was discovered that a change in solvent has a dramatic impact on the reaction outcome. A cleaner conversion of 1 to 4 was observed in methanol, affording 77% yield of product in 6 h (Table 2, entry 6). The desired reaction rate appeared to be quite sluggish in non-polar solvents such as chloroform, toluene and dichloromethane. Remarkably, the polar solvents are found to be more efficient and ethanol is proved to be the best solvent generating 79% of product exclusively in 6 h. Initially, the reaction was carried out under conventional heating on a magnetic stirrer. As expected, the catalyst shows good efficiency and the reaction generated the desired product with good yield (up to 79%). However, there is increased interest in technologies and concepts that facilitate more rapid synthesis and screening of chemical substances to identify compounds with appropriate qualities. One such high speed technology is microwave-assisted organic synthesis. Taking advantage of this phenomenon we tried to speed up the synthesis of steroidal pyridines through microwave irradiation by performing the reaction in a microwave synthesizer in the presence of MgO NPs. The reaction proceeded smoothly and was greatly accelerated when irradiated with microwaves giving higher yields of up to 89% within 20 min (Table 2, entry 14).
From the comparison of the results obtained by performing reactions through both the conventional heating method and the microwave-assisted method (Table 3), it is evident that the microwave-assisted synthesis was more efficient in terms of reaction time and product yield. Among the steroidal ketones 1–3, the best result was obtained with 3β-acetoxy-5α-cholestan-6-one (1) giving the desired products 4 and 7 in 89% and 87% yield respectively. Furthermore, the steroidal ketone 10 was reacted with four different aromatic aldehydes (11a–d) and the yields of the products were in the range of 84–89% demonstrating that substitution on the aromatic aldehyde has little effect on the formation of the final product. However, compounds 12b and 13b display maximum yields of 87% and 89% respectively in short reaction time, depicting better reactivity of 3-nitrobenzaldehyde. Interestingly, higher yields were observed in the case of steroidal ketone 10 as compared to 1–3 with short reaction time, probably due to steric hindrance in the case of substituted cholestan-6-one (1–3) where the carbonyl group is present at position 6 of the steroidal skeleton. In contrast, the carbonyl group of pregnenolone acetate 10 is present on the side chain and not on the ring carbon and therefore preferably undergoes faster reaction.
| S. no. | Products | Conventional methoda | Microwave irradiationb | ||
|---|---|---|---|---|---|
| Timec (h) | Yieldd (%) | Timec (min) | Yieldd (%) | ||
| a Reaction conditions: steroidal ketone (1–3 and 10) (1 mmol), malononitrile/methylcyanoacetate (1 mmol), substituted aromatic aldehyde 11a–d (1 mmol), ethanol (15 mL), MgO NPs (0.16 g), 70 °C. b Reaction conditions: steroidal ketone (1–3 and 10) (1 mmol), malononitrile/methylcyanoacetate (1 mmol), substituted aromatic aldehyde 11a–d (1 mmol), ethanol (15 mL), MgO NPs (0.16 g), MW-70 °C. c Reaction progress monitored by TLC. d Isolated yield of products. | |||||
| 1 |

|
6 | 79 | 20 | 89 |
| 2 |

|
8 | 74 | 25 | 82 |
| 3 |

|
8 | 70 | 25 | 80 |
| 4 |

|
6 | 78 | 20 | 87 |
| 5 |

|
7.5 | 75 | 20 | 84 |
| 6 |

|
8 | 68 | 25 | 80 |
| 7 |

|
7.5 | 74 | 20 | 84 |
| 8 |

|
6 | 79 | 20 | 87 |
| 9 |

|
7 | 76 | 20 | 86 |
| 10 |

|
8 | 72 | 25 | 85 |
| 11 |

|
7.5 | 70 | 20 | 86 |
| 12 |

|
6 | 78 | 20 | 89 |
| 13 |

|
7.5 | 76 | 20 | 85 |
| 14 |

|
8 | 71 | 25 | 84 |
We performed a series of experiments under microwave irradiation (Table 4) to study the effect of temperature on the model reaction. Notably, the product yield increases and reaction time decreases with increase in temperature and the maximum yield of 89% is obtained at 70 °C in 20 min. Further increment in temperature does not have any effect on the yield of product; therefore, we take 70 °C as a standard temperature for our synthetic protocol.
|
|
|||
|---|---|---|---|
| Entry | Temp. (°C) | Timeb (min) | Yieldc (%) |
| a Reaction conditions: steroidal ketone 1 (1 mmol), malononitrile (1 mmol), benzaldehyde 11a (1 mmol), ammonium acetate (1 mmol), MgO NPs (0.16 g), ethanol (15 mL), 70 °C-MW. b Reaction progress monitored by TLC. c Isolated yield of products. | |||
| 1 | 45 | 75 | 50 |
| 2 | 55 | 45 | 75 |
| 3 | 65 | 30 | 79 |
| 4 | 70 | 20 | 89 |
| 5 | 80 | 20 | 89 |

The formation of the product can be rationalized by initial formation of imine I by the reaction of the steroidal ketone (1–3 and 10) and ammonium acetate and simultaneous formation of arylidene II by standard Knoevenagel condensation of the substituted aromatic aldehyde (11a–d) and malononitrile/methylcyanoacetate. This is followed by Michael-type addition of imine I to the activated double bond of the arylidene intermediate. Intramolecular cyclization of these adducts and subsequent oxidation and aromatization yielded the target products (4–9, 12a–d and 13a–d) (Schemes 2 and 3). MgO NPs have been known as a highly effective heterogeneous base catalyst for Michael addition and Knoevenagel condensation reaction with Mg2+ (Lewis acid) and O2− (Lewis base) sites along with various cationic and anionic vacancies in the lattice.57 These sites are well exposed during the reaction which enables the reactant to stabilize onto these charged sites and promote facile attack of reactants so as to afford the desired product in high yields.
 | ||
| Scheme 2 Plausible reaction mechanism for the formation of steroidal pyridines 4–9. | ||
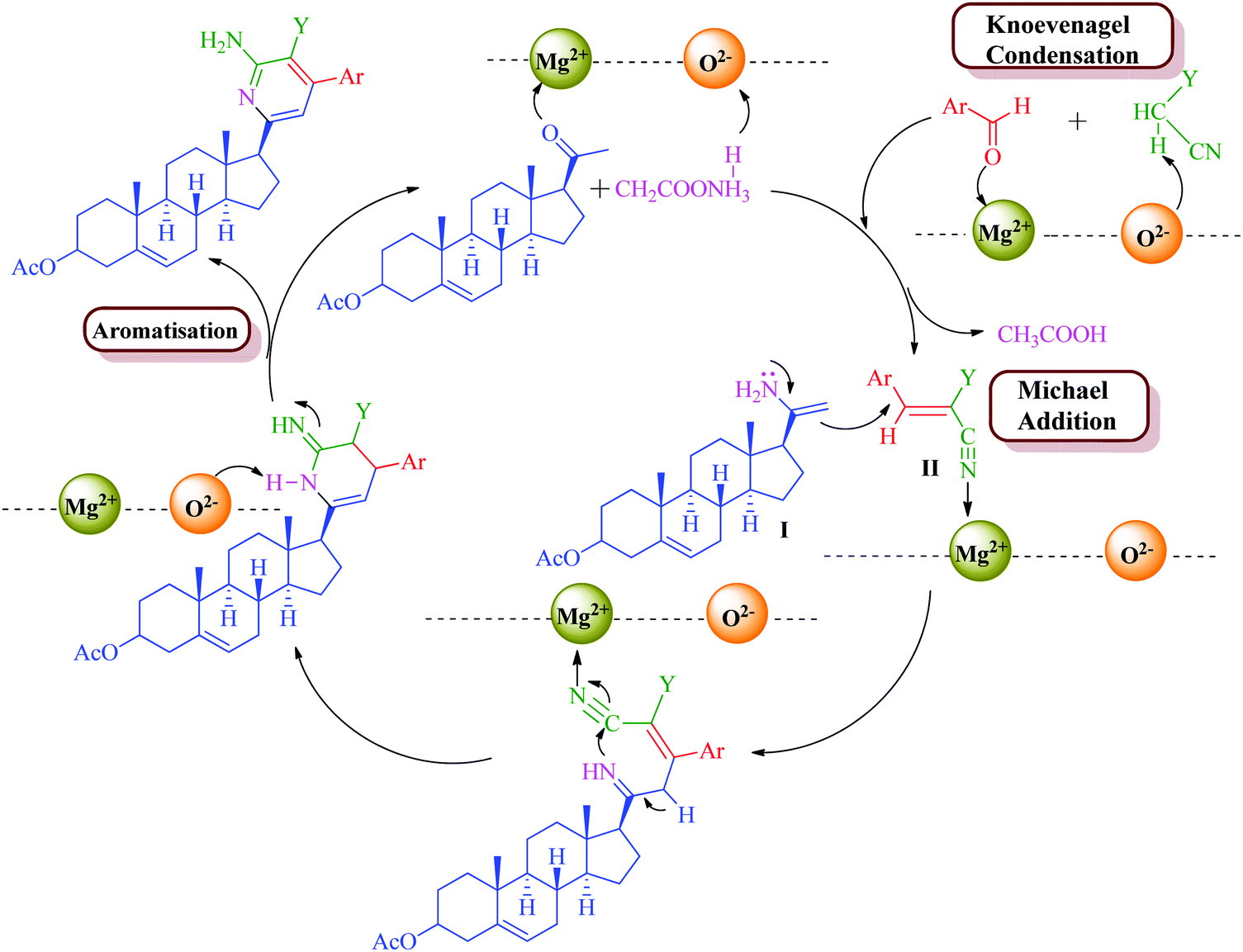 | ||
| Scheme 3 Plausible reaction mechanism for the formation of steroidal pyridines 12a–d and 13a–d. | ||
The synthesized steroidal pyridines (4–9, 12a–d and 13a–d) were characterized by IR, 1H NMR and 13C NMR. The IR spectra of the products showed NH2 stretching in the region 3325–3350 cm−1 and CN stretching vibration of the nitrile group appeared in the region 2250–2245 cm−1. In the 1H NMR spectra, the multiplet at δ 7.7–7.9 was attributed to aromatic protons, while the presence of a singlet at δ 7.3 was assigned to NH2 proton. Besides the signals of aromatic carbons, the 13C NMR spectra of the synthesized compounds show signal at δ 166.6–170.2, 140.8–148.4, and 130.1–138.9 for OC–OCH3, C–NH and CN, respectively.
MgO is a mild base and has the lowest solubility among the alkaline earth oxides and so can potentially be reused.58 The reusability of the catalyst was also examined and it is an important advantage of the heterogeneous catalyst that, being insoluble in the reaction mixture, it is easily recovered by simple filteration and can be reused. The recovery of MgO NPs was accomplished easily by filteration of the reaction mixture, and then they were washed with ethanol, air-dried and used directly for the next round of reactions. The catalyst was then reused for subsequent cycles and was found to exhibit good catalytic activity for five reaction cycles (Fig. 4).
 | ||
| Fig. 4 Recyclability of MgO NPs for the synthesis of model compound 4. | ||
4. Conclusion
In summary, we have synthesized a series of highly substituted steroidal pyridines using MgO NPs as a heterogeneous and recyclable catalyst by an efficient one-pot multicomponent reaction under microwave irradiation. As anticipated, the use of microwave irradiation considerably shortened the reaction time in comparison with conventional heating conditions. More interestingly, the use of a heterogeneous catalyst which can be simply synthesized, easily recovered and reused is valuable. This facile technique can be readily used to synthesize a diverse range of compounds for high-throughput screening in medicinal chemistry.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We sincerely thank Chairman, Department of chemistry, Aligarh Muslim University, Aligarh, India, for providing the necessary research facilities. A. Ansari is also thankful to UGC, New Delhi for awarding (MANF-JRF) a research fellowship. Centre for Nanoscience and Nanotechnology, Jamia Millia Islamia, New Delhi is acknowledged for XRD analysis. The authors would also like to acknowledge SAIF, Panjab University, Chandigarh for providing spectral data.References
- Y.-L. Zhang, Y.-F. Li, Y.-K. Shi, B. Yu, G.-C. Zhang, P.-P. Qi, D.-J. Fu, L.-H. Shan and H.-M. Liu, Steroids, 2015, 104, 1–7 CrossRef CAS PubMed.
- Shamsuzzaman, H. Khanam, A. Mashrai, M. Asif, A. Ali, A. Barakat and Y. N. Mabkhot, J. Taibah Univ. Sci., 2017, 11, 141–150 CrossRef.
- H. N. Bhatti and R. A. Khera, Steroids, 2012, 77, 1267–1290 CrossRef CAS PubMed.
- I. Cerný, M. Budesínský, V. Pouzar and P. Drasar, Steroids, 2009, 74, 88–94 CrossRef PubMed.
- L.-H. Shan, H.-M. Liu, K.-X. Huang, G.-F. Dai, C. Cao and R.-J. Dong, Bioorg. Med. Chem. Lett., 2009, 19, 6637–6639 CrossRef CAS PubMed.
- S. E. Denmark, E. Hartmann, D. J. P. Kornfilt and H. Wang, Nat. Chem., 2014, 6, 1056–1064 CrossRef CAS PubMed.
- P. P. Kaishap, S. Baruah, K. Shekarrao, S. Gogoi and R. C. Boruah, Tetrahedron Lett., 2014, 55, 3117–3121 CrossRef CAS.
- B.-L. Zhang, E. Zhang, L.-P. Pang, L.-X. Song, Y.-F. Li, B. Yu and H.-M. Liu, Steroids, 2013, 78, 1200–1208 CrossRef CAS PubMed.
- B. Yu, P.-P. Qi, X.-J. Shi, L.-H. Shan, D.-Q. Yu and H.-M. Liu, Steroids, 2014, 88, 44–52 CrossRef CAS PubMed.
- J. Zhou, B. Li, F. Hu and B.-F. Shi, Org. Lett., 2013, 15, 3460–3463 CrossRef CAS PubMed.
- E. G. Paronikyan, S. S. Dashyan, A. S. Noravyan, R. A. Tamazyan, A. G. Ayvazyan and H. A. Panosyan, Tetrahedron, 2015, 71, 2686–2691 CrossRef CAS.
- J. B. Gujar, M. A. Chaudhari, D. S. Kawade and M. S. Shingare, Tetrahedron Lett., 2014, 55, 6939–6942 CrossRef CAS.
- I. L. C. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873–886 CrossRef CAS PubMed.
- F. G. Cirujano, I. Luz, M. Soukri, C. Van Goethem, I. F. J. Vankelecom, M. Lail and D. E. De Vos, Angew. Chem., Int. Ed., 2017, 56, 13302–13306 CrossRef CAS PubMed.
- M. A. Aramendia, J. A. Benitez, V. Borau, C. Jimenez, J. M. Marinas, J. R. Ruiz and F. Urbano, Langmuir, 1999, 15, 1192–1197 CrossRef CAS.
- V. K. Das and A. J. Thakur, Tetrahedron Lett., 2013, 54, 4164–4166 CrossRef CAS.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- Y. Ding, G. Zhang, H. Wu, B. Hai, L. Wang and Y. Qian, Chem. Mater., 2001, 13, 435–440 CrossRef CAS.
- J. A. Wang, O. Novaro, X. Bokhimi, T. Lopez, R. Gomez, J. Navarrete, M. E. Llanos and E. Lopez-Salinas, J. Phys. Chem. B, 1997, 101, 7448–7451 CrossRef CAS.
- S. D. Stefanidis, S. A. Karakoulia, K. G. Kalogiannis, E. F. Iliopoulou, A. Delimitis, H. Yiannoulakis, T. Zampetakis, A. A. Lappas and K. S. Triantafyllidis, Appl. Catal., B, 2016, 196, 155–173 CrossRef CAS.
- S. V. Patil, N. D. Gaikwad and V. D. Bobade, Arabian J. Chem., 2016, 9, S1649–S1653 CrossRef CAS.
- D. Kumar, V. B. Reddy, S. Sharad, U. Dube and S. Kapur, Eur. J. Med. Chem., 2009, 44, 3805–3809 CrossRef CAS PubMed.
- I. Mohammadzadeh and H. Sheibani, Chin. Chem. Lett., 2012, 23, 1327–1330 CrossRef CAS.
- D. Kumar, V. B. Reddy, B. G. Mishra, R. K. Rana, M. N. Nadagouda and R. S. Varma, Tetrahedron, 2007, 63, 3093–3097 CrossRef CAS.
- S.-Y. Peng, Z.-N. Xu, Q.-S. Chen, Z.-Q. Wang, Y. Chen, D.-M. Lv, G. Lu and G.-C. Guo, Catal. Sci. Technol., 2014, 4, 1925–1930 CAS.
- J. I. Di Cosimo, V. K. Diez, C. Ferretti and C. R. Apesteguia, Catalysis, 2014, 26, 1–28 CAS.
- J. M. Collins and N. E. Leadbeater, Org. Biomol. Chem., 2007, 5, 1141–1150 CAS.
- C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51–63 CrossRef CAS PubMed.
- M. M. Heravi, N. Karimi, H. Hamidi and H. A. Oskooie, Chin. Chem. Lett., 2013, 24, 143–144 CrossRef CAS.
- Shamsuzzaman, A. Mashrai, H. Khanam, M. Asif, A. Ali, A. Sherwani and M. Owais, J. King Saud Univ., Sci., 2015, 27, 1–6 CrossRef.
- Shamsuzamman, A. Ali, M. Asif, A. Mashrai and H. Khanam, Eur. Chem. Bull., 2014, 3(9), 939–945 Search PubMed.
- Z. Nasir, A. Ali, M. Shakir, R. Wahab, Shamsuzzaman and Lutfullah, New J. Chem., 2017, 41, 5893–5903 RSC.
- H. Khanam, A. Ali and M. Asif, Eur. J. Med. Chem., 2016, 124, 1121–1141 CrossRef CAS PubMed.
- M. Shoeb, M. Mobin, A. Ali, Shamsuzzaman and A. H. Naqvi, Appl. Organomet. Chem., 2017, 1–18 Search PubMed.
- A. Ansari, A. Ali, M. Asif and Shamsuzzaman, New J. Chem., 2017, 41, 16–41 RSC.
- K. Krishnamoorthy, J. Y. Moon, H. B. Hyun, S. K. Cho and S.-J. Kim, J. Mater. Chem., 2012, 24610–24617 RSC.
- N. C. S. Selvam, R. T. Kumar, L. J. Kennedy and J. J. Vijaya, J. Alloys Compd., 2011, 509, 9809–9815 CrossRef CAS.
- C. M. Janet, B. Viswanathan, R. P. Viswanath and T. K. Varadarajan, J. Phys. Chem. C, 2007, 111, 10267–10272 CAS.
- A.-T. Vu, S. Jiang, K. Ho, J.-B. Lee and C.-H. Lee, Chem. Eng. J., 2015, 269, 82–93 CrossRef CAS.
- N. F. Dummer, L. Joyce, H. Ellicott and Y. Jiang, Catal. Sci. Technol., 2016, 6, 1903–1912 CAS.
- R. Pagadala, S. Maddila, V. Moodley, W. E. Van Zyl and S. B. Jonnalagadda, Tetrahedron Lett., 2014, 55, 4006–4010 CrossRef CAS.
- H. Sheibani, K. Saidi, M. Abbasnejad, A. Derakhshani and I. Mohammadzadeh, Arabian J. Chem., 2016, 9, S901–S906 CrossRef CAS.
- A. Abadi, O. Al-Deeb, A. Al-Afify and H. El-Kashef, Farmaco, 1999, 54, 195–201 CrossRef CAS PubMed.
- R. Pagadala, S. Maddila, V. D. B. C. Dasireddy and S. B. Jonnalagadda, Catal. Commun., 2014, 45, 148–152 CrossRef CAS.
- D. N. K. Reddy, K. B. Chandrasekhar, Y. S. S. Ganesh, B. S. Kumar, R. Adepu and M. Pal, Tetrahedron Lett., 2015, 56, 4586–4589 CrossRef CAS.
- Shamsuzzaman, K. A. A. A. Baqi, A. Ali, M. Asif, A. Mashrai, H. Khanam, A. Sherwani, Z. Yaseen and M. Owais, J. Mol. Struct., 2015, 1085, 104–114 CrossRef CAS.
- M. Asif, A. Ali, A. Zafar, M. Farhan, H. Khanam, S. M. Hadi and Shamsuzzaman, J. Photochem. Photobiol., B, 2017, 166, 104–115 CrossRef CAS PubMed.
- Shamsuzzaman, K. A. A. A. Baqi, M. Asif, A. Ali, H. Khanam, A. Mashraf, A. Ahamd and A. U. Khan, Eur. Chem. Bull., 2015, 4, 154–164 CAS.
- Shamsuzzaman, M. Asif, A. Ali, A. Mashrai and H. Khanam, Eur. Chem. Bull., 2014, 3(11), 1075–1080 Search PubMed.
- R. K. Callow and V. H. T. James, J. Chem. Soc., 1956, 4744–4749 RSC.
- A. H. Milburn and E. V. Truter, J. Chem. Soc., 1956, 341, 1736–1739 RSC.
- G. W. Dauben and K. H. Takemura, J. Am. Chem. Soc., 1953, 75, 6302–6304 CrossRef.
- A. Ali, M. Asif, H. Khanam, A. Mashrai, M. A. Sherwani, M. Owais and Shamsuzzaman, RSC Adv., 2015, 5, 75964–75984 RSC.
- A. Ali, M. Asif, P. Alam, M. Jane Alam, M. Asif, R. Hasan and S. Ahmad, Bioorg. Chem., 2017, 73, 83–99 CrossRef CAS PubMed.
- H. Duński, W. K. Jóźwiak and H. Sugier, J. Catal., 1994, 146, 166–172 CrossRef.
- M. Calatayud, A. Markovits, M. Menetrey, B. Mguig and C. Minot, Catal. Today, 2003, 85, 125–143 CrossRef CAS.
- H. T. B. Bui, Q. T. K. Ha, W. K. Oh, D. D. Vo, Y. N. T. Chau, C. T. K. Tu, E. C. Pham, P. T. Tran, L. T. Tran and H. Van Mai, Tetrahedron Lett., 2016, 57, 887–891 CrossRef CAS.
- C. Xu, J. K. Bartley, D. I. Enache, D. W. Knight, M. Lunn, M. Lok and G. J. Hutchings, Tetrahedron Lett., 2008, 49, 2454–2456 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj03742b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |