
Antitumor effect of sikokianin C, a selective cystathionine β-synthase inhibitor, against human colon cancer in vitro and in vivo†
Weining
Niu‡
 *,
Fei
Chen‡
,
Jun
Wang
,
Jing
Qian
and
Shasha
Yan
*,
Fei
Chen‡
,
Jun
Wang
,
Jing
Qian
and
Shasha
Yan
School of Life Sciences, Northwestern Polytechnical University, Xi'an, 710072, China. E-mail: niuweining@nwpu.edu.cn
First published on 16th November 2017
Abstract
Cystathionine β-synthase (CBS) overexpression is related to the proliferation and migration of human colon cancers. Targeted therapy that inhibits CBS has achieved promising effects in colon cancer treatments, but no selective inhibitor of CBS is available. In our previous study, a natural biflavonoid compound, sikokianin C, was identified as a potent and selective inhibitor of CBS. However, the mode of action of this compound and its antitumor efficacy in vivo remain unknown. In the present study, we have demonstrated that sikokianin C selectively inhibits CBS activity in a competitive manner, and the five key residues involved in the binding of sikokianin C to the substrate channel of CBS protein were identified via a combination of molecular docking and site-directed mutagenesis. Additionally, we analyzed the antitumor efficacy of sikokianin C against human colon cancer cells in vitro and in vivo. Sikokianin C greatly suppressed the proliferation of HT29 colon cancer cells with an IC50 value of 1.6 μM, and CBS is the target of sikokianin C in mammalian cells, as evidenced by CBS knockdown analyses. Moreover, sikokianin C induced the apoptosis of HT29 cancer cells in a dose dependent manner. Treating mice with sikokianin C dramatically reduced the tumor volume and the weight of the colon cancer xenograft in vivo. These results indicate that the selective CBS inhibitor sikokianin C can potentially be used for the treatment of colon cancer.
Introduction
Cystathionine β-synthase (CBS), a unique pyridoxal 5′-phosphate (PLP)-dependent heme protein, is the central enzyme of the transsulfuration pathway that converts homocysteine into cysteine,1–3 which is the limiting reagent in the synthesis of the antioxidant glutathione.4,5 Alternatively, CBS, which is one of three key enzymes involved in the biosynthesis of hydrogen sulfide (H2S), can catalyze the condensation of homocysteine and cysteine or the condensation of two moles of cysteine to produce H2S,6,7 a recently recognized endogenous gasotransmitter that mediates diverse biological functions, including growth, differentiation, movement and cell death.8,9Previous studies have indicated that CBS is highly expressed in colon cancers compared to that in normal colonic mucosa.10 Overexpression of the CBS protein has also been observed in multiple colon cancer cell lines including HT29, HCT116, and LoVo.10,11 Additionally, in breast, ovarian and bladder cancers, a high level of CBS expression has also been detected.12–15 Inhibition of CBS activity with inhibitors reduces the proliferation and migration of cancer cells and reduces the growth of tumors in vivo, suggesting that CBS may be an anticancer drug target.10–15 Therefore, the development of selective and potent CBS inhibitors may be of great value in treating CBS-related diseases.
However, currently, aminooxyacetic acid (AOAA) is the only small molecule compound that has been widely used to inhibit CBS activity in vivo, despite its poor selectivity for human CBS over other PLP-dependent enzymes, including another H2S-producing enzyme cystathionine γ-lyase (CSE), and its poor potency, as a higher dose of AOAA (1–10 mM) is required to inhibit CBS activity under physiological conditions.16 In previous reports, several selective CBS inhibitors from commercially available libraries have been identified through high-throughput screening assays.17–20 However, the antitumor efficacy of only one compound, benserazide, was assessed against the growth of HT29 cell xenografts in nude mice, with a high dose of 50 mg kg−1 d−1.19
More recently, we have identified four selective CBS inhibitors (which do not inhibit the activity of CSE) by screening 6491 natural compounds.20 These four inhibitors are categorized as biflavonoids, and among them, sikokianin C isolated from the medicinal herb Stellera chamaejasme had the most potent cytotoxic activity against HT29 cells, with an IC50 value of 1.5 μM.20 However, the mechanism behind the interaction between the CBS protein and sikokianin C is unknown. Additionally, the antitumor efficacy of sikokianin C against human colon cancer in vivo needs to be further evaluated. In this study, the binding mode between CBS and sikokianin C was investigated by molecular docking simulations, and the key residues involved in the binding of sikokianin C to the CBS protein were identified via a combination of molecular docking and site-directed mutagenesis. We also investigated the in vitro antiproliferation effect of sikokianin C on HT29 colon cancer cells and tested the in vivo antitumor efficacy of this compound in a human colon cancer xenograft.
Experimental
Materials
Aminooxyacetic acid (AOAA), DL-propargylglycine (PAG) and D,L-homocysteine were purchased from Sigma-Aldrich (Missouri, USA); HT29 cells (human colon adenocarcinoma cell line) were obtained from the American Type Culture Collection (ATCC, USA); Cell Counting Kit-8 (CCK-8) was purchased from Dojindo (Kumamoto, Japan); sikokianin C was obtained from BioBioPha (Kunming, China); the anti-cystathionine β-synthase (anti-CBS) antibody was obtained from ABclonal (Wuhan, China); the anti-cystathionine γ-lyase (anti-CSE) antibody was purchased from Abcam (Cambridge, USA). Unless otherwise specified, all chemicals were used as received.Protein expression and purification
Mutagenesis of CBS was carried out using a Quikchange II XL Site-directed Mutagenesis Kit (Agilent Technologies, USA). The mutations were verified by DNA sequencing (Sangon, Shanghai, China). The expression and purification of the wild-type and variant versions of human CBS were performed as described previously for the wild-type proteins.20 The purity of all protein preparations was judged to be >90% by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) analysis.H2S production assays
H2S determination was performed by using a lead sulfide assay as described previously with some modifications.21 For measuring the CBS enzyme, an enzyme reaction mixture containing 50 mM HEPES buffer (pH 7.4), 0.4 mM lead nitrate, 2 mM homocysteine and CBS was preincubated at 37 °C for 3 min, and then, the reaction was started by the addition of 2 mM cysteine. An extinction coefficient of 5500 M−1 cm−1 at 390 nm was used to estimate the lead sulfide concentration.To prepare the cell lysate, HT29 cells were washed thrice with ice-cold PBS buffer and then scraped from 10 cm plates. The cells were separated by centrifugation for 5 min, and 500 μL of HEPES buffer (50 mM, pH 7.4) was added (500 μL buffer per plate cell). The cell suspension was frozen and thawed thrice using a liquid nitrogen/water bath. The methylene blue assay was employed to measure the H2S production with the following modifications.16 The reaction solution (200 μL) contained the cell lysate, 2 mM homocysteine and 2 mM cysteine. The inhibitors DL-propargylglycine (2 mM) and aminooxyacetic acid (1 mM) were added to the mixture 10 min prior to the addition of cysteine. After 30 min of incubation at 37 °C, the reaction was terminated by the addition of 1% Pb(NO3)2 to trap H2S, followed by the addition of 15% trichloroacetic acid (TCA) to precipitate proteins. Then, N,N-dimethyl-p-phenylenediamine sulfate in 7.2 M HCl was added to the reaction mixture and the addition of FeCl3 in 1.2 M HCl immediately followed. The absorbance of the solution was measured at 670 nm. The H2S content was calculated against a calibration curve of standard H2S solutions.
Molecular docking
Molecular docking was conducted using the GOLD 5.3 program,22 and the crystal structure of CBS was obtained from the Protein Data Bank (PDB ID: 1JBQ). The 3D model of CBS was carried out with 5000 minimization steps using the ff99SB force field in Amber12.23 The 3D structure of sikokianin C was sketched using Maestro (Schrodinger Inc.) and optimized in 2000 steps in Amber12 using a GAFF force field before the docking.24 Due to the superiority of binding pose prediction, the ChemPLP score from the GOLD program was used to finally produce the best binding model.25 It should be noted that all docking simulations were conducted at pH 7.0. The details of the molecular docking followed those of previous studies.26–28 Visualization of the structures was performed using PyMol (DeLano Scientific, Palo Alto, USA.). The molecular mechanics Poisson–Boltzmann surface area method in Amber12 was employed to calculate the binding free energy of the constructed CBS–sikokianin C complex.29–31 The detailed process was run according to previous studies.27,32Effects of sikokianin C on the activity of wild-type and mutant CBS proteins
The dose-dependent effects of sikokianin C on the activity of wild-type and mutant CBS were determined by using the lead sulfide assay described above. The IC50 values of sikokianin C against wild-type and mutant CBS were measured in the presence of 2 mM homocysteine, 2 mM cysteine, 5 μg of CBS protein and 0.4 mM lead nitrate in a 200 μL total volume (5% DMSO and 50 mM HEPES (pH 7.4)). A 5% DMSO reaction mixture without sikokianin C was used as a control. The purity of sikokianin C was 95% or more. Additionally, the dose-dependent effects of AOAA on the activity of wild-type CBS were also determined according to the methods mentioned above.To test whether sikokianin C self-associates into colloidal aggregates that non-specifically inhibit CBS, the effects of sikokianin C on the activity of wild-type CBS were determined in the presence of detergent (0.01% Triton X-100, vol/vol) according to a previous report.33
Measurement of anti-proliferative activity
HT29 cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum and 1% penicillin–streptomycin solution (Sigma) at 37 °C with 5% CO2. The IC50 values of sikokianin C and AOAA against HT29 cells were determined using the CCK-8 assay, respectively. During the exponential growth phase, HT29 cells were inoculated in a 96-well plate at a density of 1 × 104 cells per well. After 24 hours of culture, fresh medium containing varying concentrations of sikokianin C and AOAA was added to the cells for an additional 24 h incubation. Subsequently, the CCK-8 solution (10 μL per well) was added to each well, followed by an additional 2 h incubation at 37 °C. The optical density at 450 nm (OD450) was measured using a microplate reader, and the medium without sikokianin C and AOAA was used as the control group. The cell viability rate is presented as the percentage of the OD450 of the experimental group compared to that of the control group, which was considered to be 100%.Measurement of apoptosis
HT29 cells (5 × 105 cells per well) were seeded in 6-well plates and treated with 0.5 μM, 1 μM and 2 μM sikokianin C for 24 h, respectively. Then, they were trypsinized, centrifuged and washed with cold PBS, and re-suspended in 195 μL of the provided binding buffer. 10 μL FITC-labeled Annexin V and 5 μL PI (Beyotime, Shanghai, China) were added to the samples and mixed gently. After 15 min incubation in the dark at room temperature, the samples were immediately run on a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA) with a total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells collected and analyzed with the Annexin V-FITC/PI apoptosis method. Cytographs were analyzed using BD CellQuest Pro Software (BD Biosciences, San Jose, CA, USA). The results were represented as the mean ± SD from three independent experiments.
000 cells collected and analyzed with the Annexin V-FITC/PI apoptosis method. Cytographs were analyzed using BD CellQuest Pro Software (BD Biosciences, San Jose, CA, USA). The results were represented as the mean ± SD from three independent experiments.
Knockdown of CBS using small interfering RNA (siRNA)
The following siRNA sequences specific to human CBS were used: 5′-GGAAGAAGUUCGGCCUGAATT-3′, 5′-GGAACUACAUGACCAAGUUTT-3′ and 5′-CCAUUGACUUGCUGAACUUTT-3′. A random siRNA was used as the negative control siRNA, and 6-FAM (6-carboxyfluorescein) labeling of random siRNA was used to monitor the transfection efficiency. All siRNAs were designed and synthesized by Sangon (Shanghai, China). HT29 cells at 30–40% confluency were transfected with 70 μmol of siRNA using Lipofectamine 2000 (Invitrogen) in 60 mm dishes and then incubated for 48 h according to the manufacturer's protocol.Western blotting
HT29 cells were washed thrice with ice-cold PBS and then lysed in lysis buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP40, and protease inhibitors). The cell lysates were centrifuged at 12000 rpm for 10 min at 4 °C, and the supernatants were collected for protein concentration determination. The proteins were resolved by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and then transferred to a PVDF membrane. The membranes were incubated with anti-CBS (1:2000) and anti-CSE (1:10000) antibodies, followed by incubation with anti-horseradish peroxidase-linked secondary antibodies. The blots were visualized using chemiluminescence (Tanon, Shanghai, China) and were quantified using ImageJ (National Institute of Health).
In vivo study
Five-week-old male BALB/c nude mice were purchased from Shanghai Lingchang Biological Technology Co., Ltd. (Shanghai, China). All experiments were performed in accordance with the official recommendations of the Chinese animal community. Mice were injected subcutaneously (s.c.) in the right armpit (100 μL of HT29 cells, 1 × 107 cells per mL). The tumor volume was measured periodically using calipers to monitor tumor growth (volume = 0.5 × length × width2). When the tumor volume reached approximately 100 mm3, the mice were randomly assigned to the control or treatment groups. Control groups were given a vehicle containing 1% DMSO (dimethyl sulfoxide) and 0.5% carboxymethylcellulose sodium in PBS buffer. AOAA (10 mg kg−1 d−1), sikokianin C at 3 mg kg−1 d−1 or sikokianin C at 10 mg kg−1 d−1 was provided to the animals 6 d per week. t-Test comparisons of the tumor volumes and final weights at harvest were performed. This experiment was approved by the Institutional Animal Care and Use Committee of Northwestern Polytechnical University (permit number: 2017012) and was performed in accordance with the National Institutes of Health guidelines for the care and use of experimental animals.Statistical analysis
Data are presented as the means ± SD. Statistical analysis was carried out using GraphPad Prism version 6.0 for Windows. P < 0.05 was considered to be statistically significant.Results and discussion
Sikokianin C selectively inhibits CBS activity via a competitive mechanism
In our recent study, sikokianin C (Fig. 1A), a natural biflavonoid compound isolated from the medicinal herb Stellera chamaejasme, was identified as a selective inhibitor of recombinant CBS.20 Moreover, it does not inhibit the activity of human cystathionine γ-lyase (CSE), which is another H2S-producing enzyme in mammals. To determine the mode of action of this selective inhibitor, the assay for monitoring the CH3SH-producing activity of CBS was employed to investigate the effect of varying concentrations of sikokianin C on CBS activity using S-methylcysteine as a single substrate.20 The results indicated that sikokianin C is a competitive inhibitor of CBS (Fig. S1†). Indeed, an IC50 value of 0.9 ± 0.2 μM was obtained in the presence of 2 mM cysteine and 2 mM homocysteine by using a standard H2S-producing assay (Fig. 1B), and the IC50 value increased to 5.5 ± 1.2 μM in the presence of 20 mM cysteine and 2 mM homocysteine (Fig. S2†), which is similar to our previous report.20 Given the low concentration of cysteine (<0.2 mM) in cells,34 the IC50 value of 0.9 ± 0.2 μM determined using 2 mM cysteine is more reasonable under physiological conditions. | ||
| Fig. 1 Dose-dependent inhibition of the activity of purified human wild-type CBS by sikokianin C and AOAA. (A) Chemical structure of sikokianin C. (B and C) The IC50 values of sikokianin C and AOAA against wild-type CBS were measured in the presence of 2 mM homocysteine, 2 mM cysteine, 5 μg of CBS protein, 0.4 mM lead nitrate, and varying concentrations of sikokianin C and AOAA in a 200 μL mixture (5% DMSO and 50 mM HEPES (pH 7.4)). The graph represents the relative activity of CBS in the presence of different concentrations of inhibitors compared with the activity of untreated CBS (control), and each data point is the mean ± SD (n = 3). GraphPad Prism 6 software was used to fit the data. | ||
We next investigated the effect of AOAA, a commonly used pharmacological CBS inhibitor, on the H2S-producing activity of CBS at different substrate concentrations. An IC50 value of 7.5 ± 0.6 μM was obtained in the presence of 2 mM substrate (Fig. 1C), and the IC50 value was 8.6 ± 0.5 μM in the presence of 20 mM substrate (Fig. S3†), which is consistent with a previous report.16 Unlike the inhibitor sikokianin C, changes in substrate concentration do not significantly affect the IC50 value of AOAA, suggesting that AOAA is a non-competitive inhibitor of CBS.
Additionally, previous studies indicated that at micromolar concentrations, many small molecules self-associate into colloidal aggregates that non-specifically inhibit enzymes and other proteins, and the aggregates can be dissociated by the addition of detergent.33 To rule out the possibility of non-specific inhibition, the effect of sikokianin C on the activity of CBS was determined in the presence of detergent (0.01% Triton X-100, vol/vol). As shown in Fig. S4,† an IC50 value of 1.4 μM was obtained, which is similar to the IC50 value (0.9 μM) in the absence of the detergent. These results indicated that sikokianin C specifically inhibits the activity of CBS.
Key residues involved in the sikokianin C binding site on CBS
To understand the mechanisms underlying the selectivity of this inhibitor, we further investigated the sikokianin C binding site on CBS using structural modelling. The crystal structure of human CBS, which has been determined in the absence of a substrate, was employed for docking analysis. The docking complex revealed that the sikokianin C compound appears to bind at the entrance of the substrate access channel on the CBS protein (Fig. S5†). According to the binding free energy calculation, five amino acid residues (T193, N194, H203, Y223 and Y308) contribute >2 kcal mol−1 to the total interaction energy, as shown in Fig. 2A and B. These five residues, H203, Y308, Y223, N194 and T193, which are listed in descending order by the total binding energy contribution, were predicted to be involved in sikokianin C binding to CBS in the simulated CBS–sikokianin C complex. In this structure, the phenolic hydroxyl groups and the carbonyl group of sikokianin C form five hydrogen bonds with these five residues. | ||
| Fig. 2 Interaction analysis for the simulated CBS–sikokianin C complex. (A) Average conformation of CBS–sikokianin C complex. (B) Total energy contribution of each amino acid residue in the CBS protein. | ||
To further verify the reliability of the molecular docking analysis, these five residues were replaced with alanine by site-directed mutagenesis. The specific activities of the wild-type and five mutant CBS proteins (T193A, N194A, H203A, Y223A and Y308A) are shown in Fig. 3A. The H2S producing activity of the N194A and Y308A CBS mutants was similar to that of wild-type CBS (24.8 ± 0.59 μmol h−1 mg−1), while the activity of the T193A, H203A and Y223A CBS mutants decreased dramatically. We next investigated the effect of sikokianin C on the activity of these five mutants by measuring the IC50 values of sikokianin C against the mutant CBS proteins. As shown in Fig. 3B–F, the binding affinity of sikokianin C was weakened to various degrees by the individual mutation of selected residues (T193, N194, H203, Y223 and Y308), with IC50 values ranging from 11.2 ± 1.8 μM to 43.0 ± 2.3 μM, compared with the high affinity of sikokianin C for wild-type CBS, as evidenced by an IC50 value of 0.9 ± 0.2 μM (Fig. 1B). Of the five mutant proteins, H203A and Y308A CBS were unfavorable for the binding of sikokianin C, with IC50 values of 43.0 ± 2.3 μM and 24.0 ± 1.0 μM, respectively, which are ∼48 and 27 times higher than the IC50 values determined for wild-type CBS. For the other three residues, namely, T193, N194 and Y223, their mutations moderately impacted the binding affinity of sikokianin C, with IC50 values of 11.2 ± 1.8 μM, 18.7 ± 1.1 μM and 16.9 ± 2.7 μM, respectively. These experimental results are consistent with the total binding energy contributions determined in the docking analysis.
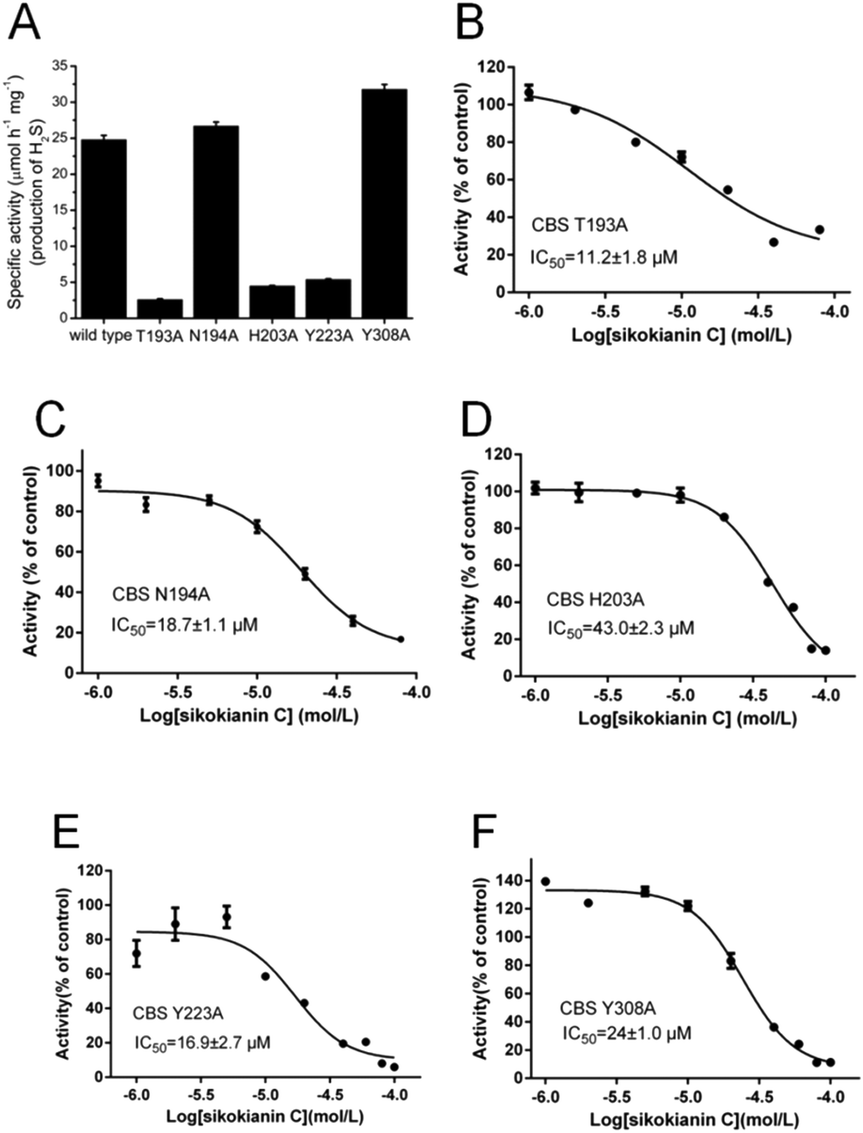 | ||
| Fig. 3 Dose–response curves of sikokianin C with the five mutant CBS proteins. (A) The activities of the wild-type and five mutant CBS proteins were determined by using the lead sulfide assay. (B–F) Dose–response curves of sikokianin C with the T193A CBS, N194A CBS, H203A CBS, Y223A CBS and Y308A CBS mutants. The mutation of each amino acid residue reduces the binding ability between CBS and sikokianin C to various degrees. | ||
Sikokianin C is cytotoxic against HT29 colon cancer cells
A previous study indicated that, in colorectal cancers, a substantially high level of human CBS expression plays a key role in promoting the proliferation and migration of cancer cells.10 We next sought to determine the CBS expression profile and the effect of sikokianin C on proliferation in HT29 cells. Consistent with a previous report, a high level of CBS expression and a very low level of CSE expression, which is another enzyme involved in the biosynthesis of H2S in mammalian cells, were detected in HT29 cells (Fig. 4A). Additionally, the H2S-producing activity of CBS and CSE in the HT29 cell lysate was determined by using the methylene blue assay in the presence of AOAA (an inhibitor of both CBS and CSE) or PAG (an inhibitor of CSE). As shown in Fig. 4B, compared with the H2S-producing activity of the control group (27 ± 0.39 nmol h−1 mg−1), the activity of the cell lysate in the presence of 1 mM AOAA or 2 mM PAG was 5.4 ± 0.16 nmol h−1 mg−1 or 24.6 ± 0.89 nmol h−1 mg−1, respectively. Therefore, the amount of CSE-derived H2S was 2.4 nmol h−1 mg−1, and that of CBS-derived H2S was 19.2 nmol h−1 mg−1. The calculated amount of CBS-derived H2S was 8-fold higher than that of CSE-derived H2S in HT29 colon cancer cells. Notably, sikokianin C inhibited the proliferation of HT29 cells with an IC50 value of 1.6 ± 0.2 μM (Fig. 5A). AOAA showed a weak inhibition of HT29 cells with an IC50 value of 159.2 ± 22.3 μM (Fig. S6†), which is consistent with previous studies.19,20 | ||
| Fig. 4 Small interfering RNA (siRNA)-mediated knockdown of CBS in HT29 cells. (A) A high expression of the H2S producing enzyme CBS, not cystathionine γ-lyase (CSE), was detected in HT29 cells by immunoblotting analysis. (B) The H2S-producing activities of CBS and CSE were measured by using the methylene blue assay described in the “Experimental” section. 200 μL of the reaction mixture (50 mM HEPES, pH 7.4) contained the cell lysate, 2 mM homocysteine, 2 mM cysteine and either 2 mM DL-propargylglycine (an inhibitor of CSE) or 1 mM aminooxyacetic acid (an inhibitor of CBS and CSE). (C) HT29 cells were transfected with a negative control siRNA (NC siRNA) or a CBS-targeted siRNA (CBS siRNA) for 48 h; then, cell lysates were used for immunoblotting analysis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the loading control. | ||
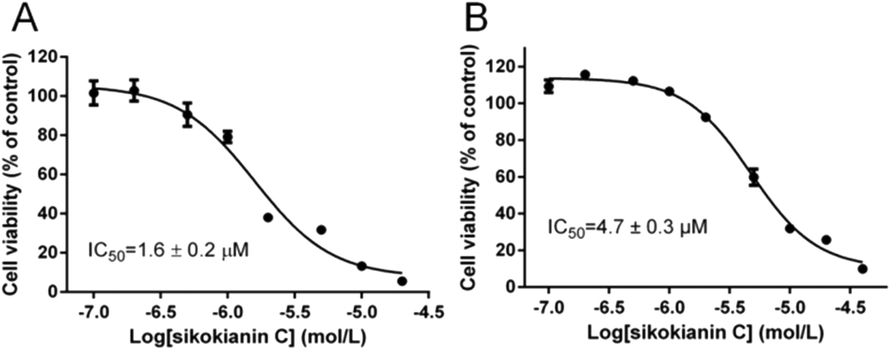 | ||
| Fig. 5 Effect of sikokianin C on the viability of HT29 cells with or without siRNA-mediated knockdown of CBS. (A) The IC50 value of sikokianin C against HT29 cells was determined using the CCK-8 assay. (B) The IC50 value of sikokianin C against HT29 cells after the siRNA-mediated knockdown of CBS was determined using the CCK-8 assay. The data represent the cell viability of the experimental group compared with that of the control group (no sikokianin C) and are presented as the means ± SD (n = 4). GraphPad Prism 6 software was used to fit the data. | ||
To investigate whether sikokianin C specifically targeted CBS in mammalian cells, CBS was knocked down in HT29 cells using siRNA. As shown in Fig. 4C, the protein expression level of CBS decreased to 41% of that of the control group, as determined by immunoblotting analysis. Consistent with the previous study,10 siRNA-mediated knockdown of CBS inhibits the proliferation of HT29 cells (Fig. S7†). The dose–response curve of sikokianin C was measured to obtain the IC50 value for inhibiting proliferation in HT29 CBS-knockdown cells. As shown in Fig. 5B, the IC50 value is 4.7 ± 0.3 μM, which is approximately 3 times higher than the IC50 value measured in the untreated HT29 cells. Expectedly, the IC50 value of AOAA against HT29 CBS-knockdown cells also increased to 286.7 ± 44.5 μM compared to 159.2 ± 22.3 μM determined in HT29 cells (Fig. S6†). Compared with our previous report, the IC50 value of sikokianin C against the human nonmalignant colonic epithelial NCM356 cell line (which overexpresses CSE but has very low levels of CBS10) is 19 μM.20 The discrepancy between these two IC50 values in the CBS-knockdown HT29 cells and the NCM356 cells may result from the low siRNA knockdown efficiency (only 59% of CBS protein was silenced in HT29 cells). These results indicated that CBS is the target of sikokianin C in mammalian cells. However, it should be noted that sikokianin C probably has other targets in vivo in addition to the CBS protein.
Additionally, to explore the mode of cell death for HT29 cancer cells, sikokianin C was employed to induce HT29 cancer cell apoptosis, which was then examined by the Annexin V-FITC/PI assay. As shown in Fig. 6, the percentages of apoptosis for HT29 cells treated with sikokianin C at 0.5, 1, and 2 μM for 24 hours were 17.1, 22.0, and 31.5%, respectively. These results indicated that sikokianin C induced the apoptosis of HT29 cancer cells in a dose dependent manner.
 | ||
| Fig. 6 Flow cytometric analysis of HT29 cell apoptosis after being treated with different concentrations of sikokianin C. (A) HT29 cells (untreated and sikokianin C treated) were stained with Annexin V/PI. (B) The percentages of Annexin V-positive cells of HT29 cells treated with different concentrations of sikokianin C. Data were represented as means ± SD (n = 3). **p < 0.01 vs. control; ***p < 0.001 vs. control. | ||
In vivo antitumor activity of sikokianin C in HT29 colon cancer xenografts
Given its encouraging activity in vitro, we next investigated the antitumor efficacy of sikokianin C in vivo. Here, we established a colon xenograft tumor model derived from HT29 cells in nude mice. When the average tumor volume reached approximately 100 mm3, daily s.c. treatments of sikokianin C (3 mg kg−1 d−1), sikokianin C (10 mg kg−1 d−1), AOAA (10 mg kg−1 d−1) or vehicle control were provided.A previous study indicated that tumor growth was significantly reduced by 9 mg kg−1 d−1 AOAA, but not at lower doses in nude mice bearing subcutaneous colon cancer cell xenografts.35 Therefore, in the present study, AOAA (10 mg kg−1 d−1) was used as a positive control. No major adverse effects on body weight were observed in the sikokianin C and AOAA groups compared with that of the control group (Fig. S8†). The animals were sacrificed on day 28 (after 12 treatments), and tumors were excised and weighed. Both sikokianin C (3 mg kg−1 d−1 and 10 mg kg−1 d−1) and AOAA significantly inhibited the growth of xenograft tumors compared with the control (Fig. 7A–C). Moreover, sikokianin C dose-dependently inhibited the growth of tumors. The tumor weights in the sikokianin C (10 mg kg−1 d−1) and AOAA groups were reduced to 46% and 44%, respectively, compared to that of the control group (Fig. 7D), suggesting that the antitumor efficacy of sikokianin C is identical to that of AOAA at a dose of 10 mg kg−1 d−1.
 | ||
| Fig. 7 Sikokianin C inhibits tumor growth in human colon cancer xenograft models. Nude mice with established subcutaneous HT29 human colon tumors were injected s.c. with aminooxyacetic acid (AOAA, positive control), sikokianin C or the vehicle control. All regimens were administered daily for 6 d with a 1 d drug holiday for a total number of 12 treatments. (A) The macroscopic appearance of tumors in BALB/c nude mice 12 days after treatment (n = 6 per group). Antitumor effect of sikokianin C and AOAA against established xenograft tumors in nude mice. On day 28, all the mice were killed, and tumors were excised. (B and C) Sikokianin C treatment significantly reduced the tumor volume after the s.c. injection of HT29 cells in human tumor xenograft models. Each data point represents the mean ± SD (n = 6). (D) Mean tumor weight (±SD). ***p < 0.001. | ||
Currently, AOAA is the only small molecule compound that has been widely used as an inhibitor of CBS in vivo. However, a previous study indicated that AOAA is also an effective CSE inhibitor, probably leading to side effects in the cardiovascular system where CSE is the major enzyme responsible for producing H2S. Therefore, AOAA should no longer be used to selectively inhibit CBS in vitro and in vivo.16 Thus, there has been increasing interest in developing a selective and potent CBS inhibitor. Collectively, the results of our study demonstrate that sikokianin C, a selective CBS inhibitor, has significant antitumor activity against colon cancer in vitro and in vivo. However, it should be noted that sikokianin C probably has other targets in vivo in addition to the CBS protein.
Conclusions
In summary, our results show that sikokianin C is a selective, competitive inhibitor of CBS. The key residues involved in the binding of sikokianin C to CBS were identified via a combination of molecular docking and site-directed mutagenesis. Moreover, our findings clearly show that sikokianin C significantly inhibits colon cancer cell proliferation and survival in vitro and in vivo. These results suggest that sikokianin C can be used as a selective inhibitor of CBS in vivo.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Number 31471086), the Key Science and Technology Program of Shaanxi Province, China (Grant Number 2017GY-143), the Fundamental Research Funds for the Central Universities of China (Grant Number: G2017KY0001) and the Seed Foundation of Innovation and Creation for Graduate Students in Northwestern Polytechnical University (Grant Number Z2017233).References
- R. Banerjee and C. G. Zou, Arch. Biochem. Biophys., 2005, 433, 144–156 CrossRef CAS PubMed.
- E. W. Miles and J. P. Kraus, J. Biol. Chem., 2004, 279, 29871–29874 CrossRef CAS PubMed.
- R. Banerjee, R. Evande, O. Kabil, S. Ojha and S. Taoka, Biochim. Biophys. Acta, 2003, 1647, 30–35 CrossRef CAS.
- X. Chen, K. H. Jhee and W. D. Kruger, J. Biol. Chem., 2004, 279, 52082–52086 CrossRef CAS PubMed.
- S. Singh, D. Padovani, R. A. Leslie, T. Chiku and R. Banerjee, J. Biol. Chem., 2009, 284, 22457–22466 CrossRef CAS PubMed.
- O. Kabil and R. Banerjee, J. Biol. Chem., 2010, 285, 21903–21907 CrossRef CAS PubMed.
- O. Kabil and R. Banerjee, Antioxid. Redox Signaling, 2014, 20, 770–782 CrossRef CAS PubMed.
- O. Kabil, N. Motl and R. Banerjee, Biochim. Biophys. Acta, 2014, 1844, 1355–1366 CrossRef CAS PubMed.
- H. Kimura, Antioxid. Redox Signaling, 2010, 12, 1111–1123 CrossRef CAS PubMed.
- C. Szabo, C. Coletta, C. Chao, K. Módis, B. Szczesny, A. Papapetropoulos and M. Hellmich, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12474–12479 CrossRef CAS PubMed.
- M. Hellmich, C. Coletta, C. Chao and C. Szabo, Antioxid. Redox Signaling, 2015, 22, 424–448 CrossRef CAS PubMed.
- S. Bhattacharyya, S. Saha, K. Giri, I. R. Lanza, K. S. Nair, N. B. Jennings, C. Rodriguez-Aguayo, G. Lopez-Berestein, E. Basal, A. L. Weaver, D. W. Visscher, W. Cliby, A. K. Sood, R. Bhattacharya and P. Mukherjee, PLoS One, 2013, 8, e79167 Search PubMed.
- S. Sen, B. Kawahara, D. Gupta, R. Tsai, M. Khachatryan, S. Roy-Chowdhuri, S. Bose, A. Yoon, K. Faull, R. Farias-Eisner and G. Chaudhuri, Free Radical Biol. Med., 2015, 86, 228–238 CrossRef CAS PubMed.
- J. W. Gai, W. Qin, M. Liu, H. F. Wang, M. Zhang, M. Li, W. H. Zhou, Q. T. Ma, G. M. Liu, W. H. Song, J. Jin and H. S. Ma, Urol. Oncol., 2016, 34, 166 Search PubMed.
- C. Szabo, Nat. Rev. Drug Discovery, 2016, 15, 185–203 CrossRef CAS PubMed.
- A. Asimakopoulou, P. Panopoulos, C. Chasapis, C. Coletta, Z. Zhou, G. Cirino, A. Giannis, C. Szabo, G. Spyroulias and A. Papapetropoulos, Br. J. Pharmacol., 2013, 169, 922–932 CrossRef CAS PubMed.
- M. Thorson, T. Majtan, J. Kraus and A. Barrios, Angew. Chem., Int. Ed., 2013, 52, 4641–4644 CrossRef CAS PubMed.
- Y. Zhou, J. Yu, X. Lei, J. Wu, Q. Niu, Y. Zhang, H. Liu, P. Christen, H. Gehring and F. Wu, Chem. Commun., 2013, 49, 11782–11784 RSC.
- N. Druzhyna, B. Szczesny, G. Olah, K. Módis, A. Asimakopoulou, A. Pavlidou, P. Szoleczky, D. Gerö, K. Yanagi, G. Törö, I. López-García, V. Myrianthopoulos, E. Mikros, J. Zatarain, C. Chao, A. Papapetropoulos, M. Hellmich and C. Szabo, Pharmacol. Res., 2016, 113, 18–37 CrossRef CAS PubMed.
- W. Niu, P. Wu, F. Chen, J. Wang, X. Shang and C. Xu, MedChemComm, 2017, 8, 198–201 RSC.
- W. Niu, P. K. Yadav, J. Adamec and R. Banerjee, Antioxid. Redox Signaling, 2015, 22, 350–361 CrossRef CAS PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- G. Hummer, J. C. Rasaiah and J. P. Noworyta, Nature, 2001, 414, 188–190 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- O. Korb, T. Stutzle and T. E. Exner, J. Chem. Inf. Model., 2009, 49, 84–96 CrossRef CAS PubMed.
- Z. Tian, J. Liu and Y. Zhang, Sci. Rep., 2016, 6, 22336 CrossRef CAS PubMed.
- J. Liu, X. Yang and Y. Zhang, Appl. Microbiol. Biotechnol., 2014, 98, 8947–8962 CrossRef CAS PubMed.
- Z. Tian, J. Liu, Y. Zhang and J. Agric, Food Chem., 2016, 64, 7994–8001 CrossRef CAS PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan and W. Wang, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Comput. Chem., 2011, 32, 866–877 CrossRef CAS PubMed.
- X. Q. Yang, J. Y. Liu, X. C. Li, M. H. Chen and Y. L. Zhang, J. Chem. Inf. Model., 2014, 54, 1356–1370 CrossRef CAS PubMed.
- B. Y. Feng and B. K. Shoichet, Nat. Protoc., 2006, 1, 550–553 CrossRef CAS PubMed.
- M. Fu, W. Zhang, L. Wu, G. Yang, H. Li and R. Wang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2943–2948 CrossRef CAS PubMed.
- C. Chao, J. R. Zatarain, Y. Ding, C. Coletta, A. A. Mrazek, N. Druzhyna, P. Johnson, H. Chen, J. L. Hellmich, A. Asimakopoulou, K. Yanagi, G. Olah, P. Szoleczky, G. Törö, F. J. Bohanon, M. Cheema, R. Lewis, D. Eckelbarger, A. Ahmad, K. Módis, A. Untereiner, B. Szczesny, A. Papapetropoulos, J. Zhou, M. R. Hellmich and C. Szabo, Mol. Med., 2016, 22, 361–379 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00484b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |