
Novel cell-penetrating-amyloid peptide conjugates preferentially kill cancer cells
John R.
Veloria
 a,
Luxi
Chen
b,
Lin
Li
a,
Gail A. M.
Breen
a,
Jiyong
Lee
b and
Warren J.
Goux
*b
a,
Luxi
Chen
b,
Lin
Li
a,
Gail A. M.
Breen
a,
Jiyong
Lee
b and
Warren J.
Goux
*b
aDepartment of Biological Sciences, The University of Texas at Dallas, 800 W. Campbell Rd, Richardson, TX 75080, USA
bDepartment of Chemistry and Biochemistry, The University of Texas at Dallas, 800 W. Campbell Rd, Richardson, TX 75080, USA. E-mail: wgoux@utdallas.edu
First published on 5th December 2017
Abstract
The goal of this study was to develop a peptide which could use the toxic effects of amyloid, a substance which is the hallmark of over 25 known human diseases, to selectively kill cancer cells. Here we demonstrate that two separate amyloid-forming hexapeptides, one from the microtubule associated protein Tau involved in formation of paired helical filaments of Alzheimer's disease, and the other an amyloid forming sequence from apolipoprotein A1, when conjugated to a cell penetrating peptide (CPP) sequence, form toxic oligomers which are stable for up to 14 h and able to enter cells by a combination of endocytosis and transduction. The amyloid peptide conjugates showed selective cytotoxicity to breast cancer, neuroblastoma and cervical cancer cells in culture compared to normal cells. Fluorescence imaging experiments showed the CPP–amyloid peptide oligomers formed intracellular fibrous amyloid, visible in the endosomes/lysosomes, cytosol and nucleus with thioflavin S (ThS) staining. Further experiments with rhodamine-conjugated Dextran, propidium iodide (PI), and acridine orange (AO) suggested the mechanism of cell death was the permeability of the lysosomal membrane brought about by the formation of amyloid pores. Cytotoxicity could be abrogated by inhibitors of lysosomal hydrolases, consistent with a model where lysosomal hydrolases leak into the cytosol and induce cytotoxicity in subsequent downstream steps. Taken together, our data suggest that CPP–amyloid peptide conjugates show potential as a new class of anti-cancer peptides (ACPs).
Introduction
“Cancer” is a general term that refers to a family of diseases affecting many different tissues and cell types. While some localized types of cancer can be treated with surgical intervention or radiation therapy, more advanced forms involving metastatic disease are more successfully treated with chemotherapy. Many of these conventional small molecule chemotherapeutic agents affect healthy cells leading to deleterious side-effects. In addition, cancer cells frequently become resistant to chemotherapy as a consequence of their ability to adapt by detoxifying agents using intracellular enzymes or their ability to quickly repair DNA damage and defects in the machinery that mediates apoptosis.1Cell permeating peptides (CPPs) are short arginine-rich peptides that are able to transport cargo into cells, examples of which include human immunodeficiency virus (HIV)-1 trans-activator of transcription (Tat) (48-60), Drosophila Antennapedia (Antp)-(43-58) and synthetic oligoarginine peptides.2–4 CPPs show increased affinity to cancer cells, likely a result of a simple receptor-independent attraction between the positively charged CPP and the high density of negatively charged O-glycosylated mucins found in their outer membrane.1,5–7 Because changes in membrane properties are generic to all cancers, specially designed “anti-cancer peptides” (ACPs), which resemble CPPs in having a high content of positively charged residues, have been successfully used in treating slower growing cancers,8 as well as faster growing or metastatic cancers, without developing resistance.1,6 More recent studies demonstrate that resistance may be acquired in mammalian cancer cell lines expressing high levels of membrane sialic acid, although these cells may retain sensitivity to conventional small molecule chemotherapeutics.9
ACPs may be grouped according to their method of entry into the cell and the mechanism by which they affect cell viability. Perhaps the simplest are those having a high content of hydrophobic residues in addition to positively charged residues. These peptides are able to integrate into and disrupt plasma membrane integrity, leading to cell death by means of necrosis,5,7,8,10–13 being similar to antimicrobial peptides (AMPs) in their mechanism of action. On the other end of the spectrum are “smart” ACPs which are made up of a CPP, allowing for entry by transduction or endocytosis, one or more homing entities (peptides or small molecules) which home to cancer antigens in the vasculature or on the cell surface, and a killing peptide or drug designed to function once inside the cell.14–18 In some cases specificity of these peptides may be increased by using phage libraries to design CPPs that target specific cancer lines19–21 or CPPs which become activated by cancer-specific proteases in the lysosomes or embedded in the cancer cell membrane.22–25
Amyloid is a form of insoluble fibrous protein deposits which characterizes over 25 human diseases, the most notable of which include Alzheimer's disease (AD) and Parkinson's disease.26–32 Recent studies suggest that the fibrous forms of amyloid, either disease-related or prepared as de novo peptides, have limited toxicity and that it is the oligomeric form, composed of only a few protein monomers, that confers the greatest toxicity.33,34 In an earlier report we covalently linked an oligoarginine peptide CPP to an amyloid forming hexapeptide, homologous to an amyloid forming sequence in the microtubule binding region of human Tau protein (306VQIVYK311), and showed that this peptide (AcPHF6R9) was able to enter cells and form intracellular amyloid, creating a cell culture model for Alzheimer's disease.35–39 At the time we found AcPHF6R9 was toxic to neurons and neuroblastoma cells but had limited toxicity in embryonic kidney cells. Our hypothesis to explain these findings was that the positively charged AcPHF6R9 peptide showed higher permeability in neurons and neuroblastoma cells compared to embryonic kidney cells, owing to their high content of negatively charged gangliosides and O-glycosylated mucins in their outer membrane. We recently realized that the positively charged CPP might be able to target the negatively charged lipids or O-glycosylated mucins found in the outer membrane of cancer cells, while the conjugated amyloid might act to disrupt the external or internal membrane structure, killing unwanted cell populations. In the present study, we tested the cytotoxicity of AcPHF6R9 on cancer cells and normal cells in culture. To test the generality of our approach, we also tested a second CPP–amyloid peptide conjugate, where the amyloid sequence was homologous to an N-terminal sequence of apolipoprotein A1. While this hexapeptide has not been proven to be related to any disease, it has been shown to form filaments in vitro and displays all of the properties of amyloid.40 We found that both peptides formed long lived oligomers and showed selective toxicity to cancer cells including breast cancer, neuroblastoma, and cervical cancer cells compared to normal cells. Our data suggests that our conjugate peptides form a new class of ACPs where amyloid oligomers confer toxicity by mechanisms including disruption of lysosomal membranes. We propose that these new amyloid-based ACPs may add to the existing arsenal of ACPs as a line of treatment for overcoming multiple-drug resistance in cancer.
Results and discussion
Physical properties of peptides used in this study
We studied two CPP–amyloid peptide conjugates AcPHF6R9, which contains an amyloid forming sequence taken from the microtubule binding region of human Tau protein and AcLAV6R9, with an amyloid forming sequence predicted by computer scanning of apolipoprotein A1.40 Aggregation kinetics for these peptides and for corresponding peptides lacking the CPP R9 sequence (AcPHF6 and AcLAV6) are shown in Fig. 1A and kinetic parameters extrapolated from fits to the data are summarized in Table 1. While the hexapeptides AcPHF6 and AcLAV6 aggregated rapidly with no measurable lag times, amyloid peptide–CPP conjugates AcLAV6R9 and AcPHF6R9 aggregated more slowly and showed lag times of nearly 2 h and 14 h. This contrasts with much shorter lag times we previously determined for AcPHF6R9 in the presence of heparin, a known inducer of amyloid filament formation.35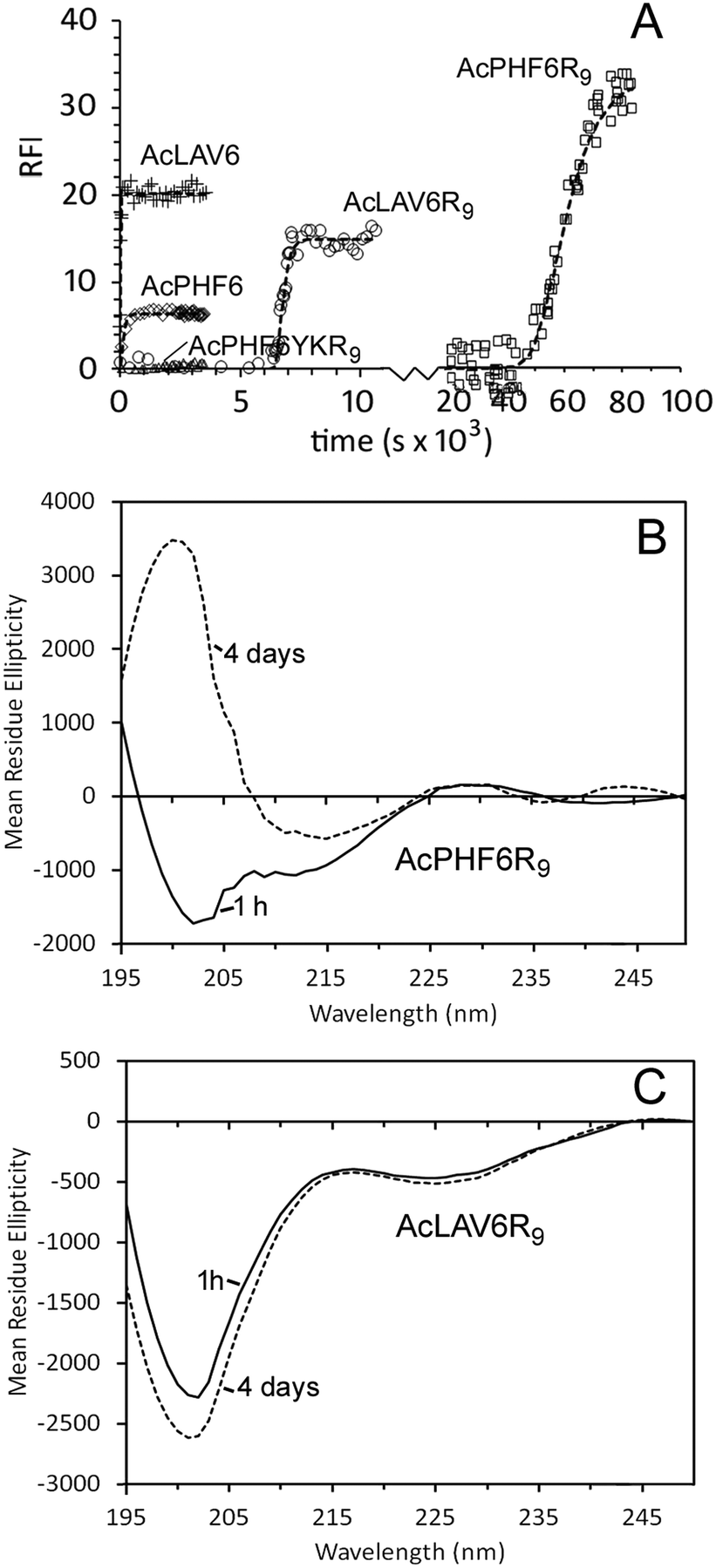 | ||
| Fig. 1 Self-aggregation kinetics of AcPHF6R9, AcLAV6R9, AcPHF6, AcLAV6 and AcPHF6YKR9 in buffer A at 30 °C. Amyloid formation was followed using thioflavin S (ThS) fluorescence (A). Solid lines represent non-linear best fits to Gompertz growth curves.60 All base-line corrected measurements were carried out in triplicate. Data points were collected at 15 s or 1 min intervals. For the purpose of simplifying the display some data points have been omitted. Far UV CD of AcLAV6R9 (B) and AcPHF6R9 (C) in buffer A immediately after dissolution (solid line) and after aging for 4 days (dashed line) (figure B and some data contained in figure A were reprinted (adapted) with permission from ref. 39. Copyright 2017 American Chemical Society). | ||
| Kinetic propertiesa | ||||||
|---|---|---|---|---|---|---|
| No. | Peptide | Abbreviation | Source | A | k 1 (s−1 x 103) | LT (h) |
| a Kinetics data, corrected for the blank, were fitted to Gompertz growth curves,60 yielding the pre-exponential factor (A), the first order rate constant (k1) and lag time (LT). | ||||||
| 1 | Ac-VQIVYK-NH2 | AcPHF6 | Human tau res. 306–311 | 6.3 ± 0.1 | 9.0 ± 1.5 | 0 |
| 2 | Ac-VQIVYK(R)9-NH2 | AcPHF6R9 | de novo from (1) | 39.7 ± 1.8 | 0.12 ± 0.06 | 14.4 ± 0.6 |
| 3 | Ac-VQIVKK(R)9-NH2 | AcPHF6YKR9 | de novo from (2) | ND | ND | ND |
| 4 | Ac-LAVLFL-NH2 | AcLAV6 | Apolipoprotein A1 Res. 8–13 | 19.6 ± 0.1 | 62.5 ± 4.7 | 0 |
| 5 | Ac-LAVLFL(R)9-NH2 | AcLAV6R9 | de novo from (4) | 14.9 ± 1.1 | 4.8 ± 1.1 | 1.8 ± 0.1 |
Fig. 1B and C show CD spectra of AcPHF6R9 and AcLAV6R9 peptides in buffer 1 h and 4 days after dissolution. AcLAV6R9 peptide displayed a low intensity negative band at 225 nm and a more intense negative band at 201 nm, independent of aging. A similar spectrum was also observed for AcPHF6R9 prior to aging, in agreement with our previous findings.39 This type of spectrum is consistent with some βII proteins having a high fraction of random coil and truncated or twisted β-sheet structure and has also been observed for some β-amyloid oligomers.41–45 Upon aging, the spectrum of AcPHF6R9 developed a positive band at 202 nm and a well-defined negative band at 215 nm, more typical of βI proteins and amyloidogenic peptides which form extensive fibrous networks.36,42,44
Fig. 2A and C show the TEM of AcPHF6R9 and AcLAV6R9 in buffer before aging while Fig. 2B and D show the same peptides following aging for 4 days. 1 h following dissolution both peptides form oligomers. The average oligomer diameter was found to be 55.0 ± 6.5 nm for AcPHF6R9 and 15.5 ± 3.8 nm for AcLAV6R9. Upon aging, AcPHF6R9 formed abundant straight filaments 16.9 ± 2.1 nm in width while AcLAV6R9 formed large aggregates 200–500 nm in diameter.
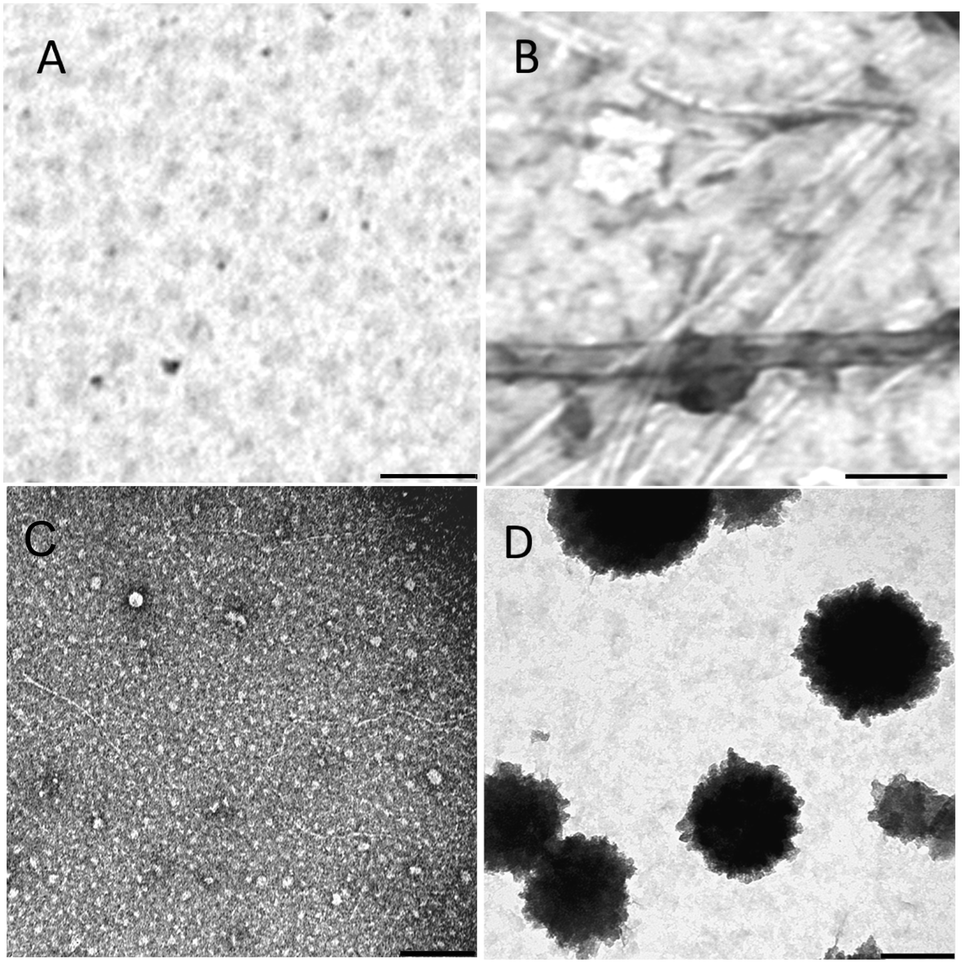 | ||
| Fig. 2 TEM of AcPHF6R9 and AcLAV6R9 immediately after dissolution (A and C) and after 4 days of aging (B and D). Scale bar = 200 nm. | ||
In summary, our physical measurements predict that both AcPHF6R9 and AcLAV6R9 form oligomers following dissolution and that these oligomers persist for several hours. As illustrated by our kinetic data and the TEMs of AcPHF6R9 and AcLAV6R9 before and after aging, oligomers formed during the lag period are ThS negative while fibers or large aggregates are ThS positive. These results are in qualitative agreement with those for β-amyloid oligomers.46,47
Cell permeability to CPP–amyloid peptide conjugates and subsequent intracellular amyloid formation
In order to investigate the uptake of our CPP–amyloid conjugates in cells we labelled the N-termini of the peptides with rhodamine. Fig. 3 shows neuoroblastoma (HT-22), cervical cancer (HeLa), and human embryonic kidney (HEK293) cells following a 3 h treatment with RhPHF6R9, and compares them to treatment with RhPHF6, a peptide without a CPP entity. Punctate staining in and around the nucleus and diffuse staining of the cytosol was observed in all cell types treated with RhPHF6R9. The polyarginine tag, acting as a nuclear localization signal, has been shown to rapidly transport other CPPs into the nucleus and may account for the additional labelling of nuclear organelles.3,48 In contrast, much of RhPHF6 appeared as punctate staining closely associated with the cell membrane. Both RhPHF6 and RhPHF6R9 appear to accumulate in HT-22 cells in the first 3 h followed by disappearance of fluorescence at later times (results not shown), suggesting these peptides are degraded by the proteolytic enzymes. Integrated intensities of the cells over time showed that 3 times more RhPHF6R9 entered cells in the first 3 h relative to RhPHF6, demonstrating that the CPP entity was quite effective in increasing the membrane permeability of the labelled CPP–amyloid peptide.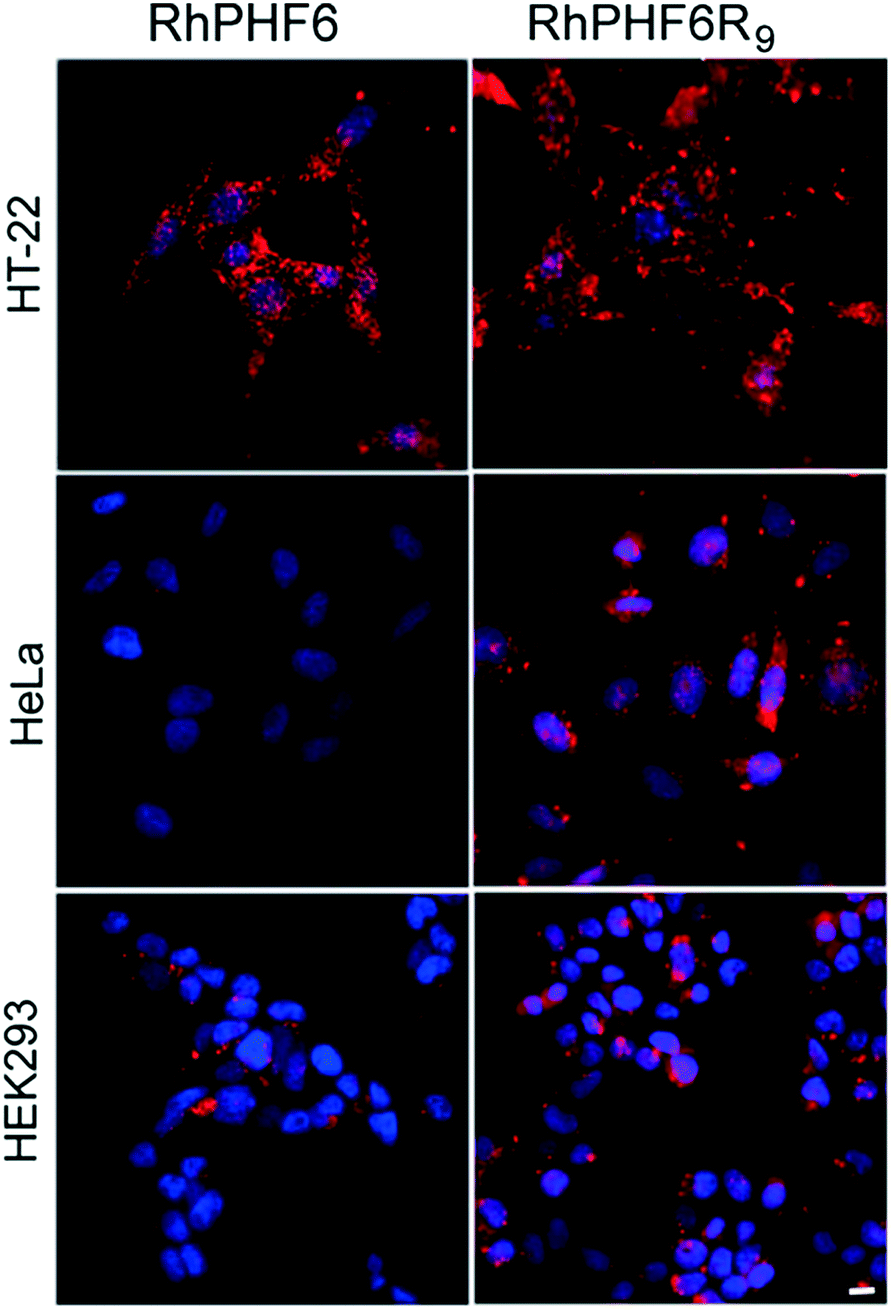 | ||
| Fig. 3 Fluorescence microscopy images of HT-22, HeLa and HEK293 cells following a 3 h incubation with peptides RhPHF6 and RhPHF6R9. Red in the images arises from the Rh-peptides in the cytoplasm and nucleus and the blue arises from DAPI staining of the nuclei. Scale bar = 10 μm. | ||
Fig. 4A shows fluorescent images of HT-22 cells treated with peptides and stained with ThS to highlight amyloid formation within the cells. There is no ThS staining in cells treated only with vehicle (control) or in cells treated with AcPHF6YKR9, a peptide we have previously shown does not form amyloid.35,39 In contrast, cells treated with AcPHF6R9 showed both punctate and diffuse ThS staining in the cytosol, qualitatively mirroring the punctate and diffuse fluorescence seen for RhPHF6R9 (Fig. 3), while cells treated with AcPHF6 showed much less intense diffuse staining. The presence of ThS staining suggests that at least some of AcPHF6R9 oligomers are converted to fibrous amyloid46,47 and since some of the staining appears to localize in the endosomes, it is likely that the conversion occurred during or before endocytosis. Since the oligomers present are not visible it is not possible to quantitate how much of the AcPHF6R9 remains oligomeric. In future studies we plan to quantitate the fraction of AcPHF6R9 present as oligomers using an oligomer specific antibody. We treated cells with AcLAV6R9 and AcPHF6R9 and stained with DAPI, ThS, and propidium iodide (PI), a dye which only is able to enter cells with a compromised plasma membrane (Fig. 4B). Some of the cells treated with AcLAV6R9 accumulate fibrous amyloid prior to cell death, as illustrated by the ThS stained cells not stained by PI (highlighted with arrow heads). In contrast, nearly all of the cells treated with AcPHF6R9 appear PI permeable, suggesting that cell death may be occurring more rapidly once this peptide enters cells. There are two alternative interpretations of this data: (1) there is rapid disruption of the plasma membrane by a mechanism similar to that used by antimicrobial peptides,5,7,8,10–13,26–30,33 facilitating the entry of amyloid into the cell or (2) intracellular amyloid formation has induced cell death by an internal apoptotic mechanism,18,49 which in turn leads to increased permeability of the plasma membrane. The presence of living cells having amyloid inclusions suggests that CPP–amyloid peptide conjugates are transported into cells prior to disruption of the plasma membrane, or that amyloid forms within the cell, subsequently causing death. This is also consistent with the rhodamine peptide conjugates seen in the endosome fraction in living cells (Fig. 3).
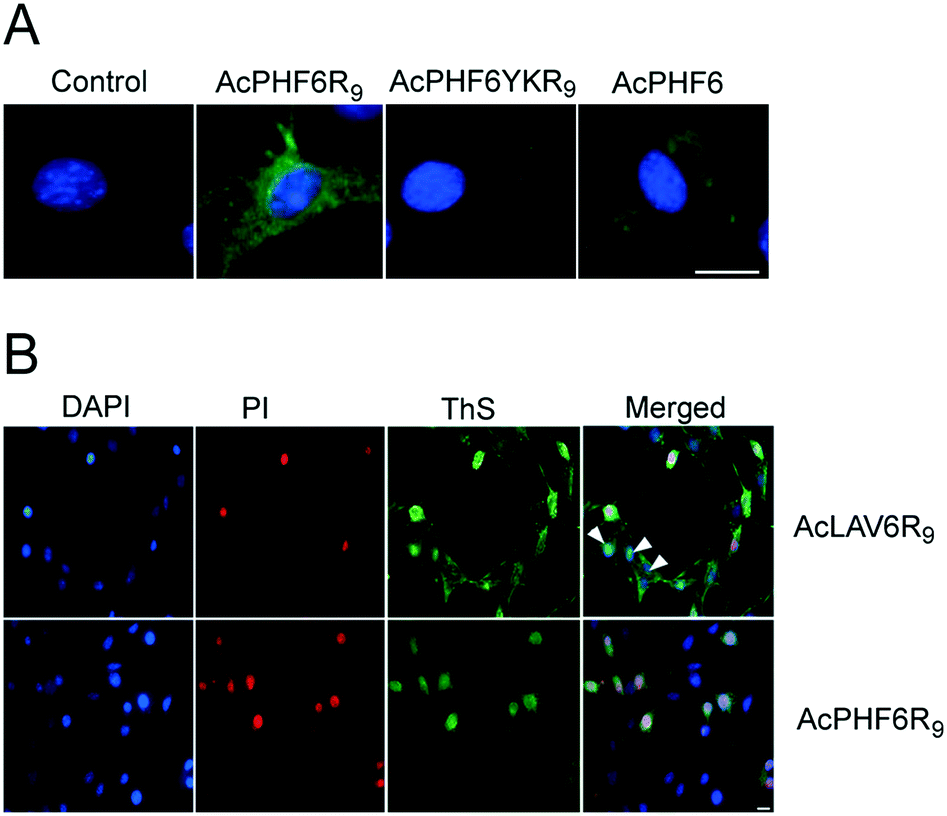 | ||
| Fig. 4 Fluorescent images of HT-22 cells treated with AcPHF6R9 or AcLAV6R9 and stained with DAPI and ThS to highlight amyloid formation within the cells (A). Propidium iodide (PI), DAPI and ThS staining of cells treated with AcPHF6R9 or AcLAV6R9 (B). Some cells treated with AcLAV6R9 accumulate amyloid prior to cell death as illustrated by cells not stained by PI (highlighted with arrow heads). Scale bar = 10 μm for all images. | ||
Cytotoxicity of CPP–amyloid peptide conjugates
Fig. 5A shows viability of HeLa cells treated for 24 h with the peptides used in this study. The results show the most toxic peptide is AcLAV6R9 with an ED50 of 3 μM while AcLAV6 peptide, the corresponding amyloid peptide without the CPP entity, has an ED50 nearly 5 times as great. AcPHF6R9 is much less toxic to these cells compared to AcLAV6R9. However, like AcLAV6R9, AcPHF6R9 is much more toxic than the AcPHF6, as a result of the CPP entity. Most noteworthy is the lack of cytotoxicity of AcPHF6R9 to the human embryonic kidney cell line (HEK293), which suggests that the positively charged peptides selectively target cancer cells, much like other ACPs.5–8,10–13 Our control peptide, AcPHF6YKR9, does not form amyloid, and showed little cytotoxicity, suggesting that toxicity is sequence dependent and relies upon the amyloid forming potential of the peptide.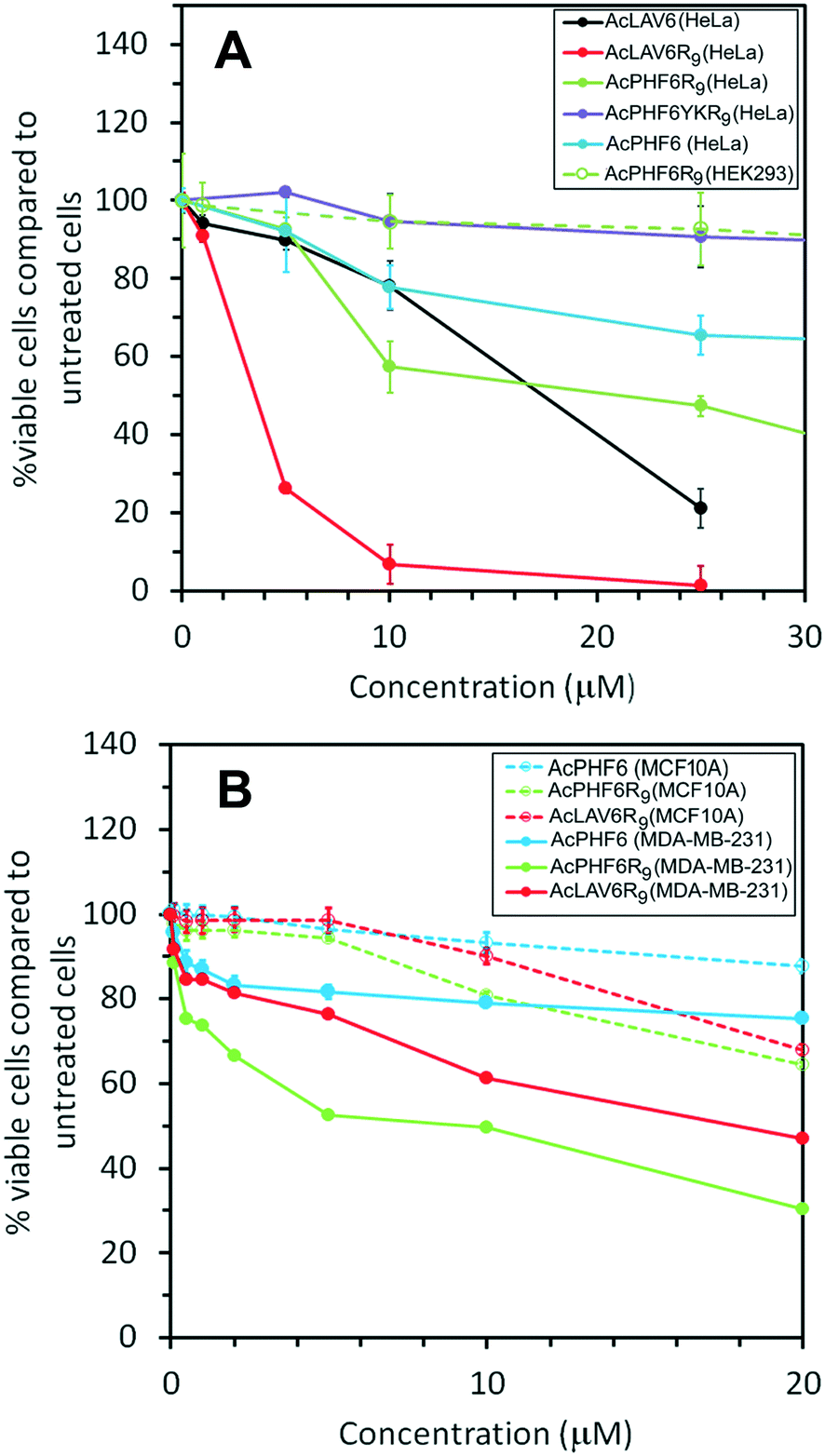 | ||
| Fig. 5 Survival of HeLa cells following treatment with AcLAV6, AcLAV6R9, AcPHF6, AcPHF6R9 and AcPHF6YKR9. Also shown is the dose dependent survival of HEK293 cells following treatment with AcPHF6R9 (A). Dose dependent survival curves for breast cancer MDA-MB-231 cells following treatment with AcPHF6, AcPHF6R9 and AcLAV6R9. Also shown are survival curves for normal breast cells MCF10A (B). Viability was determined with MTT assay after 24 h. | ||
To further investigate the selective cytotoxicity of our CPP–amyloid conjugate peptides for cancer cells, we tested toxicity of AcPHF6, AcPHF6R9 and AcLAV6R9 peptides on a breast cancer cell line, MDA-MB-231, which represents a highly metastatic mesenchymal stem-like (MSL) subtype of triple-negative breast cancer (TNBC). Of the peptides tested AcPHF6R9 was the most toxic (ED50 of 5 μM), followed by AcLAV6R9 (ED50 of 20 μM). AcPHF6, without the CPP moiety, was the least toxic. All three peptides showed little cytotoxicity towards a normal breast epithelial cell line, MCF10A.
It has been suggested that there is an inverse correlation between oligomer size and toxicity.33,34 Taking into account our physical characterization of the CPP–amyloid peptide conjugates, one would predict that AcLAV6R9 peptide, having smaller sized oligomers, would be the more toxic if a high fraction of oligomers are in fact present in the cell. This appears to be the case when HeLa cells are treated with this peptide but for breast cancer MDA-MB-231 AcPHF6R9 is the more toxic of the two CPP–amyloid peptide conjugates we tested (Fig. 5). Both CPP–amyloid peptide conjugates were more toxic than the corresponding short amyloid peptides without the CPP entity, in qualitative agreement with the amount of amyloid entering the cell (Fig. 3 and 4A). Most cells were killed within 6 h, possibly as a result of proteolytic degradation of these peptides over longer treatment times. The attractive interaction between the positively charged CPP–amyloid peptide conjugates and the negatively charged membranes of cancer cells most likely accounts for their selectivity relative to non-cancerous cells. The cytotoxicity and selectivity we observed for our peptides are comparable to that of a similar peptide containing only a CPP and mitochondrial membrane disrupting peptide.18 This peptide was toxic to a wide range of cancers including HeLa, lung, neuroblastoma, glioblastoma and colon cancers.
Localization of CPP–amyloid peptides to endosomes/lysosomes where they mediate lysosomal membrane permeability (LMP)
The punctate staining of HT-22 cells seen when treated with AcPHF6R9 (Fig. 4A) suggests that AcPHF6R9 enters cells through the endosome/lysosome system. Furthermore, the observation that cells treated with AcLAV6R9 accumulate amyloid prior to disruption of the plasma membrane (Fig. 4B (top row)) is evidence that amyloid formation is an early event. To investigate the uptake mechanism of our CPP–amyloid peptides, we co-labelled cells with rhodamine-conjugated dextran (Rh-Dextran), a glycan that is preferentially taken up through liquid-phase endocytosis, and fluorescein-conjugated PHF6R9 (Fl-PHF6R9). We found some of the Fl-PHF6R9 in perinuclear vesicles colocalized with the Rh-Dextran-labelled endosomes/lysosomes (Fig. 6A). Fl-PHF6R9 visible in the cytosol likely arises from peptide entering by means of transduction or by being released from the endosomes/lysosomes.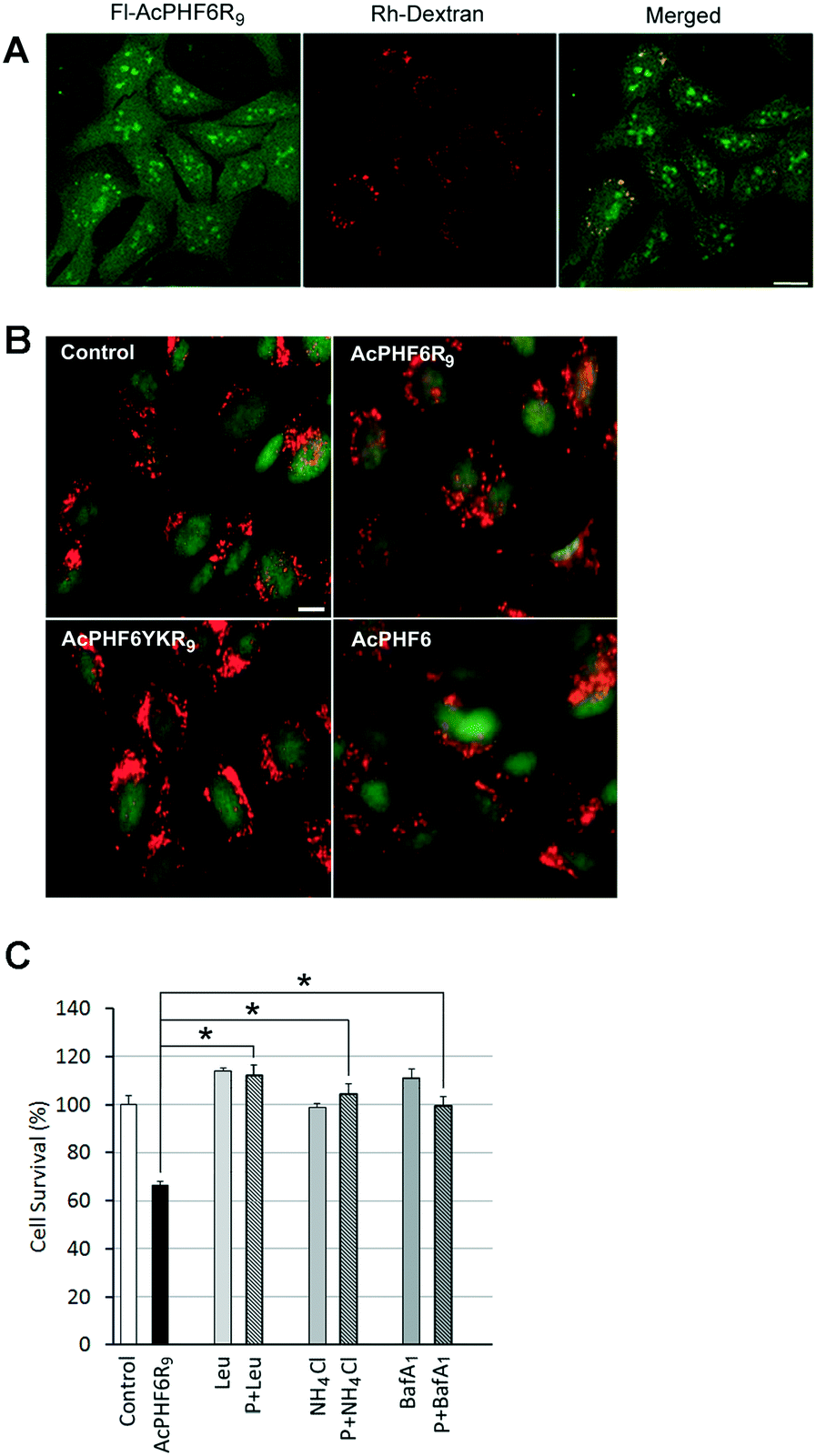 | ||
| Fig. 6 HT-22 cells were labelled with Rh-Dextran together with Fl-PHF6R9 (A). Images were captured using a confocal microscope. The yellow color in the merged image indicates colocalization of Fl-PHF6R9 with Rh-Dextran in late endosomes/lysosomes. HT-22 cells were treated with either vehicle (control), AcPHF6R9, AcPHF6YKR9 or AcPHF6, together with acridine orange (B). A shift from punctate red fluorescent staining to a more diffuse cytoplasmic red fluorescent staining was observed in AcPHF6R9-treated cells, indicating permeabilization of the endosomal/lysosomal membrane. Note the red cytoplasm of a dead/dying cell treated with AcPHFR9 (arrow). Scale bars = 10 μm. Effect of lysosomal inhibitors leupeptin (Leu), NH4Cl and Bafilomycin A1 (BafA1) on cell survival (C). HT-22 cells were pre-treated with either leupeptin (Leu) for 24 h, NH4Cl for 24 h, or BafA1 for 40 min, before treatment with either vehicle (control), AcPHF6R9 (P), or AcPHF6R9 together with the inhibitors for 24 h. *p < 0.05. Reprinted (adapted) with permission from ref. 39. Copyright 2017 American Chemical Society. | ||
To investigate whether LMP is induced by AcPHF6R9 treatment, cells were labelled using the ionizable lysosomotropic dye, acridine orange.39,49 In vehicle-treated control cells stained with acridine orange punctate red fluorescent vesicles indicative of acidic organelles, such as endosomes/lysosomes, were observed in the cytoplasm (Fig. 6B). Compared to cells treated with vehicle only, AcPHF6 or non-amyloid forming peptide AcPHF6YKR9, AcPHF6R9 treated cells showed a loss of the punctate red fluorescent staining, coupled with an increase in diffuse red and green staining in the cytoplasm, indicating increased permeability of the endosomal/lysosomal membrane. Taken together, these results suggest that AcPHF6R9 entered the endocytic pathway and induced endosomal/lysosomal leakage. Such lysosomal permeability would allow lysosomal proteases as well as amyloid to be released into the cytoplasm, perhaps accounting for the observed diffuse ThS staining or fluorescence of fluorescently tagged AcPHF6R9 (Fig. 3 and 4A). The development of LMP is consistent with “the amyloid channel” hypothesis by which pores in membranes results from the ability of oligomers to integrate into the membrane forming channels.34,50,51 This phenomenon is consistent with separate but related studies showing that amyloid formed by [Aβ]1–42 or α-synuclein induced leakage from the lysosomes, leading to apoptotic cell death.49–52 To further investigate the hypothesis of LMP induced cell death we treated cells with lysosomal protease inhibitors, leupeptin and Bafilomycin A1, as well as ammonium chloride, a base which increases the pH in the lysosome lumen, resulting in a deactivation of proteases. Consistent with our previous results,39 we found that all three of these agents reduced cytotoxicity of AcPHF6R9 (Fig. 6C), suggesting that either the formation of lysosomal amyloid or the mechanism which allowed for disruption of the lysosomal membrane could be prevented by these agents. In contrast, lysosomal inhibitors were not able to rescue cells from LAV6R9-mediated toxicity (data not shown), further confirming that cytotoxicity is not mediated in a similar manner to AcPHF6R9. However, our observation that cells treated with AcPHF6R9 can be rescued using lysosomal inhibitors, is consistent with the idea that peptide oligomers are transported into living cells prior to their death, consistent with amyloid staining patterns show in Fig. 4B.
Conclusions
In the past, “smart” ACPs have been designed to release a drug, growth inhibitor or peptide which disrupts internal membranes, initiating apoptosis.14–19,53,54 Some of the more sophisticated systems of this type also contain a peptide entity which is able to home to markers associated with cancer cells or tumor vasculature14,15,17,53 or contain a CPP with becomes activated by cancer associated cell surface proteases.23–25,54 The goal of this study was to develop an ACP which could enter cancer cells and use the toxic effects of amyloid, a substance which is the hallmark of over 25 known human diseases, to perturb homeostasis and selectively kill unwanted cell populations. We chose to initially investigate (1) whether CPP–amyloid peptide conjugates could generate amyloid oligomers, the most toxic form of amyloid, (2) whether CPP–amyloid peptide conjugates could enter cells, (3) whether CPP–amyloid peptide conjugates could selectively kill cancer cells in culture, and (4) the mechanism by which cell death occurs. We showed that two separate amyloid-forming hexapeptides conjugated to cell penetrating R9 peptide formed oligomers which were stable for hours and were able to enter cells, likely by a combination of endocytosis and transduction. Furthermore, these amyloid peptide conjugates showed greater toxicity to cancer cells relative to non-cancer cells, likely a result of the attraction of the positively charged polyarginine entity of the conjugates to negatively charged lipids or O-glycosylated mucins found in their outer membrane of cancer cells.1,5–7 Our experiments with PI and acridine orange suggest the mechanism of cell death to be permeability of biological membranes brought about by the formation of amyloid pores and is in agreement with our previously obtained results.39 A simplified model of oligomeric peptides entering the cell and inducing cytotoxicity is shown in Fig. 7.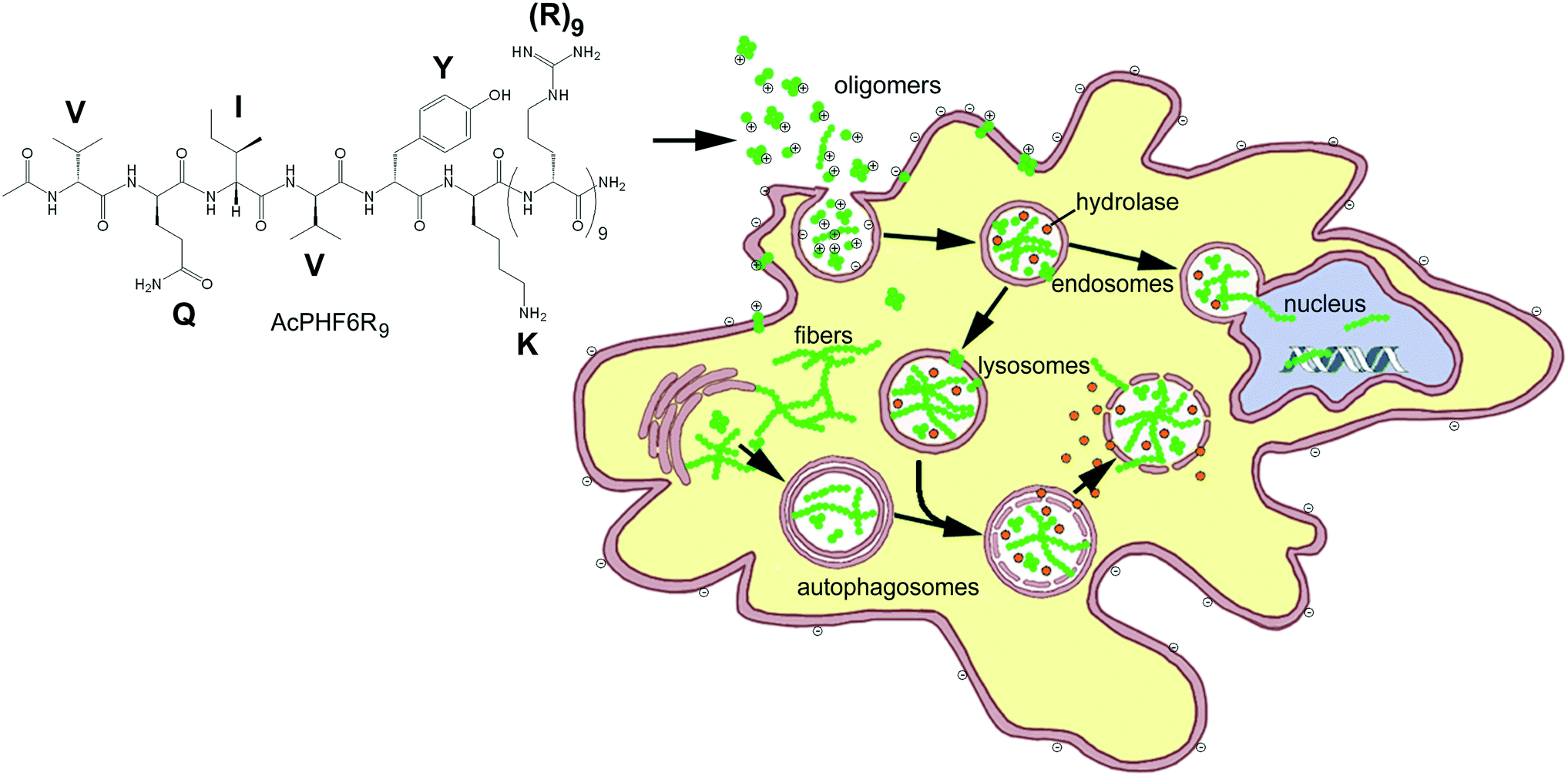 | ||
| Fig. 7 Simplified model showing AcPHF6R9 forming positively charged toxic amyloid oligomers and fibers. Positively charged amyloid is attracted to the negatively charged phosphoslipids and glycomucins expressed in cancer cells and is taken into the cells by endocytosis or transduction where it localizes intracellularly in the endosomes and lysosomes or is transported into the nucleus. The lysosome membrane is made permeable by the formation of amyloid pores, allowing lysosomal hydrolases and amyloid conjugate peptide to escape into the cytosol. Amyloid oligomers and fibers in the cytosol may be recycled back into lysosomes by autophagosomes. | ||
In summary, our data suggests that our conjugate amyloid peptides form a new class of ACPs where amyloid oligomers confer cytotoxicity by various mechanisms, including membrane disruption. We believe that the selectivity of these ACPs may be enhanced by including entities which enhance residence time in the tumor and home to tumor vasculature. This work compliments our previous studies where we showed that AcPHF6R9 could induce amyloid formation in cells of neuronal origin.39 We propose that these new amyloid-based ACPs may add to the existing arsenal of ACPs as a line of treatment for overcoming multiple-drug resistance in cancer. Additionally, identifying new ACPs which internalize into cells and induce cell death from within, as observed with AcPHF6R9, may be a novel approach to combat the acquired resistance to plasma membrane targeting.
Experimental
Materials
All reagents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. Cell culture reagents, including Dulbecco's modified Eagle's medium (DMEM), and L-glutamine were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was from Hyclone (Denver, CO), Bafilomycin A1 from Tocris Bioscience (Minneapolis, MN), and Fluoromount-G mounting fluid from Southern Biotech (Birmingham, AL).Peptides
N-Acetyl peptide amides were synthesized manually with solid phase peptide synthesis (SPPS) on a 0.1 mmol scale using standard 9-fluorenylmethoxycarbonyl (Fmoc) chemistry.55 The extremely hydrophobic peptide Ac-LAVLFL-NH2 (AcLAV6) was purified by water precipitation,56 while the remaining peptides including Ac-VQIVYK-NH2 (AcPHF6), Ac-VQIVYK(R)9-NH2 (AcPHF6R9), Ac-VQIVKK(R)9-NH2 (AcPHF6YKR9) and Ac-LAVLFL(R)9-NH2 (AcLAV6R9) were purified by HPLC, using a preparative Phenomenex Luna (Torrance, CA) semipreparative C18 column (250 × 10 mm), as previously described.35 For fluorescently labelled peptides, the succinic acid–piperazine conjugate of rhodamine B was prepared according to published methods57 and was conjugated to the peptide through two N-terminal 6-aminohexanoic acid “linker” residues using standard Fmoc solid-phase peptide synthesis methods. A 5′(6)-carboxyfluorescein derivative of AcPHF6R9 was similarly prepared.35 Purity of all peptides was checked by MALDI-TOF MS. Stock concentrations of fluorescein- and rhodamine-conjugated peptides were confirmed using the molar extinction coefficients of carboxyfluorescein (ε495nm = 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and rhodamine (ε567nm = 96000 M−1 cm−1).
000 M−1 cm−1) and rhodamine (ε567nm = 96000 M−1 cm−1).
Transmission electron microscopy (TEM)
Samples were dissolved in 5 mM MOPS buffer (with 0.15 M NaCl, 0.1% NaN3), pH 7.2 (buffer A), to a concentration of 0.1 mg mL−1. After preparation, samples were left to incubate at room temperature for 1 h to several days before preparing the TEM grids. Samples were loaded onto carbon-coated Formvar copper grids (200 mesh) and stained with 2% wt/wt uranyl acetate. A JEOL 1200 EX scope interfaced to a digital camera was used to visualize samples.Far UV-CD spectroscopy
Purified peptides were accurately weighed and dissolved in 1 mL (∼1 mg mL−1) of buffer A. Concentrations determined by weight, assuming the TFA salt of the purified peptide, agreed within 5% of concentrations determined using the reported molar extinction coefficients for tyrosine (ε276 of 1390 M−1 cm−1 (ref. 58)) or for the peptide bond (ε192 of 7110 M−1 cm−1 (ref. 59)). Stock solutions were diluted 1:4 with buffer and left to age at room temperature from 1 h to 4 days. Spectra were acquired with a 1 mm or a 0.2 mm path cell using an AVIV 202 CD spectrophotometer. Spectra were acquired at 21 °C, between 195 and 250 nm with a spectral resolution of 1 nm and an averaging time of 2 s per data point. The spectra reported represent an average of 8 acquisitions and have been smoothed using boxcar averaging (5 data point average).
Kinetic analysis
Polymerization kinetics were measured by monitoring the increase in Thioflavin S (ThS) fluorescence at 490 nm (440 nm excitation), as described previously.37–39 For peptides other than AcLAV6, a 1 mg mL−1 sample stock solution was filtered through a Millipore Ultrafree-MC 100000 NMWL filter unit (Billerica, MA) by centrifuging for 10 minutes at 14500 rpm, using an Eppendorf MiniSpin plus benchtop centrifuge (Westbury, NY). AcLAV6 was dissolved in DMSO at a concentration of 10 mg mL−1. Immediately following preparation of monomer stock, aliquots of the peptides were pipetted into wells of a 96-well clear-bottom ELISA plate containing Buffer A, ThS (final concentration of 100 μM) and a 1/8′′ PTFE Grade 2 polished mixing bead (Orange Products, Allentown, PA) to yield an assay solution 100 μM in peptide and ThS. Data was collected in triplicate, using a Molecular Devices SPECTRA Max Gemini XPS spectrofluorimeter (Sunnyvale, CA). Measurements were taken at 15 s (AcLAV6) or 1 min intervals, mixing in between readings. Kinetics data, corrected for the blank, were fitted to a Gompertz growth curve60 (eqn (1))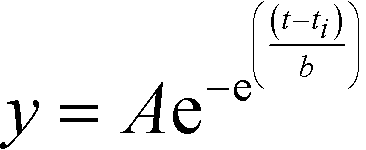 | (1) |
Neuroblastoma, human embryonic kidney and human HeLa cell culture and imaging
Mouse HT-22 hippocampal neuroblastoma, human embryonic kidney (HEK293) and human HeLa cervical adenocarcinoma cells were grown at 37 °C in a humidified incubator with 5% CO2. For peptide treatments, cells were treated at approximately 50–70% confluence in serum-free media for cell viability assays and 40–50% confluence for imaging.Cells grown on glass coverslips were mounted on a glass slide using Fluoromount-G. Images were captured using NIS Elements software with a Photometrics Cool-Snap ES2 camera attachment on a Nikon Eclipse TE300 Inverted Microscope with a Nikon TE-EM epi-fluorescence attachment using Nikon Plan Fluor 40× air (0.75 NA) and Nikon Plan Apo 60× oil immersion (1.4 NA) objectives, unless otherwise noted. Confocal images were captured using the Leica Application Suite with a Leica TCS-SP2 confocal microscope using a Leica Plan Apo 63× oil immersion objective with immersion oil (1.4 NA). Images were an average of at least 25 successive z-scans from a single plane. Images were captured at room temperature and quantified and merged using ImageJ software (http://rsbweb.nih.gov/ij/). Fluorescence intensity per cell was calculated in ImageJ using integrated density after adjusting for background fluorescence.
To carry out epifluorescence imaging, HT-22 cells were incubated with 3.5 μM Rh-PHF6R9 or Rh-PHF6 for 30 min, 3 h, 6 h, and 24 h at 37 °C. Cells were washed twice with pre-warmed DMEM, once with PBS, and then fixed with 4% paraformaldehyde (PFA) in PBS for 20 min. Nuclei were stained with DAPI, cells were washed once with water, and then mounted.
For experiments involving endocytotic uptake, HT-22 cells were washed with DMEM, labelled with Rhodamine-conjugated Dextran (Rh-Dextran; 1 mg ml−1 in DMEM) for 30 min at 37 °C, washed three times with pre-warmed DMEM, and then labelled with 7.5 μM Fl-PHF6R9 for 2.5 h. Cells were washed once with DMEM, three times with PBS, and fixed with 4% paraformaldehyde (PFA). After fixation, cells were washed once with water and mounted. Images were captured using a confocal microscope.
Human mammary epithelial and human breast cancer cell culture
Human mammary epithelial cell line MCF10A was cultured in LONZA Mammary Epithelial Cell Growth Medium (MEGM) plus the Bulletkit (CC-3150). Human breast invasive ductal carcinoma cells MDA-MB-231 were cultured in HyClone Dulbecco's modified Eagle medium (DMEM) with high glucose content. All cell culture media were supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine and 0.5% penicillin/streptomycin antibiotic. The cultured cells were maintained at 37 °C in a humidified incubator with 5% CO2. MCF10A and MDA-MB-231 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). For peptide/drug treatments, cells were treated at approximately 50–60% confluence for cell viability assays.Thioflavin-S staining of intracellular amyloid aggregates
To detect intracellular amyloid, cells were treated with either vehicle alone (control) or 15 μM AcPHF6R9, AcPHF6YKR9, or AcPHF6 for 1 h. Cells were fixed using 4% PFA for 20 min, washed once with water and then stained using 0.005% (w/v) ThS in water for 10 min. After ThS staining, the cells were washed three times with absolute ethanol for 5 min each, washed once with water, counterstained with DAPI, and then mounted.For AcLAV6R9 peptide, cells were treated with either vehicle alone (control) or 7.5 μM AcLAV6R9 peptide for 3 h. Cells were incubated with 20 μg mL−1 propidium iodide for 10 min, washed with three times with PBS, and then fixed using 4% PFA for 20 min. Cell were then washed once with water and then stained using 0.005% (w/v) ThS in water for 10 min. After ThS staining, the cells were washed three times with absolute ethanol for 5 min each, washed once with water, counterstained with DAPI, and then mounted.
Lysosomal membrane permeabilization assays
Lysosomal membrane permeabilization was investigated using the basic cell-permeable dye, acridine orange, as described previously.39,49 HT22 cells were treated with either vehicle alone (control), 15 μM AcPHF6R9, 15 μM AcPHF6YKR9 or 15 μM AcPHF6, together with 150 μM acridine orange for 1 h. Cells were then washed extensively with PBS and mounted and imaged using epifluorescence microscopy.Cell viability assays
For neuroblastoma and HeLa cells, viability was measured using MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Thermo, Fisher Scientific, MA), assays as previously described.35 For human mammary epithelial cells, 1.5 × 104 cells were seeded on 48-well cell culture plate, and were incubated for at least 24 h prior to drug treatment. The MTT reagent, pre-solubilized in sterile HyClone Dulbecco's phosphate-buffered saline (DPBS) (Fisher Scientific, MA) to make stock of 5 mg mL−1, was added to the culture medium 3 h prior to the end of drug/compound incubation. The reaction took place in dark in the 37 °C incubator with 5% CO2 for 3 h. Cell culture medium was then removed from the wells. The plates were left to air-dry for 15 min. Cells with the insoluble purple formazan were dissolved with 99.7% pure DMSO for 10 min at room temperature, 0.5 mL per well. The solution was aliquot to 96-well plate for absorbance reading. Absorbance was measured using BioTek Synergy H1 microplate reader (Winooski, VT) at 595 nm. Results are given as mean ± standard deviation. Cell viability was expressed as a percentage of the untreated cells. Experiments were performed in triplicate for at least three independent experimental trials.Treatment with inhibitors
HT-22 cells were pre-treated with various inhibitors and inhibitors were present during the subsequent treatment with AcPHF6R9 at the same concentration used in the pre-treatments, unless otherwise indicated. Leupeptin was used at a concentration of 10 μg mL−1 and NH4Cl at a concentration of 10 mM, and cells were pre-treated for 24 h. Cells were pre-treated with 200 nM Bafilomycin A1 for 40 min and Bafilomycin A1 was present at a concentration of 2 nM during the incubation with AcPHF6R9.Statistical analysis of data
For statistical significance, data was evaluated with a 0.05 level of significance using a two-tailed Student's t-test, with data values run as unpaired.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from The University of Texas at Dallas Office of Research.Notes and references
- D. W. Hoskin and A. Ramamoorthy, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 357–375 CrossRef CAS PubMed.
- S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS PubMed.
- T. B. Potocky, A. K. Menon and S. H. Gellman, J. Biol. Chem., 2003, 278, 50188–50194 CrossRef CAS PubMed.
- A. Ziegler, P. Nervi, M. Dürrenberger and J. Seelig, Biochemistry, 2005, 44, 138–148 CrossRef CAS PubMed.
- K. J. Lim, B. H. Sung, J. R. Shin, Y. W. Lee, D. J. Kim, K. S. Yang and S. C. Kim, PLoS One, 2013, 8, e66084 CAS.
- S. Riedl, B. Rinner, M. Asslaber, H. Schaider, S. Walzer, A. Novak, K. Lohner and D. Zweytick, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2638–2645 CrossRef CAS PubMed.
- N. Papo and Y. Shai, Biochemistry, 2003, 42, 9346–9354 CrossRef CAS PubMed.
- N. Papo, A. Braunstein, Z. Eshhar and Y. Shai, Cancer Res., 2004, 64, 5779–5786 CrossRef CAS PubMed.
- K. Ishikawa, S. H. Medina, J. P. Schneider and A. J. S. Klar, Cell Chem. Biol., 2017, 24, 149–158 CrossRef CAS PubMed.
- H. Schröder-Borm, R. Bakalova and J. Andrä, FEBS Lett., 2005, 579, 6128–6134 CrossRef PubMed.
- N. Papo, D. Seger, A. Makovitzki, V. Kalchenko, Z. Eshhar, H. Degani and Y. Shai, Cancer Res., 2006, 66, 5371–5378 CrossRef CAS PubMed.
- C. Sinthuvanich, A. S. Veiga, K. Gupta, D. Gaspar, R. Blumenthal and J. P. Schneider, J. Am. Chem. Soc., 2012, 134, 6210–6217 CrossRef CAS PubMed.
- D. Gaspar, A. S. Veiga, C. Sinthuvanich, J. P. Schneider and M. A. R. B. Castanho, Biochemistry, 2012, 51, 6263–6265 CrossRef CAS PubMed.
- M. Mäe, H. Myrberg, S. El-Andaloussi and Ü. Langel, Int. J. Pept. Res. Ther., 2009, 15, 11–15 CrossRef.
- H. Myrberg, L. Zhang, M. Mäe and Ü. Langel, Bioconjugate Chem., 2008, 19, 70–75 CrossRef CAS PubMed.
- M. Lindgren, K. Rosenthal-Aizman, K. Saar, E. Eiríksdóttir, Y. Jiang, M. Sassian, P. Östlund, M. Hällbrink and Ü. Langel, Biochem. Pharmacol., 2006, 71, 416–425 CrossRef CAS PubMed.
- L. Agemy, D. Friedmann-Morvinski, V. R. Kotamraju, L. Roth, K. N. Sugahara, O. M. Girard, R. F. Mattrey, I. M. Verma and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17450–17455 CrossRef CAS PubMed.
- I. D. Alves, M. Carré, M. P. Montero, S. Castano, S. Lecomte, R. Marquant, P. Lecorché, F. Burlina, C. Schatz, S. Sagan, G. Chassaing, D. Braguer and S. Lavielle, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2087–2098 CrossRef CAS PubMed.
- E. Kondo, K. Saito, Y. Tashiro, K. Kamide, S. Uno, T. Furuya, M. Mashita, K. Nakajima, T. Tsumuraya, N. Kobayashi, M. Nishibori, M. Tanimoto and M. Matsushita, Nat. Commun., 2012, 3, 951 CrossRef PubMed.
- M. Higa, C. Katagiri, C. Shimizu-Okabe, T. Tsumuraya, M. Sunagawa, M. Nakamura, S. Ishiuchi, C. Takayama, E. Kondo and M. Matsushita, Biochem. Biophys. Res. Commun., 2015, 457, 206–212 CrossRef CAS PubMed.
- T. Teesalu, K. N. Sugahara, V. R. Kotamraju and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16157–16162 CrossRef CAS PubMed.
- S. M. J. van Duijnhoven, M. S. Robillard, K. Nicolay and H. Grull, J. Nucl. Med., 2011, 52, 279–286 CrossRef CAS PubMed.
- E. N. Savariar, C. N. Felsen, N. Nashi, T. Jiang, L. G. Ellies, P. Steinbach, R. Y. Tsien and Q. T. Nguyen, Cancer Res., 2013, 73, 855–864 CrossRef CAS PubMed.
- J. L. Crisp, E. N. Savariar, H. L. Glasgow, L. G. Ellies, M. A. Whitney and R. Y. Tsien, Mol. Cancer Ther., 2014, 13, 1514–1525 CrossRef CAS PubMed.
- T. Jiang, E. S. Olson, Q. T. Nguyen, M. Roy, P. A. Jennings and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17867–17872 CrossRef CAS PubMed.
- K. E. Marshall, R. Marchante, W.-F. Xue and L. C. Serpell, Prion, 2014, 8, 192–196 CrossRef CAS.
- M. Andreasen, N. Lorenzen and D. Otzen, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1897–1907 CrossRef CAS PubMed.
- M. Stefani, Prog. Neurobiol., 2012, 99, 226–245 CrossRef CAS PubMed.
- R. Kayed and C. A. Lasagna-Reeves, Adv. Alzheimer's Dis., 2012, 3, 67–78 CrossRef PubMed.
- R. Kayed, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson, C. Cecchi and F. Chiti, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- U. Sengupta, A. N. Nilson and R. Kayed, EBioMedicine, 2016, 6, 42–49 CrossRef PubMed.
- M. J. Guerrero-Muñoz, D. L. Castillo-Carranza and R. Kayed, Biochem. Pharmacol., 2014, 88, 468–478 CrossRef PubMed.
- K. Zhao, G. Ippolito, L. Wang, V. Price, M. H. Kim, G. Cornwell, S. Fulenchek, G. A. Breen, W. J. Goux and S. R. D'Mello, J. Neurosci. Res., 2010, 88, 3399–3413 CrossRef CAS PubMed.
- W. J. Goux, L. Kopplin, A. D. Nguyen, K. Leak, M. Rutkofsky, V. D. Shanmuganandam, D. Sharma, H. Inouye and D. A. Kirschner, J. Biol. Chem., 2004, 279, 26868–26875 CrossRef CAS PubMed.
- C. L. Moore, M. H. Huang, S. A. Robbennolt, K. R. Voss, B. Combs, T. C. Gamblin and W. J. Goux, Biochemistry, 2011, 50, 10876–10886 CrossRef CAS PubMed.
- F. A. Rojas Quijano, D. Morrow, B. M. Wise, F. L. Brancia and W. J. Goux, Biochemistry, 2006, 45, 4638–4652 CrossRef CAS PubMed.
- J. R. Veloria, L. Li, G. A. M. Breen and W. J. Goux, ACS Chem. Neurosci., 2017 DOI:10.1021/acschemneuro.7b00275.
- M. T. Pastor, N. Kümmerer, V. Schubert, A. Esteras-Chopo, C. G. Dotti, M. López de la Paz and L. Serrano, J. Mol. Biol., 2008, 375, 695–707 CrossRef CAS PubMed.
- M. C. Manning, M. Illangasekare and R. W. Woody, Biophys. Chem., 1988, 31, 77–86 CrossRef CAS PubMed.
- N. Sreerama and R. W. Woody, Protein Sci., 2003, 12, 384–388 CrossRef CAS PubMed.
- J. Wu, J. T. Yang and C. S. C. Wu, Anal. Biochem., 1992, 200, 359–364 CrossRef CAS PubMed.
- R. Woody, Theory of circular dichroism of proteins, in Circular Dichroism and the Conformational Analysis of Biomolecules, NY, 1996 Search PubMed.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, B. George, D. B. Teplow, G. Bitan, M. D. Kirkitadze, A. Lomakint, S. Sabrina, G. B. Benedekt and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330–335 CrossRef CAS PubMed.
- A. L. Clos, C. A. Lasagna-Reeves, B. Kelly, R. Wagner, M. Wilkerson, G. R. Jackson and R. Kayed, J. Am. Acad. Dermatol., 2011, 65, 1023–1031 CrossRef CAS PubMed.
- R. Kayed, E. Head, F. Sarsoza, T. Saing, C. W. Cotman, M. Necula, L. Margol, J. Wu, L. Breydo, J. L. Thompson, S. Rasool, T. Gurlo, P. Butler and C. G. Glabe, Mol. Neurodegener., 2007, 2, 18 CrossRef PubMed.
- E. Vives, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS PubMed.
- P. Boya, K. Andreau, D. Poncet, N. Zamzami, J.-L. Perfettini, D. Metivier, D. M. Ojcius, M. Jäättelä and G. Kroemer, J. Exp. Med., 2003, 197, 1323–1334 CrossRef CAS PubMed.
- K. Ditaranto, T. L. Tekirian and A. J. Yang, Neurobiol. Dis., 2001, 8, 19–31 CrossRef CAS PubMed.
- A. J. Yang, D. Chandswangbhuvana, L. Margol and C. G. Glabe, J. Neurosci. Res., 1998, 698, 691–698 CrossRef.
- D. Freeman, R. Cedillos, S. Choyke, Z. Lukic, K. McGuire, S. Marvin, A. M. Burrage, S. Sudholt, A. Rana, C. O'Connor, C. M. Wiethoff and E. M. Campbell, PLoS One, 2013, 8, e62143 CAS.
- M. Tan, K. H. Lan, J. Yao, C. H. Lu, M. Sun, C. L. Neal, J. Lu and D. Yu, Cancer Res., 2006, 66, 3764–3772 CrossRef CAS PubMed.
- M. Mae, O. Rautsi, J. Enback, M. Hallbrink, K. R. Aizman, M. Lindgren, P. Laakkonen and U. Langel, Int. J. Pept. Res. Ther., 2012, 18, 361–371 CrossRef.
- Fmoc Solid Phase Peptide Synthsis: A Practical Approach, ed. W. Chan and P. White, Oxford University Press, USA, Cary, NC, 1st edn, 2000 Search PubMed.
- M. M. Condron, B. H. Monien and G. Bitan, Open Biotechnol. J., 2008, 2, 87–93 CrossRef CAS PubMed.
- T. Nguyen and M. Francis, Org. Lett., 2003, 5, 3245–3248 CrossRef CAS PubMed.
- H. Edelhock, Biochemistry, 1967, 6, 1948–1954 CrossRef.
- S. Gill and P. von Hippel, Anal. Biochem., 1989, 182, 319–326 CrossRef CAS PubMed.
- M. Necula and J. Kuret, J. Biol. Chem., 2004, 279, 49694–49703 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |