
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotosensitizer-free visible light-mediated gold-catalysed cis-difunctionalization of silyl-substituted alkynes†
Jie-Ren
Deng
ab,
Wing-Cheung
Chan
b,
Nathanael
Chun-Him Lai
ab,
Bin
Yang
ab,
Chui-Shan
Tsang
b,
Ben
Chi-Bun Ko‡
b,
Sharon
Lai-Fung Chan‡
ab and
Man-Kin
Wong
 *ab
*ab
aThe Hong Kong Polytechnic University, Shenzhen Research Institute, Shenzhen, People’s Republic of China. E-mail: mankin.wong@polyu.edu.hk
bState Key Laboratory of Chirosciences, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hum, Hong Kong
First published on 4th September 2017
Abstract
A new photosensitizer-free visible light-mediated gold-catalysed cis-difunctionalization reaction is developed. The reaction was chemoselective towards silyl-substituted alkynes with excellent regioselectivity and good functional group compatibility, giving a series of silyl-substituted quinolizinium derivatives as products. The newly synthesized fluorescent quinolizinium compounds, named JR-Fluor-1, possessed tunable emission properties and large Stokes shifts. With unique photophysical properties, the fluorophores have been applied in photooxidative amidations as efficient photocatalysts and cellular imaging with switchable subcellular localization properties.
Introduction
Over the past years, homogeneous gold catalysis has become a powerful tool for the synthesis of complex organic molecules, particularly in the activation of C–C multiple bonds with gold catalysts acting as strong soft π-Lewis acids.1 In recent years, instead of accessing hydrofunctionalized products, a promising cross-coupling strategy involving Au(I)/Au(III) catalytic cycles has been developed to afford difunctionalized products.2 With a significantly high redox potential of the Au(I)/Au(III) couple, strong oxidants are required to access the oxidation of Au(I) species, while the functional group compatibility could be hampered.3a A complementary approach to overcome this barrier was developed by Glorius3 and Toste4 through merging gold catalysis with photoredox catalysis, which promoted oxidation of Au(I) species employing photosensitizers and aryl radicals generated in situ under irradiation. This approach inspired the development of diverse organic transformations including difunctionalization of alkenes,3b,c,4b allenes5a and alkynes,3e,5b,c as well as C–C3d,4d,5d–f and C–P4c cross-coupling reactions. Recently, Hashmi and co-workers reported a visible light-mediated gold-catalysed oxyarylation of alkynes6a and an aryl–aryl coupling reaction6b which could be conducted without addition of photosensitizers.For alkyne difunctionalization reactions undergoing an anti-nucleophilic addition, trans-difunctionalized products are afforded in the majority of the examples.3c,e,5c,6a However, visible light-mediated cis-difunctionalization of alkynes still remains largely unexplored.7 Inspired by a stereo- and regioselective gold-catalysed hydroamination of internal alkynes reported by Stradiotto et al.,8 we set out to combine visible light mediated Au(I)/Au(III) catalysis with a plausible syn insertion pathway to achieve alkyne cis-difunctionalization products (Scheme 1).7b,c
 | ||
| Scheme 1 Literature works and our strategy for visible light-mediated gold-catalysed alkyne cis-difunctionalization. | ||
Development of organic fluorescent materials has become an emerging and important research area due to their wide applications in chemistry, biology and materials science.9 Compared to the intensive investigations on scaffolds such as fluorescein, BODIPY and, recently, Seoul-Fluor,10 only a few examples of cationic fluorophores have been reported, although the unique properties of fluorophores containing positive charge have been demonstrated in photocatalysis and cellular imaging.11 Quinoliziniums, cationic aromatic heterocycles bearing a quaternary bridgehead nitrogen, were firstly investigated in alkaloid chemistry and recently employed as efficient DNA intercalators.12 However, structure photophysical property relationship (SPPR) studies and applications in photocatalysis and cellular imaging of quinolizinium compounds still remain elusive.13
Along with our ongoing interest in gold-catalysed organic transformations,14 herein, we report a new photosensitizer-free visible light-mediated gold-catalysed alkyne cis-difunctionalization reaction affording a series of silyl-substituted fluorescent quinolizinium compounds. Spectroscopy experiments and DFT calculations were conducted to study the SPPR of the quinolizinium derivatives. Applications of the fluorescent quinolizinium compounds as efficient photocatalysts for photooxidative amidation and as fluorescent dyes for live cell imaging have also been conducted.
Results and discussion
We initiated the reaction screening by treatment of quinoline-substituted aryl diazonium 1a (0.12 mmol, 1.2 equiv.), and (4-fluorophenylethynyl)trimethylsilane 2a (0.10 mmol, 1 equiv.) with Ph3PAuCl 3a (10 mol%) to afford silyl-substituted quinolizinium 4a in 71% yield (Table 1, entry 1). To our surprise, no cross-coupling product 4aa (reported by Toste et al. using phenyl diazoniums as substrates) was obtained in our experiments.4d Besides, the reaction was exceptionally regioselective giving no regioisomer 4a′. The gold(I) catalyst 3b bearing comparatively electron-rich phosphine led to a lower yield of product formation (entry 2), while catalysts 3c–d bearing electron-poor phosphine gave higher yields of 4a (entries 3–4). No desired product was obtained when Ph3PAuBF43e was employed as a catalyst (entry 5). Using gold(I) catalyst 3f possessing a diphosphine ligand led to product formation in 21% yield (entry 6). Only a trace amount of product was observed when simple AuCl 3g was used (entry 7). In addition, employing gold(III) catalysts 3h–j resulted in no product formation (entries 8–10). Without the addition of gold catalyst, no desired product was observed (entry 11), indicating that the gold(I) phosphine chloride catalyst played a crucial role in this transformation. Addition of the transition metal-based photocatalysts Ru(bpy)3Cl2, Ru(bpy)3(BF4)2 and Ir(ppy)3 or the organic photocatalyst Rose bengal led to lower yields of the desired product (entries 12–15). A control experiment in the dark gave no product formation (entry 16). These results suggested that this reaction was conducted under photosensitizer-free reaction conditions. Additionally, only a trace amount of product was detected when the reaction was conducted in toluene, dichloromethane or methanol as the solvent (entries 17–19), and a lower yield (35%) of product was afforded when the reaction was carried out in open air (entry 20).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Au cat. | Photo cat. | Light | N2/air | Solvent | Yieldb [%] |
| a Reaction conditions: treatment of 1a (0.12 mmol), 2a (0.10 mmol) and gold catalyst 3a–j (10 mol%) with or without photocatalyst (5 mol%) in 5 mL of solvent under N2 at room temperature for 16 h. b Yield of 4a was determined by 19F-NMR using fluorobenzene as the internal standard. c Blue LEDs (λmax = 469 nm) were employed as a light source. d n.d.: product formation could not be detected. | ||||||
| 1 | 3a | — | Bluec | N2 | CH3CN | 71 |
| 2 | 3b | — | Bluec | N2 | CH3CN | 58 |
| 3 | 3c | — | Bluec | N2 | CH3CN | 75 |
| 4 | 3d | — | Bluec | N2 | CH3CN | 83 |
| 5 | 3e | — | Bluec | N2 | CH3CN | n.d.d |
| 6 | 3f | — | Bluec | N2 | CH3CN | 21 |
| 7 | 3g | — | Bluec | N2 | CH3CN | Trace |
| 8 | 3h | — | Bluec | N2 | CH3CN | n.d.d |
| 9 | 3i | — | Bluec | N2 | CH3CN | n.d.d |
| 10 | 3j | — | Bluec | N2 | CH3CN | n.d.d |
| 11 | — | — | Bluec | N2 | CH3CN | n.d.d |
| 12 | 3a | Ru(bpy)3Cl2 | Bluec | N2 | CH3CN | 9 |
| 13 | 3a | Ru(bpy)3(BF4)2 | Bluec | N2 | CH3CN | Trace |
| 14 | 3a | Ir(ppy)3 | Bluec | N2 | CH3CN | Trace |
| 15 | 3a | Rose bengal | Bluec | N2 | CH3CN | 65 |
| 16 | 3a | — | Dark | N2 | CH3CN | n.d.d |
| 17 | 3a | — | Bluec | N2 | Toluene | Trace |
| 18 | 3a | — | Bluec | N2 | CH2Cl2 | Trace |
| 19 | 3a | — | Bluec | N2 | CH3OH | Trace |
| 20 | 3a | — | Bluec | Air | CH3CN | 35 |
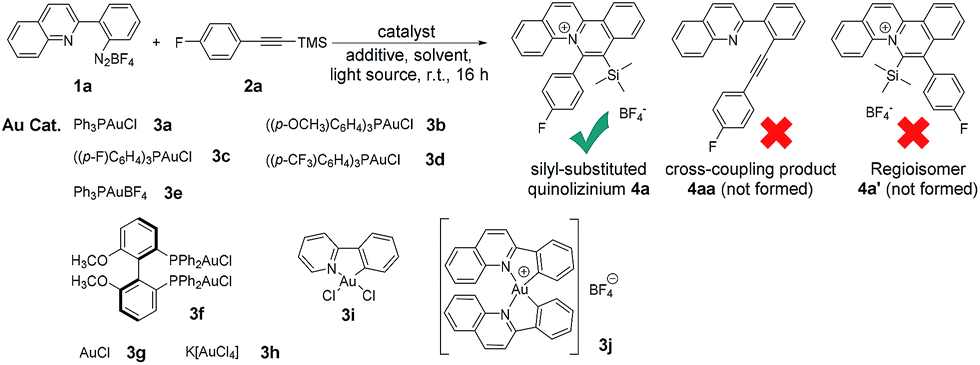
With the optimized reaction conditions, we expanded the scope of this reaction by using various diazoniums (0.60 mmol, 1.2 equiv.) and alkynes (0.50 mmol, 1.0 equiv.) as substrates. Silyl-substituted alkynes 2a–q bearing ether, alkyl, halogen, aldehyde, carboxylic acid, cyanide, trifluoromethyl, nitro and hetero-aromatics as substituents were well tolerated with the reactions giving quinolizinium products 4a–q in 31–69% yield. Increasing the steric bulkiness using ortho-disubstituted phenylethynylsilane 2r gave a trace amount of product. Using a triethylsilyl group gave quinolizinium 4u in slightly lower yield (45%). Further increasing the bulkiness employing tert-butyldiphenylsilyl-substituted alkyne 2t gave no product 4v. Interestingly, employing terminal alkyne phenylacetylene 2u, cyclohexylacetylene 2v or internal alkyne 1,2-diphenylethyne 2w, 1-phenyl-1-propyne 2x or 1-phenyl-1-butyne 2y gave no desired product 7a–e, suggesting that the reaction was chemoselective towards silyl-substituted alkynes. To support our hypothesis, alkyne 2z bearing diphenylethynyl and trimethylsilylethynyl groups was used, and only the trimethylsilylethynyl cis-difunctionalization product 4w was obtained in 46% yield, which convinced us of the chemoselectivity of this gold-catalysed transformation. Further expansion of the scope employing diazoniums 1b–d bearing different structure skeletons and heterocycles gave the desired products 5a–c and 6a in up to 60% yield. These results indicated that the reaction was very compatible with various diazoniums and highly selective towards silyl-substituted alkynes (Table 2).

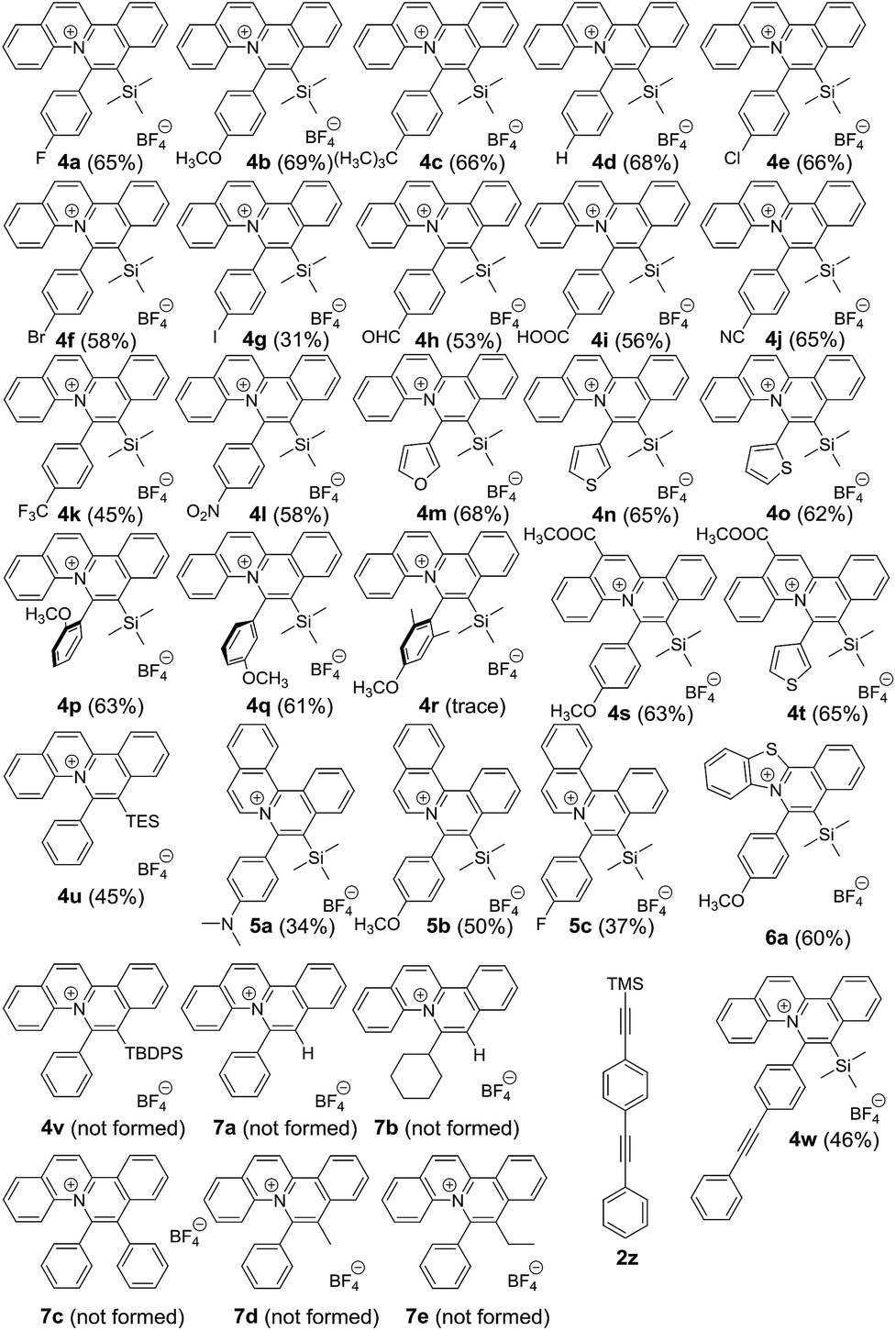
To provide insight into this visible light-mediated gold-catalysed cis-difunctionalization reaction, stoichiometric reactions were set up by treatment of aryl diazonium 1a (0.10 mmol) with Ph3PAuCl 3a (0.10 mmol) under irradiation for 0 to 240 min. 1H-NMR monitoring revealed that aryl diazonium 1a was consumed and new signals appeared gradually from 0 to 60 min, suggesting the formation of plausible intermediates (Fig. 1a). Irradiation for a longer time gave a slight decomposition of the plausible intermediates. These results were consistent with those observed by 31P-NMR analysis, which indicated the gradual appearance of new signals at 31.0, 44.2 and 45.3 ppm (Fig. 1b). The reaction mixtures were further treated with silyl-substituted alkyne 2a (1.5 equiv.) for 60 min in the dark, resulting in the disappearance of the newly observed signals after irradiation and concomitant formation of the signals of the quinolizinium product 4a (Fig. S3a and b in the ESI†). The yield of the product formed was monitored by 19F-NMR analysis through the addition of fluorobenzene as the internal standard (Fig. S3c in the ESI†). In addition, regeneration of Ph3PAuCl 3a could be recovered as a precipitate from the reaction mixtures.
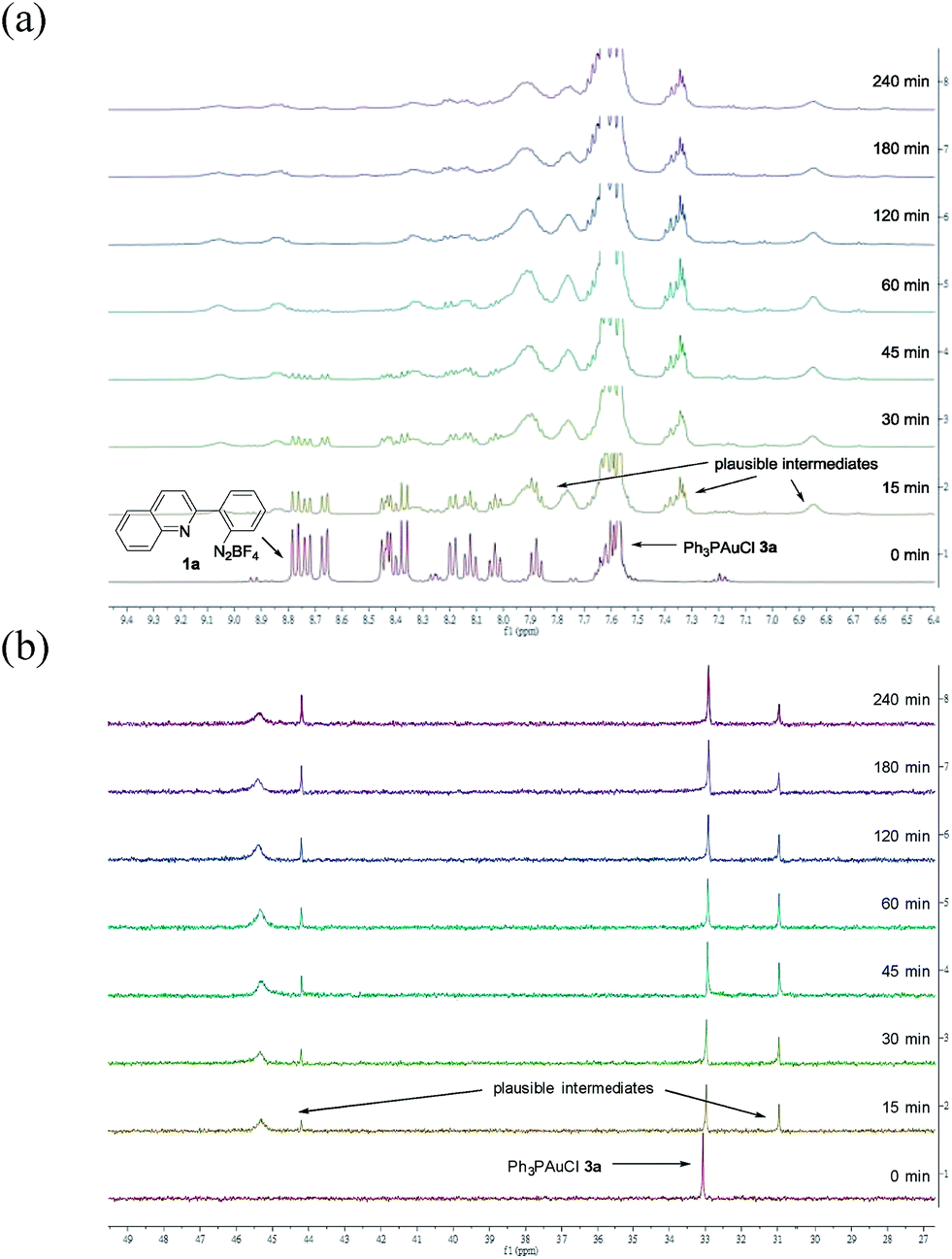 | ||
| Fig. 1 (a) 1H-NMR studies on reaction mixtures X0–240 min in CD3CN; (b) 31P-NMR studies on reaction mixtures X0–240 min in CD3CN. | ||
Due to difficulties in the isolation of the reaction intermediates, we sought to investigate the plausible Au(III) species through ESI-MS analysis by treatment of aryl diazonium 1a (0.12 mmol) with Ph3PAuCl 3a (0.10 mmol) under irradiation for 1 h. The experimental results indicated that rather than the formation of A (m/z = 204.0792) by the displacement of N2 of 1a in ESI-MS analysis, plausible Au(III) intermediates B (m/z = 698.1014) and B′ (m/z = 960.1910) were found (Fig. 2a). Further treatment of the afforded reaction mixture Y with silyl-substituted alkyne 2a (1.5 equiv.) in the dark for 1 h afforded reaction mixture Y′. ESI-MS analysis of reaction mixture Y′ revealed that no signal for B or B′ was observed, and the signal of the quinolizinium product 4a appeared, suggesting that both B and B′ were consumed and reacted with 2a to form the quinolizinium product 4a (Fig. 2b). For a detailed understanding of the plausible Au(III) intermediates B and B′, ESI-MS/MS analysis of species B (precursor ion m/z = 698) and B′ (precursor ion m/z = 910) was conducted. Product phosphonium ions B-I (m/z = 459.0483) and Au(I) species B-II (m/z = 466.1634) were found in the MS/MS analysis of species B (Fig. S4a in the ESI†). Formation of B-I was assumed to be ascribed to the reductive elimination of the Au(III) species B, which was previously reported as a feasible deactivation pathway of phosphine-supported aryl Au(III) complexes.15 In MS/MS analysis of species B′, product ions of B (m/z = 698.0994), B-I (m/z = 466.1672) and B-II (m/z = 459.0518) were found (Fig. S3b in the ESI†). Results suggested that species B′ was composed of Au(III) species B and triphenylphosphine and presumably formed by possible transmetallation.16 A control experiment under the same reaction conditions without irradiation led to no formation of the Au(III) species B, B′ or product 4a, suggesting that a light source was necessary for promotion of the Au(I)/Au(III) transformation in this reaction.
 | ||
| Fig. 2 (a) ESI-MS analysis of the reaction mixture Y; (b) ESI-MS analysis of the reaction mixture Y′. | ||
To study the photosensitizer-free reaction conditions, we measured the UV/Vis absorption properties of aryl diazonium 1a and Ph3PAuCl 3a. Spectroscopic analysis revealed that no absorption peak of Ph3PAuCl 3a was observed at λabs > 395 nm (Fig. S6 in the ESI†), indicating that Ph3PAuCl 3a might not be directly excited by the light source in this reaction which was consistent with literature work indicating that the photosensitizer was necessary to initiate the reaction.3–5 On the contrary, a tail of the lowest energy absorption peak of aryl diazonium 1a was found at λabs > 395 nm (Fig. 3a), suggesting that aryl diazoniums could be firstly excited to initiate the reaction.17 To further support our hypothesis, we measured the difference of the fluorescence intensity before and after mixing Ph3PAuCl 3a (1 equiv.) with 1a. Spectroscopic analysis indicated that the fluorescence of 1a could be quenched (Iq/I0 = 0.68) after the addition of Ph3PAuCl 3a, providing strong evidence for electron transfer from Ph3PAuCl 3a to aryl diazonium 1a under irradiation (Fig. 3b). In addition, the estimated excited state reduction potential (Ered*) of aryl diazonium 1a was 3.28 V, which was much higher than the redox potential of the Au(I)/Au(III) couple (E0 = 1.41 V),18 indicating that Ph3PAuCl 3a could be oxidized by the aryl diazonium 1a in the first step of the reaction.
 | ||
| Fig. 3 (a) UV/Vis absorption spectrum of aryl diazonium 1a; (b) fluorescence quenching of aryl diazonium 1a with Ph3PAuCl 3a. | ||
Based on the aforementioned experimental results, a reaction mechanism is proposed as shown (Scheme 2). Under irradiation, the aryl diazonium compound is firstly excited and subsequently reduced by single electron transfer from Au(I) catalyst 3a to form an aryl radical with the generation of a Au(II) species. The Au(II) species further recombines with the aryl radical to give Au(III) intermediate B. The oxidation and addition on Au(I) catalyst 3a to form Au(III) intermediate B is also possibly a concerted reaction pathway rather than a two-step process.6a The reaction quantum yield was found to be 0.91 (less than 1), suggesting that the radical chain process was not prominent in this transformation. Then, silyl-substituted alkyne is activated by Au(III) intermediate B through π-activation to give species C. After that, the Au–N bond is regioselectively inserted by the π-activated alkyne to form cis vinyl gold species D. Then, reductive elimination provides a quinolizinium compound as the cis-difunctionalized product and regenerates the Au(I) catalyst 3a.
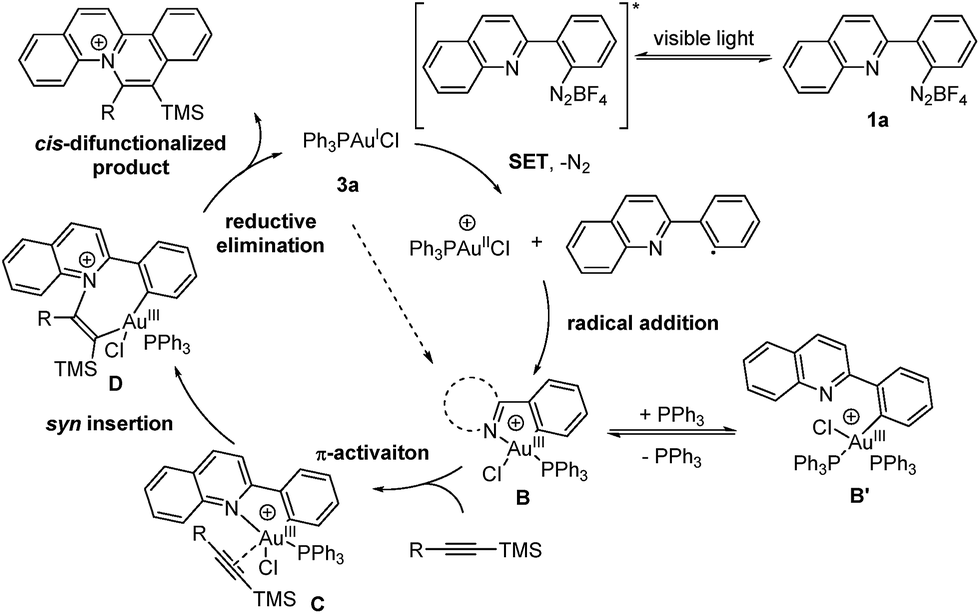 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Pioneered by Cheng and co-workers,19 rhodium-catalysed C–H bond activation followed by annulation has been used as a powerful tool for the synthesis of disubstituted quinolizinium compounds. We sought to compare the newly developed approach of gold-catalysed cis-difunctionalization with rhodium catalysis. An optimized reaction employing 2-phenylquinoline (0.1 mmol, 1 equiv.) with internal alkyne 1,2-diphenylethyne 2w (0.1 mmol, 1 equiv.), [Cp*RhCl2]2 (5 mol%) and AgBF4 (0.1 mmol, 1 equiv.) in 1,2-dichloroethane under open air at room temperature for 16 h afforded diphenyl-substituted quinolizinium 7c in 68% yield. However, when 1-phenyl-2-trimethylsilylacetylene 2d was employed as a substrate, no desired product 4d was formed. To our knowledge, the present photosensitizer-free visible light-mediated gold-catalysed alkyne cis-difunctionalization is the first example of a highly regioselective synthesis of silyl-substituted quinolizinium compounds (Scheme 3).
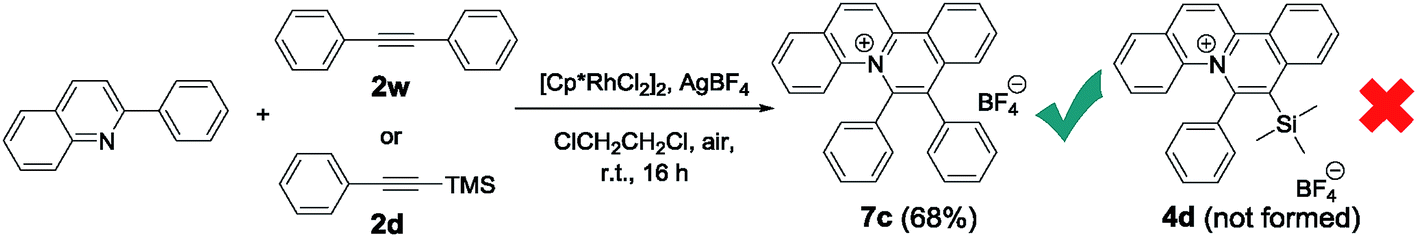 | ||
| Scheme 3 Rhodium-catalysed synthesis of quinolizinium compounds. | ||
X-ray crystal structure analysis revealed a twisted conformation of quinolizinium 4b, with a torsion angle of 107.84° (C1–C2–C18–C19), suggesting that the molecule could be divided into two parts: a slightly distorted planar quinolizinium moiety and a phenyl moiety. This twisted structure suggested that the two moieties should be weakly coupled conjugatively (Fig. 4).
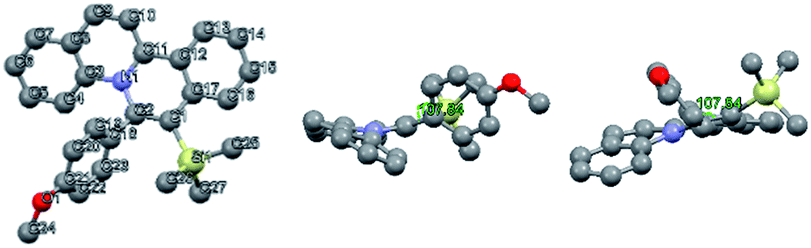 | ||
| Fig. 4 X-ray crystal structure of 4b (front view, left view and vertical view). | ||
After synthesis of the new silyl-substituted quinolizinium compounds, we moved on to study their absorption and emission properties (Table 3). Interestingly, these compounds possessed the lowest energy absorption maxima at λabs > 395 nm, full color tunable emission properties (λem = 450 to 640 nm) in the visible light region and large Stokes shifts (up to 6797 cm−1) with quantum yields up to 0.59. Incorporation of electron-donating groups at the para-position of the phenyl moiety or introduction of π-excessive heteroaromatics resulted in bathochromic shifts of the emission properties. Further bathochromic shifts were observed by the introduction of an electron-withdrawing ester group on the quinolizinium moiety. Substituents bearing heavy atoms (Cl, Br, I) led to lower quantum yields. A positive solvatochromism was found in compound 4b, which indicated an increase of dipole moment of the excited state compared to its ground state (Fig. S8 in the ESI†). An unexpectedly low quantum yield of 5a (0.02) bearing a dimethylamine substituent was observed, in which the fluorescence was proposed to be quenched by the amine group via intramolecular photo-induced electron transfer (PET). Cyclic voltammetry (CV) experiments indicated a quasireversible oxidation couple at +1.08 V (vs. SCE) of 5a which originated from the presence of the amine group and no similar peak was found in 5c (ESI†). Protonation of the amine group by measuring the emission in HCl/NaOH buffer (pH changing from 7 to 1) gave a ≥100 fold enhancement of the emission intensity at a shorter wavelength (λem = 436 nm) which supported our hypothesis (Fig. S9 in the ESI†).
| Cpd. | Absorption maximum λabs (nm) (ε (104 dm3 mol−1 cm−1)) | Emission maximum λem (nm) | Stokes shift (cm−1) | Quantum yield ΦFb |
|---|---|---|---|---|
| a Absorption and emission properties were measured in CH2Cl2 at a concentration of 1 × 10−5 M. b Quantum yields were measured using fluorescein (ΦF = 0.95 in 0.1 N NaOH buffer) as a standard. | ||||
| 4a | 423 (1.39) | 495 | 3439 | 0.49 |
| 4b | 430 (1.14) | 569 | 5681 | 0.17 |
| 4c | 428 (1.10) | 507 | 3640 | 0.44 |
| 4d | 423 (1.45) | 491 | 3275 | 0.44 |
| 4e | 424 (1.62) | 494 | 3342 | 0.37 |
| 4f | 422 (0.95) | 493 | 3413 | 0.16 |
| 4g | 424 (1.00) | 493 | 3301 | 0.03 |
| 4h | 423 (1.06) | 480 | 2807 | 0.28 |
| 4i | 417 (0.75) | 484 | 3320 | 0.44 |
| 4j | 420 (1.30) | 479 | 2933 | 0.28 |
| 4k | 420 (1.17) | 479 | 2933 | 0.34 |
| 4l | 419 (1.23) | 476 | 2858 | 0.24 |
| 4m | 423 (1.45) | 506 | 3878 | 0.18 |
| 4n | 424 (1.68) | 508 | 3900 | 0.16 |
| 4o | 428 (1.43) | 547 | 5363 | 0.02 |
| 4p | 421 (1.50) | 511 | 4183 | 0.11 |
| 4q | 423 (1.80) | 511 | 4071 | 0.16 |
| 4s | 446 (0.79) | 640 | 6797 | 0.01 |
| 4t | 446 (1.05) | 557 | 4468 | 0.08 |
| 4u | 424 (1.42) | 494 | 3342 | 0.41 |
| 4w | 427 (1.05) | 555 | 5401 | 0.07 |
| 5a | 397 (1.74) | 495 | 4987 | 0.02 |
| 5b | 405 (1.23) | 504 | 4850 | 0.30 |
| 5c | 401 (1.09) | 450 | 2840 | 0.59 |
For a detailed investigation of the SPPR of the quinolizinium compounds, compound 4d was subjected to TDDFT calculations. Results reveal that the lowest energy absorption band at 435 nm is originated from HOMO → LUMO transitions. The HOMO is composed of a π orbital of the quinolizinium and phenyl ring whereas the LUMO is composed of a π* orbital of the quinolizinium ring. The low energy absorption band can be assigned as an admixture of π → π* transitions within the quinolizinium ring and π → π* transitions from the phenyl to quinolizinium ring. Hence, bathochromic shifts were observed by the introduction of an ester group at C9 (LUMO was dominant) and the introduction of electron-donating groups at C21 (HOMO was partially dominant) (Fig. 5).
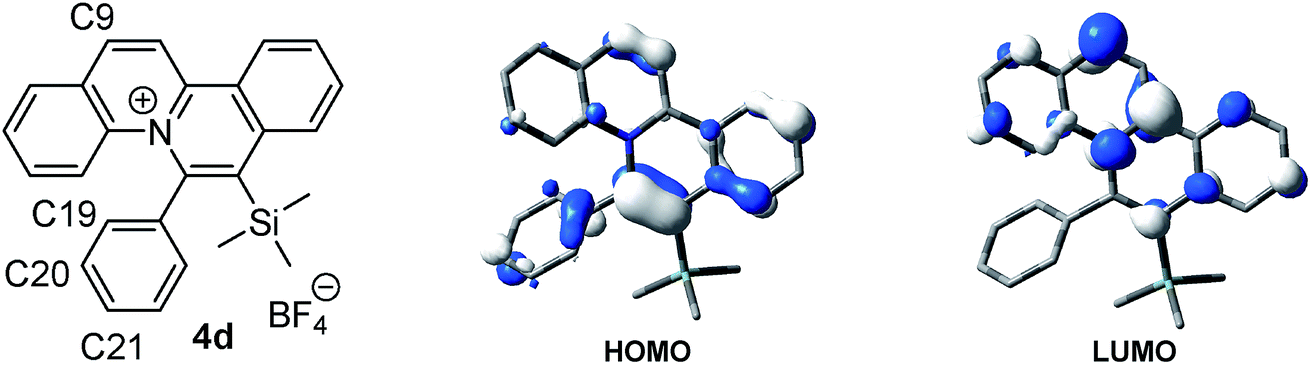 | ||
| Fig. 5 HOMO and LUMO diagrams of 4d. | ||
In line with our interest in amide synthesis,20 we envisioned that the newly developed quinolizinium fluorophores could be utilized as photocatalysts for photooxidative amidation of aldehydes and secondary amines. We first investigated the catalytic activities by treatment of 4-nitrobenzaldehyde 8a (0.1 mmol, 1 equiv.), piperidine 9a (0.2 mmol, 2 equiv.), Na2CO3 (0.2 mmol, 2 equiv.) and selected photocatalysts (5 mol%) in CH3CN under irradiation (blue LEDs) and air for 16 h. Interestingly, the catalytic activities of quinoliziniums were comparable or even better than the currently used photocatalysts (Table 4). The reaction was optimized using 4e as a photocatalyst to afford amide 10a in 86% yield after 48 h. Expansion of the scope employing different aldehydes and secondary amines gave amides 10b–g in up to 84% yield (Table 5). Further investigations revealed that the quinolizinium compounds possessed tunable and high excited state reduction potentials (Ered* = 1.97 to 2.23 V), which were comparable to the well-known photocatalyst 9-mesityl-10-methylacridinium [Acr+-Mes](BF4) developed by Fukuzumi.21 To our knowledge, this is the first example of employing quinoliziniums as photocatalysts for visible light-mediated photoredox catalysis.
|
|
||||
|---|---|---|---|---|
| Entry | Photo cat. | E red* (V) | Time (h) | Yieldb [%] |
| a Reaction conditions: treatment of 8a (0.1 mmol, 1 equiv.), 9a (0.2 mmol, 2 equiv.), Na2CO3 (0.2 mmol, 2 equiv.) and photocatalyst (5 mol%) in 5 mL of CH3CN under blue LEDs and air at room temperature. b Yield was determined by 1H-NMR using 1,3,5-trimethoxybenzene as the internal standard. c E red* refers to ref. 9a and literature cited there. d Reaction was performed by treatment of 8a (1 mmol, 1 equiv.), 9a (2 mmol, 2 equiv.), Na2CO3 (2 mmol, 2 equiv.) and photocatalyst (5 mol%) in 5 mL of CH3CN under blue LEDs and air at room temperature. e Isolated yield. | ||||
| 1 | 4a | 1.99 | 16 | 53 |
| 2 | 4b | 1.97 | 16 | 50 |
| 3 | 4e | 2.21 | 16 | 61 |
| 4 | 4f | 2.22 | 16 | 59 |
| 5 | 4j | 2.24 | 16 | 52 |
| 6 | 4p | 2.07 | 16 | 53 |
| 7 | Eosin Y | 1.23c | 16 | 48 |
| 8 | Fluorescein | 1.25c | 16 | 47 |
| 9 | Rose Bengal | 1.18c | 16 | 32 |
| 10 | [Acr+-Mes](BF4) | 2.08c | 16 | 44 |
| 11 | 4e | 2.21 | 48 | 86 |
| 12d | 4e | 2.21 | 48 | 71e |

The feasibility in cellular imaging was demonstrated by incubation of HeLa cells with 2 μM of the fluorescent quinolizinium compounds 4a–b, d, f, h, l–o, s–t, w and 5c, respectively (Experimental details in the ESI†). Confocal fluorescence microscopic images revealed that the quinoliziniums were selectively localized in cytoplasm with no transportation into the nucleus, which was different from the known examples of quinoliziniums.22 Compound 5c and 4l were chosen for colocalization studies to track the subcellular localization. Compound 5c was specifically localized in the mitochondria (Fig. 6a–c), which could be attributed to the presence of the positively charged quinolizinium skeleton, directing it towards the mitochondria membrane with −180 mV potential. Interestingly, apart from localization in the mitochondria, compound 4l bearing a nitro substituent localized mainly in the lysosome and partially in late the endosome (Fig. 6d–i). The subcellular localization of the quinoliziniums could be switched by simply modifying the substituents, and these compounds would be amenable for the design of specific molecular probes for individual organelle imaging.
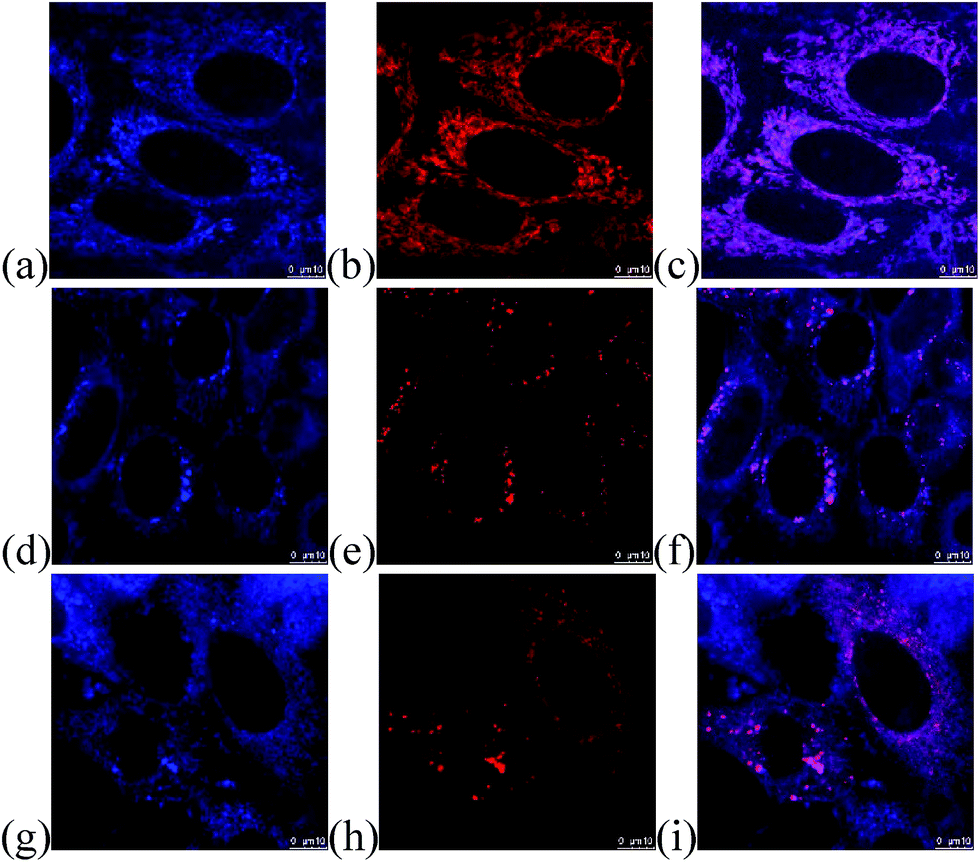 | ||
| Fig. 6 Confocal fluorescence microscopic images of HeLa cells. (a) Sub-cellular localization of 5c; (b) subcellular localization of MitoTracker® red; (c) merged images of (a) and (b); (d) subcellular localization of 4l; (e) subcellular localization of LysoTracker® deep red; (f) merged images of (d) and (e); (g) subcellular localization of 4l; (h) subcellular localization of mRFP-Rab7; (i) merged images of (g) and (h). | ||
Conclusions
In summary, we have developed the first photosensitizer-free visible light-mediated gold-catalysed cis-difunctionalization reaction with high chemoselectivity and excellent regioselectivity for modular synthesis of a series of silyl-substituted quinolizinium compounds. Control experiments as well as a combination of NMR, ESI-MS and spectroscopic analysis indicate a plausible visible light-mediated Au(I)/Au(III) catalytic transformation involving a regioselective syn insertion of silyl-substituted alkynes. Additionally, we have studied applications of the newly synthesized silyl-substituted quinolizinium compounds in photooxidative amidation and cellular imaging. The efficient modular synthesis and unique photophysical properties of the quinolizinium compounds will open up a new direction in gold catalysis, photoredox catalysis and molecular imaging.Conflicts of interest
M.-K. Wong, J.-R. Deng and N. C.-H. Lai applied patents on quinolizinium compounds 4a–q, s–u, w and 5a–c.Acknowledgements
We are grateful for the financial support of the National Natural Science Foundation of China (21272198), Hong Kong Research Grants Council (PolyU 153031/14P, 153001/17P, X-ray diffractometer-PolyU11/CRF/13E), State Key Laboratory of Chirosciences and Department of Applied Biology and Chemical Technology. We thank Prof. K.-Y. Wong for facilitating the project by providing access to Bioanalytical Systems (BAS) for cyclic voltammetry experiments and Prof. Z. Zhou and Dr W. T.-K. Chan for X-ray crystallographic analysis.Notes and references
- (a) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed; (b) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (c) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed; (d) A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS PubMed; (e) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (f) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (g) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (h) H.-S. Yeom and S. Shin, Acc. Chem. Res., 2014, 47, 966 CrossRef CAS PubMed; (i) Z. Zheng, Z. Wang, Y. Wang and L. Zhang, Chem. Soc. Rev., 2016, 45, 4448 RSC; (j) A. M. Asiri and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 4471 RSC; (k) W. Zi and F. D. Toste, Chem. Soc. Rev., 2016, 45, 4567 RSC.
- (a) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248 CrossRef CAS PubMed; (b) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236 CrossRef CAS PubMed.
- (a) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS PubMed; (b) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (c) M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794 CrossRef CAS; (d) A. Tlahuext-Aca, M. N. Hopkinson, B. Sahoo and F. Glorius, Chem. Sci., 2016, 7, 89 RSC; (e) A. Tlahuext-Aca, M. N. Hopkinson, R. A. Garza-Sanchez and F. Glorius, Chem.–Eur. J., 2016, 22, 5909 CrossRef CAS PubMed.
- (a) M. D. Levin, S. Kim and F. D. Toste, ACS Cent. Sci., 2016, 2, 293 CrossRef CAS PubMed; (b) X.-Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed; (c) Y. He, H. Wu and F. D. Toste, Chem. Sci., 2015, 6, 1194 RSC; (d) S. Kim, J. Rojas-Martinab and F. D. Toste, Chem. Sci., 2016, 7, 85 RSC.
- (a) D. V. Patil, H. Yun and S. Shin, Adv. Synth. Catal., 2015, 357, 2622 CrossRef CAS; (b) J. Um, H. Yun and S. Shin, Org. Lett., 2016, 18, 484 CrossRef CAS PubMed; (c) Z. Xia, O. Khaled, V. Mouriès-Mansuy, C. Ollivier and L. Fensterbank, J. Org. Chem., 2016, 81, 7182 CrossRef CAS PubMed; (d) T. Cornilleau, P. Hermange and E. Fouquet, Chem. Commun., 2016, 52, 10040 RSC; (e) V. Gauchot and A.-L. Lee, Chem. Commun., 2016, 52, 10163 RSC; (f) V. Gauchot, D. R. Sutherland and A.-L. Lee, Chem. Sci., 2017, 8, 2885 RSC.
- (a) L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 4808 CrossRef CAS PubMed; (b) S. Witzel, J. Xie, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2017, 359, 1522 CrossRef CAS.
- (a) M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022 CrossRef CAS PubMed; (b) F. Rekhroukh, R. Brousses, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 1266 CrossRef CAS PubMed; (c) F. Rekhroukh, C. Blons, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Chem. Sci., 2017, 8, 4539 RSC; (d) C. Gryparis, M. Kidonakis and M. Stratakis, Org. Lett., 2013, 15, 6038 CrossRef CAS PubMed; (e) C. Gryparis and M. Stratakis, Org. Lett., 2014, 16, 1430 CrossRef CAS PubMed.
- K. D. Hesp and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 18026 CrossRef CAS PubMed.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590 CrossRef CAS PubMed; (c) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed; (d) G. J. Hedley, A. Ruseckas and I. D. W. Samuel, Chem. Rev., 2017, 117, 796 CrossRef CAS PubMed.
- (a) H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620 CrossRef CAS PubMed; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed; (c) E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538 CrossRef CAS PubMed.
- (a) Z. Xu and L. Xu, Chem. Commun., 2016, 52, 1094 RSC; (b) W. Xu, Z. Zeng, J. H. Jiang, Y. T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658 CrossRef CAS PubMed.
- (a) G. Anton and I. Heiko, Synlett, 2016, 27, 1775 CrossRef; (b) D. Sucunza, A. M. Cuadro, J. Alvarez-Builla and J. J. Vaquero, J. Org. Chem., 2016, 81, 10126 CrossRef CAS PubMed; (c) A. Granzhan, H. Ihmels and G. Viola, J. Am. Chem. Soc., 2007, 129, 1254 CrossRef CAS PubMed.
- A. Barbafina, A. M. Melia, L. Latterini, G. G. Aloisi and F. Elisei, J. Phys. Chem. A, 2009, 113, 14514 CrossRef CAS PubMed.
- (a) V. K.-Y. Lo, Y. Liu, M.-K. Wong and C.-M. Che, Org. Lett., 2006, 8, 1529 CrossRef CAS PubMed; (b) H.-M. Ko, K. K.-Y. Kung, J.-F. Cui and M.-K. Wong, Chem. Commun., 2013, 49, 8869 RSC; (c) J.-F. Cui, H.-M. Ko, K.-P. Shing, J.-R. Deng, N. C.-H. Lai and M.-K. Wong, Angew. Chem., Int. Ed., 2017, 56, 3074 CrossRef CAS PubMed.
- H. Kawai, W. J. Wolf, A. G. DiPasquale, M. S. Winston and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 587 CrossRef CAS PubMed.
- W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159 CrossRef CAS PubMed.
- M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777 CrossRef CAS PubMed.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1 CrossRef CAS.
- (a) C.-Z. Luo, P. Gandeepan and C.-H. Cheng, Chem. Commun., 2013, 49, 8528 RSC; (b) C.-Z. Luo, P. Gandeepan, J. Jayakumar, K. Parthasarathy, Y.-W. Chang and C.-H. Cheng, Chem.–Eur. J., 2013, 19, 14181 CrossRef CAS PubMed; (c) C.-Z. Luo, P. Gandeepan, Y.-C. Wu, C.-H. Tsai and C.-H. Cheng, ACS Catal., 2015, 5, 4837 CrossRef CAS.
- (a) W.-K. Chan, C.-M. Ho, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 14796 CrossRef CAS PubMed; (b) A. O.-Y. Chan, C.-M. Ho, H.-C. Chong, Y.-C. Leung, J.-S. Huang, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2012, 134, 2589 CrossRef CAS PubMed; (c) G.-L. Li, K. K.-Y. Kung and M.-K. Wong, Chem. Commun., 2012, 48, 4112 RSC; (d) F. K.-C. Leung, J.-F. Cui, T.-W. Hui, K. K.-Y. Kung and M.-K. Wong, Asian J. Org. Chem., 2015, 4, 533 CrossRef CAS.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed.
- (a) R. Bortolozzi, H. Ihmels, L. Thomas, M. Tian and G. Viola, Chem.–Eur. J., 2013, 19, 8736 CrossRef CAS PubMed; (b) A. C. Shaikh, D. S. Ranade, P. R. Rajamohanan, P. P. Kulkarni and N. T. Patil, Angew. Chem., Int. Ed., 2017, 56, 757 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1545248. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02294h |
| ‡ Dr Sharon Lai-Fung Chan designed and performed the spectroscopic studies and computational experiments of this research. Dr Ben Chi-Bun Ko designed the cellular imaging experiments. |
| This journal is © The Royal Society of Chemistry 2017 |