DOI:
10.1039/C7SC02780J
(Edge Article)
Chem. Sci., 2017,
8, 7545-7551
Selective photocatalytic hydroxylation and epoxidation reactions by an iron complex using water as the oxygen source†
Received
22nd June 2017
, Accepted 2nd September 2017
First published on 4th September 2017
Introduction
The conversion of sunlight to chemical energy has been one of the great challenges for chemists in the quest for a sustainable world.1 The inspiration for such a process comes from photosynthesis, wherein sunlight is used to accomplish the energetically uphill water oxidation reaction.2 During photosynthesis, a high-valent manganese-oxo cluster has been proposed to be the active intermediate for water oxidation.3 Similarly, high-valent iron-oxo complexes have been shown to be the active intermediates for heme/non-heme enzymes and their model complexes which catalyse hydrocarbon oxidation using O2 as the oxidant.4 Combining these concepts, Inoue et al. first demonstrated that visible light can be utilized for the oxidation of organic substrates using water as the oxygen atom source.5 Gray et al. also developed a photochemical method to generate the intermediates “Compound I” and “Compound II” from peroxidases by using [RuII(bpy)3]2+ (bpy = 2,2′-bipyridine) as a photosensitizer and [CoIII(NH3)5Cl]2+ as a mild one-electron oxidant in aqueous medium.6 Using these strategies, several photochemical oxygenation systems based on metal complexes (mostly Ru and Mn) as catalysts have been evolved. Examples include various Ru-complexes7,8 and Mn-complexes such as Mn–porphyrin,9 [(R,R-BQCN)MnII(OTf)2] (BQCN = N,N′-dimethyl-N,N′-bis(8-quinolyl) cyclohexanediamine)10a,b and the Mn(V)–nitrido complex, [Mn(N)(CN)4]2−,10c which have been shown to be active catalysts for photocatalytic oxygenation reactions where water was used as the oxygen source. Although ruthenium- and manganese-based metal complexes have been explored for photochemical alcohol, olefin and sulfide oxidation reactions, they have not been shown to catalyse the oxidation of unactivated C–H bonds.7–10 The likely reason for this is that the Ru-oxo and Mn-oxo intermediates, formed during these reactions, do not cleave unactivated C–H bonds at rates fast enough for them to function as catalysts for C–H bond oxidation reactions. In contrast, iron-based metalloenzymes (sMMO,11a,b heme and non-heme enzymes4,11c,d) and their synthetic models4b,c,e exhibit excellent reactivity and selectivity towards hydrocarbon oxidation. However, for synthetic systems, very few examples are known where a cheap and environmentally benign metal like iron has been used as a catalyst for photochemical oxidation reactions. Recently, the photochemical generation of oxoiron(IV) has been demonstrated using [FeII(N4Py)]2+ [N4Py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine]12 and [FeII(MePy2tacn)]2+ [MePy2tacn = N-methyl-N,N-bis(2-picolyl)-1,4,7-triazacyclononane].13 The complex [FeII(MePy2tacn)]2+, along with [RuII(bpy)3]2+ as a photosensitizer and sodium persulfate as a sacrificial oxidant under light irradiation has been shown to catalyse sulfoxidation reactions, albeit with low yields. However, the photochemical hydroxylation of unactivated C–H bonds using the [FeII(MePy2tacn)]2+ complex described above has not been reported. Mechanistic studies on iron-catalysed hydroxylation reactions have revealed that in both chemical and biological systems, efficient and selective hydroxylation of unactivated C–H bonds has been mostly catalysed by reactive intermediates such as oxoiron(V) (synthetic systems),14,15,19 the isoelectronic oxoiron(IV) radical cation (heme enzymes)4a,11c,d and [(μ-O)2FeIV2] (methane monooxygenase11a,b). None of these intermediates have been formed in the photochemical systems reported to date using [FeII(N4Py)]2+ and [FeII(MePy2tacn)]2+ complexes, which explains the absence of iron catalysts being used to catalyse photochemical C–H bond oxidation in the literature.
We focused our attention on the peroxidase-mimicking iron(III) complex of bTAML14 (bTAML = biuret-modified tetraamidomacrocyclic ligand) synthesized in our laboratory. The highly electron-donating tetraanionic N-donors are well-known to stabilize high-valent iron-oxo species such as [(bTAML)FeV(O)]− and [(bTAML)FeIV(O)]2− (subsequently referred to as FeV(O) and FeIV(O), respectively).15 Moreover, complex 1 in combination with chemical oxidants catalyses the oxidation of unactivated alkanes, alkenes and alcohols selectively.15b,17b,18,19 The high selectivity obtained has been attributed to the presence of oxoiron(V) and [{(bTAML)FeIV}2(μ-O)]2− (2) as active intermediates in these reactions. We have also shown that the oxidant [RuIII(bpy)3]3+, generated either chemically or photochemically, is a competent oxidant to oxidize the [(bTAML)FeIII(OH2)]− (1) complex in solution to the catalytically active [{(bTAML)FeIV}2(μ-O)]2− dimer (2).16 We therefore hypothesized that complex 1, along with [RuII(bpy)3]2+ and [CoIII(NH3)5Cl]2+, can be a competent system to catalyse the photochemical oxidation of synthetically challenging reactions such as the selective hydroxylation of unactivated C–H bonds and epoxidation reactions. The development of such iron-based catalysts would be transformational in the goal to achieve green methods for the selective oxidation of C–H bonds.
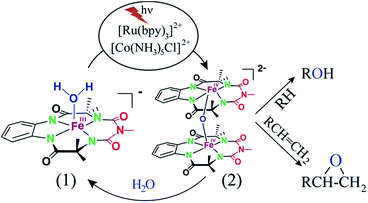
Herein, we report selective photocatalytic alkane hydroxylation and olefin epoxidation by employing [Et4N][(bTAML)FeIII(OH2)] (1) as a catalyst, [RuII(bpy)3]Cl2 as a photosensitizer, [CoIII(NH3)5Cl]Cl2 as a mild one-electron acceptor and water as the oxygen atom source. We also demonstrate that under the reaction conditions, intermediate 2 was generated and found to be the active oxidant (Scheme 1). To the best of our knowledge, this represents the first example of the use of an iron-complex to catalyse the photochemical selective oxidation of unactivated C–H bonds and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds using water as the O-atom donor.
C bonds using water as the O-atom donor.
 |
| | Scheme 1 Photocatalytic oxygenation of hydrocarbons. | |
Results and discussion
Photocatalytic oxidation of alkanes
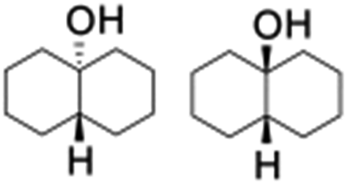
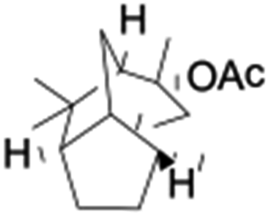
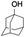
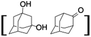
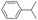
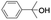
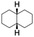
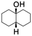
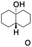
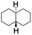
All of the photocatalytic hydroxylation reactions were performed by employing catalyst 1 (∼3–7 mol% loading), [RuII(bpy)3]Cl2 and [CoIII(NH3)5Cl]Cl2 in an acetonitrile–phosphate buffer solution mixture in the presence of light (3 W blue LED, 440 nm) under an argon atmosphere. The photocatalytic hydroxylation of alkanes with different unactivated 3° and activated 2° C–H bonds was attempted under optimized reaction conditions. Adamantane, having twelve 2° and four 3° C–H bonds, was used as a substrate probe to study the regioselectivity using the photocatalytic system. Upon completion of the reaction, 1-adamantanol was formed in good yield (88%) and the 3°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2° selectivity for adamantane oxidation was calculated to be ∼100:1 (Table 1; entry 1). In the case of cumene, which contains six 1° and one benzylic 3° C–H bonds, the 3° hydroxylated product was formed as the major product (67% yield; Table 1, entry 2). Next, cis-1,2-dimethylcyclohexane and cis-decalin were chosen as the substrates to examine the stereoselectivity of this oxidation. The reaction of cis-1,2-dimethylcyclohexane and cis-decalin displayed primarily 3° hydroxylated product (99% and 96% yields, respectively) with more than ∼97% retention of configuration under the reaction conditions (Table 1; entries 3 and 4). The high stereo-retention observed excluded the possibility of radical processes since cis-1,2-dimethylcyclohexane and cis-decalin are known to epimerize to the trans isomer if radical processes are operational. In cyclohexane derivatives, the stereochemical orientation of the 3° C–H bonds (axial or equatorial) determines the regioselective outcome of the reaction. For the catalytic hydroxylation of trans-decalin, the reaction was comparatively slower with a lower yield (60%) of the oxidized product (Table 1, entry 5) in contrast to that of the cis isomer. The oxidation of trans-decalin also exhibited oxidation at the methylenic C–H bonds unlike its cis congener, resulting in the formation of both alcohol (99% retention of configuration) and ketone at a ratio of 3:2. The difference in reactivity between the cis and trans isomers can be attributed to the strain release in the transition state for the cis-isomers.20 In short, the hydroxylated products formed after the reaction displayed very high regioselectivity of 3° over 2° C–H bonds (Table 1; entries 3, 4 and 6), where 3° hydroxylated products were formed predominantly. This result is consistent with a similar oxidation reported by us using complex 1 and mCPBA as the oxidant.19 Furthermore, the regioselective oxidation of 3° C–H bonds in the natural product derivative of cedrol, cedryl acetate, was performed. Cedrol is a sesquiterpene alcohol found in essential oil, having a very rigid structure with five 3° C–H positions. The substrate was hydroxylated very selectively with good yield, albeit with a low conversion (Table 1, entry 6). In order to find the O-atom source in the product formed, oxidation reactions of cis-1,2-dimethylcyclohexane and adamantane were carried out in a mixture of CH3CN and H218O (3:2 v/v). We observed >90% and >95% incorporation of 18O-labelled oxygen atoms in 1-adamantanol (Fig. 2D) and (1S,2R or 1R,2S)-1,2-dimethylcyclohexanol (Fig. S1†), respectively, which confirmed water as the O-atom source. This also precluded the involvement of an O2-based radical pathway during the reaction.
:2° selectivity for adamantane oxidation was calculated to be ∼100:1 (Table 1; entry 1). In the case of cumene, which contains six 1° and one benzylic 3° C–H bonds, the 3° hydroxylated product was formed as the major product (67% yield; Table 1, entry 2). Next, cis-1,2-dimethylcyclohexane and cis-decalin were chosen as the substrates to examine the stereoselectivity of this oxidation. The reaction of cis-1,2-dimethylcyclohexane and cis-decalin displayed primarily 3° hydroxylated product (99% and 96% yields, respectively) with more than ∼97% retention of configuration under the reaction conditions (Table 1; entries 3 and 4). The high stereo-retention observed excluded the possibility of radical processes since cis-1,2-dimethylcyclohexane and cis-decalin are known to epimerize to the trans isomer if radical processes are operational. In cyclohexane derivatives, the stereochemical orientation of the 3° C–H bonds (axial or equatorial) determines the regioselective outcome of the reaction. For the catalytic hydroxylation of trans-decalin, the reaction was comparatively slower with a lower yield (60%) of the oxidized product (Table 1, entry 5) in contrast to that of the cis isomer. The oxidation of trans-decalin also exhibited oxidation at the methylenic C–H bonds unlike its cis congener, resulting in the formation of both alcohol (99% retention of configuration) and ketone at a ratio of 3:2. The difference in reactivity between the cis and trans isomers can be attributed to the strain release in the transition state for the cis-isomers.20 In short, the hydroxylated products formed after the reaction displayed very high regioselectivity of 3° over 2° C–H bonds (Table 1; entries 3, 4 and 6), where 3° hydroxylated products were formed predominantly. This result is consistent with a similar oxidation reported by us using complex 1 and mCPBA as the oxidant.19 Furthermore, the regioselective oxidation of 3° C–H bonds in the natural product derivative of cedrol, cedryl acetate, was performed. Cedrol is a sesquiterpene alcohol found in essential oil, having a very rigid structure with five 3° C–H positions. The substrate was hydroxylated very selectively with good yield, albeit with a low conversion (Table 1, entry 6). In order to find the O-atom source in the product formed, oxidation reactions of cis-1,2-dimethylcyclohexane and adamantane were carried out in a mixture of CH3CN and H218O (3:2 v/v). We observed >90% and >95% incorporation of 18O-labelled oxygen atoms in 1-adamantanol (Fig. 2D) and (1S,2R or 1R,2S)-1,2-dimethylcyclohexanol (Fig. S1†), respectively, which confirmed water as the O-atom source. This also precluded the involvement of an O2-based radical pathway during the reaction.
Table 1 Photocatalytic hydroxylation of different alkanes by 1 using water as the oxygen sourcea
| Entry |
Substrate |
Products |
(Other products) |
Conversion (%) |
Yieldc (%) |
|
Reaction conditions: 1 (1.0 × 10−4 M), [Ru(bpy)3]2+ (2.0 × 10−3 M), [Co(NH3)5Cl]2+ (2.0 × 10−2 M) and substrate (3.0 × 10−3 M) in acetonitrile-aqueous phosphate buffer (3:2 v/v, 10 mM, pH 10). Photoirradiation with LED (3 W, 440 nm), at room temperature (27 °C), under argon for 40 min.
Catalyst 1 (2.0 × 10−4 M).
Yields are based on substrate conversion (the amounts of side products indicated inside the parenthesis are not included); yields and conversions were estimated by GC.
|
| 1 |

|
|
|
76 |
88 |
| 2 |
|
|
|
50 |
67 |
| 3 |
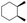
|
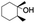
|
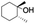
|
55 |
99 (60:1), (cis:trans) |
| 4b |
|
|
|
99 |
96 (65:1), (cis:trans) |
| 5b |
|
|
|
50 |
60 (20:1), (trans:cis) |
| 6b |
|
|
|
37 |
97 |
| 7 |
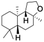
|
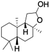
|
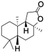
|
56 |
98 (7:12), (alcohol:ketone) |
| 8 |
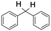
|
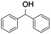
|
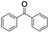
|
60 |
94 (7:10), (alcohol:ketone) |
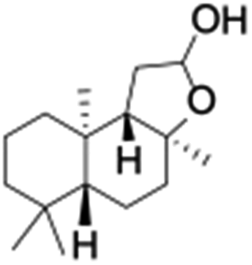
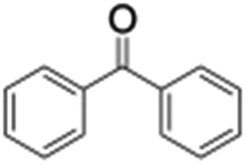
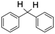
Finally, the selective oxidation of substrates bearing activated methylenic and benzylic C–H bonds was explored. In the case of the substrate ambroxide, oxidation at the α-ethereal C–H bond occurred predominantly among numerous other electronically and sterically accessible secondary and tertiary sites (Table 1; entry 7). At a lower substrate concentration, over-oxidized ketone was the major product formed (Table 1; entries 7 and 8). The alcohol to ketone product ratio increased with increasing substrate concentration (3 mM to 40 mM; Table 2) under the same reaction conditions. Similar results were found when diphenylmethane was employed as the substrate (Table 1; entry 8). This formation of ketone was attributed to the over-oxidation of the hydroxylated product, which was first formed during the hydroxylation reaction (cyclohexanol oxidation has been shown to be 400 times faster than cyclohexane oxidation18). The incorporation of ∼80% 18O-labelled oxygen atoms in the ketone product of ambroxide (Fig. S2†) also supports this hypothesis. The quantum yields for the photocatalytic oxidation of the alkanes were determined using a standard actinometer (potassium ferrioxalate) and a maximum value of 12.2% was observed in the case of the cis-decalin hydroxylation (Table S1†).
Table 2 Effect of substrate concentration on the photocatalytic hydroxylation of 2° C–H bonds by 1 using water as the oxygen sourcea
| Entry |
Substrate |
Products |
Alcohol/ketone (ratio) in different substrate concentrations (mM) |
| 3 mM |
10 mM |
20 mM |
40 mM |
|
Reaction conditions: 1 (1.0 × 10−4 M), [Ru(bpy)3]2+ (2.0 × 10−3 M) and [Co(NH3)5Cl]2+ (2.0 × 10−2 M) in acetonitrile-aqueous phosphate buffer (3:2 v/v, 10 mM, pH 10). Photoirradiation with LED (3 W, 440 nm), at room temperature (27 °C), under argon for 40 min.
|
| 1 |
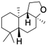
|
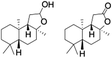
|
0.6 |
1.8 |
2.4 |
5.3 |
| 2 |
|
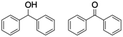
|
0.7 |
1.5 |
3.5 |
6.1 |
Photocatalytic oxidation of alkenes
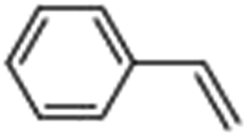
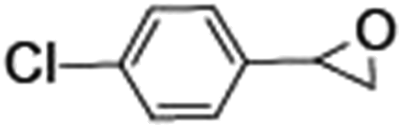
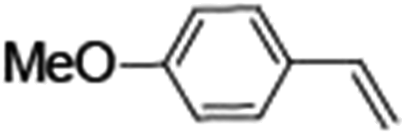
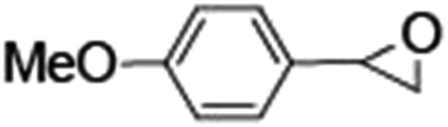
Epoxidation of various alkenes was also performed under optimized reaction conditions by employing catalyst 1 (2 mol% loading) (Fig. S3†). Analysis of the products indicated predominant formation of alkene oxides in moderate to high yields (79–84%) and only a trace amount of the side-product aldehyde (∼5–7% with respect to epoxide) was observed. At first, styrene was chosen as the substrate for the photocatalytic epoxidation reaction where styrene oxide was obtained as the predominant product. Subsequently, different para-substituted styrene derivatives such as 4-chlorostyrene and 4-methoxystyrene were investigated and their corresponding epoxides were obtained with yields of up to 92% (Table 3; entries 1, 2 and 3). The higher amount of epoxide formation for the para-substituted electron-donating group on styrene in comparison to the electron-withdrawing group supports the likely involvement of an electrophilic high-valent iron-oxo intermediate, as has been reported earlier by us.17b For cis-stilbene, a substrate which contains a sterically constrained double bond, a lower conversion of 58% and a moderate yield of 79% (Table 3; entry 4) was obtained. The cis/trans product ratio 19:2 was estimated by 1H-NMR (Fig. S4†) (note: additional stereo-scrambling in the cis/trans ratio was observed in GC run). The substrate scope was further expanded to include cyclooctene and norbornene (Table 3; entries 5 and 6) where a 94% yield of cyclooctene oxide and a 92% yield of exo-norbornene oxide indicate selectivity for the CC bond over the C–H bonds. For the epoxidation of alkenes, a maximum quantum yield of 18.7% for 4-methoxystyrene was observed (Table S2†).
Table 3 Photocatalytic epoxidation of different alkenes by 1 using water as the oxygen sourcea
| Entry |
Substrate |
Product |
Conversion (%) |
Yield (%) |
|
Reaction conditions: 1 (1.0 × 10−4 M), [Ru(bpy)3]2+ (2.0 × 10−3 M), [Co(NH3)5Cl]2+ (2.0 × 10−2 M) and substrate (5.0 × 10−3 M) in acetonitrile-aqueous phosphate buffer (3:2 v/v, 10 mM, pH 10). Photoirradiation with LED (3 W, 440 nm), at room temperature (27 °C), under air for 40 min. Yields are based on substrate conversion (the amounts of side products are not included); yields and conversions were estimated by GC.
|
| 1 |
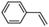
|
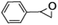
|
72 |
90 |
| 2 |
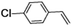
|
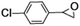
|
50 |
84 |
| 3 |
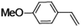
|
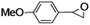
|
95 |
92 |
| 4 |
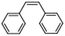
|
|
58 |
79 |
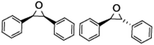
| 5 |
|
|
51 |
94 |
| 6 |

|
|
64 |
92 |
Mechanistic insight
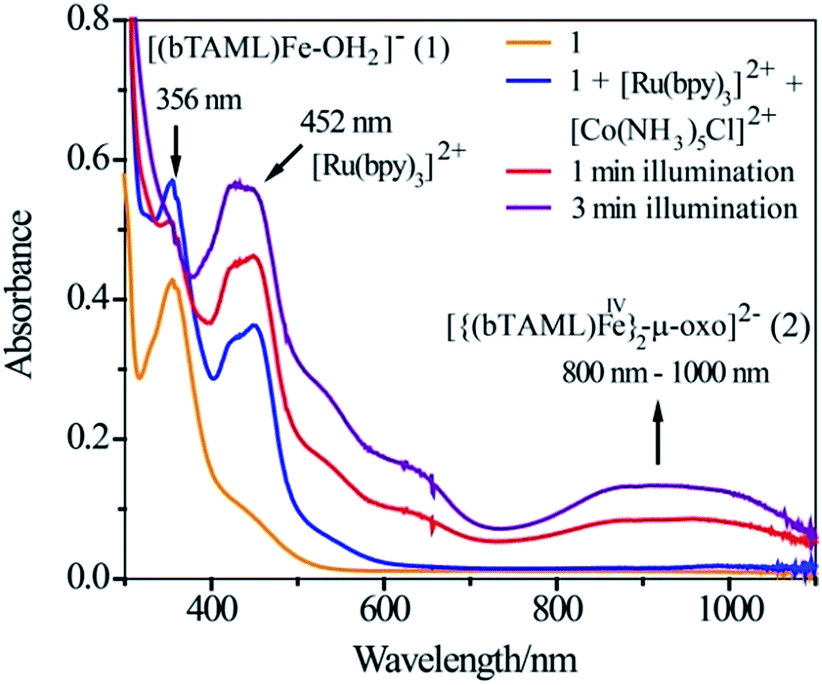
Since the operation of free radical oxidation was ruled out, the involvement of a high-valent iron-oxo intermediate during the reaction was investigated. Upon irradiation of the mixture of 1, Ru2+ and Co3+ in a 3:2 CH3CN–phosphate buffer solution mixture with blue LED light (λmax = 440 nm), a broad absorption band in the region of 800–1000 nm was observed (Fig. 1; violet coloured spectrum). This new species was assigned as the previously characterized dimer, [{(bTAML)FeIV}2(μ-O)]2− (2),15b which is consistent with the UV-vis spectrum of the chemically synthesized dimer 2 (Fig. S5†). This intermediate species was not observed in the absence of any one of the components (catalyst, Ru2+, Co3+ or light). Upon addition of the substrate (alkenes or alkanes) to this solution, 2 reacted with the substrate and regenerated the parental complex 1 (Fig. S6†), which restarted the catalytic cycle upon light irradiation with the concomitant formation of the oxygenated product (alcohol or epoxide).
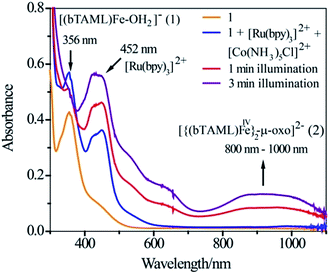 |
| | Fig. 1 UV-vis spectral scan of a photochemical reaction mixture of 1 (1.0 × 10−4 M), [RuII(bpy)3]2+ (2.0 × 10−5 M) and [CoIII(NH3)5Cl]2+ (6.0 × 10−4 M) in acetonitrile-aqueous phosphate buffer (3:2 v/v) solvent. | |
We demonstrated earlier that independently synthesized [RuIII(bpy)3]3+ was competent in oxidizing 1 to form the complex 2.16 We therefore propose that [RuIII(bpy)3]3+, which is generated due to one-electron transfer from the excited state of [RuII(bpy)3]2+ to the sacrificial oxidant [CoIII(NH3)5Cl]2+, oxidizes 1 containing an axial H2O ligand to generate a putative [(bTAML)FeIV–OH]− species by one-electron and one-proton transfer processes (PCET). This proposition is based on previous electrochemical studies reported by us.15c,d Under neutral or basic conditions (pH 10 in this case), the FeIV–OH species dimerizes immediately to form complex 2 as shown below.
| [RuII(bpy)3]2+∗ + [CoIII(NH3)5Cl]2+ → [RuIII(bpy)3]3+ + [CoII(NH3)5Cl]+ |
| [RuIII(bpy)3]3+ + 1 → [(bTAML)FeIV–OH]− + [RuII(bpy)3]2+ |
| [(bTAML)FeIV–OH]− + [(bTAML)FeIV–OH]− → 2 |
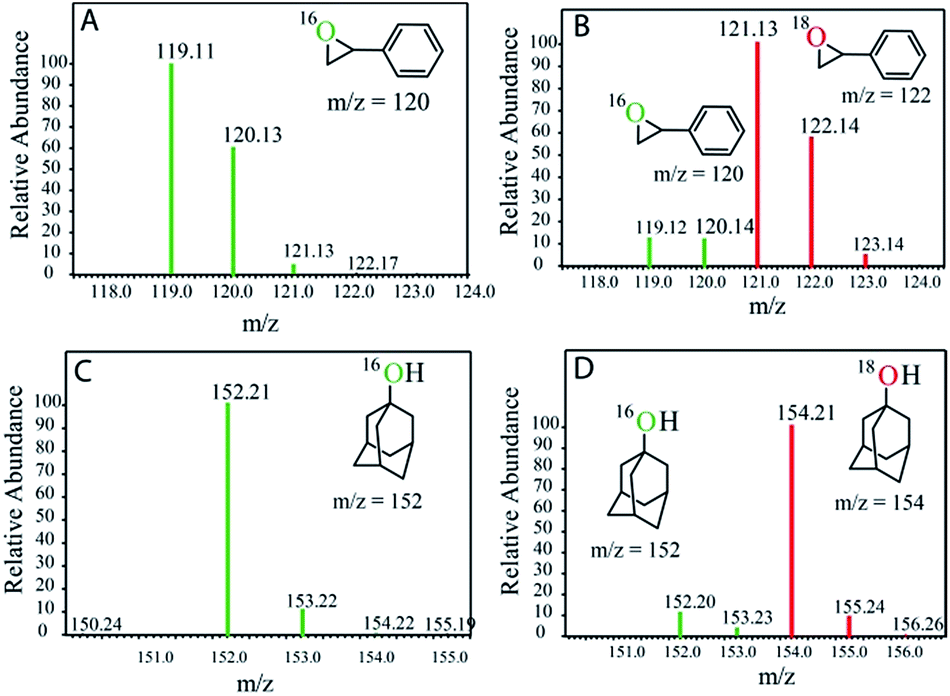
This is in contrast to the [(N4Py)FeII]2+ and [(MePy2tacn)FeII]2+ complexes where the FeIII–OH species is oxidized by [RuIII(bpy)3]3+ to form the corresponding FeIV(O).12,13 The reactivities of the FeIV complexes, i.e. the FeIV(O) and [(μ-O)FeIV2] dimer (2), differ. While the high-valent [(N4Py)FeIV(O)] and [(Me2Pytacn)FeIV(O)] [Me2Pytacn = 1-(2-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane] species are competent in cleaving strong C–H bonds selectively, the low redox potential of the FeIII–OH formed prevents the subsequent “rebound” process, thus leading to free radical auto-oxidation.22 In contrast, our investigations on the reactivity of the [{(bTAML)FeIV}2-μ-oxo]2− (2) with alkanes, alkenes and alcohols demonstrate that the dimer exists in equilibrium with the corresponding FeV(O) and FeIII (1). Such a proposal is based on our previously reported kinetic and mass spectral studies with 2.17b Upon addition of the substrate, the dimer (2) disproportionates into FeV(O) and FeIII (1), and the FeV(O) intermediate remains the active oxidant. The primary formation of the cis-hydroxylated product (Table 1; entry 3) in reactions with cis-1,2-dimethylcyclohexane also supports our reported mechanism that involves C–H bond abstraction by FeV(O) and the subsequent formation of a hydroxylated product by a “rebound” mechanism.15b H218O labelling experiments with styrene and adamantane result in the formation of more than 90% 18O-labelled epoxide and hydroxylated product, respectively, which clearly indicates that water is the primary oxygen atom source (Fig. 2).
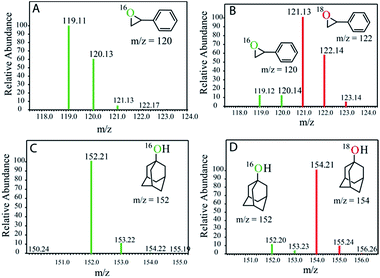 |
| | Fig. 2 GC-MS spectra of the products after photochemical reaction with (A) styrene in H216O, (B) styrene in H218O, (C) adamantane in H216O, and (D) adamantane in H218O. | |
Proposed photocatalytic reaction cycle
Finally, on the basis of product analysis, UV-vis spectroscopy, 18O-labelling experiments, mass analysis and previously reported observations, we propose the following catalytic cycle for the photochemical oxygenation reaction (Scheme 2). In water, FeIII-bTAML (1), upon irradiation with light (3 W blue LED, 440 nm) in the presence of a photosensitizer [RuII(bpy)3]2+ and an electron acceptor [CoIII(NH3)5Cl]2+, gets oxidized to [(bTAML)FeIV–OH]− which immediately gets converted into dimer 2. Upon addition of substrate, the dimer disproportionates to FeV(O) and FeIII (1). For the hydroxylation reaction, the mechanism likely involves a C–H bond abstraction followed by a rebound process to generate the corresponding alcohol. For epoxidation reactions, the formation of a radical intermediate upon electrophilic attack of the FeV(O) onto the alkene followed by a fast ring closing step is expected.17b
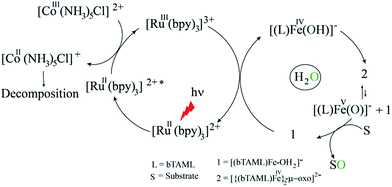 |
| | Scheme 2 Proposed reaction mechanism. | |
Before concluding, one important point is worth noting. Irradiation with visible and UV light has been shown to alter the reactivity of intermediates such as [{(corrole)FeIV}2-μ-oxo], [FeIV(O)(MePy2tacn)]2+ and cofacial bis-porphyrin-diiron(III)-μ-oxo complexes as has been reported by Newcomb et al.,21b Lloret-Fillol et al.13 and Nocera et al.21a, respectively. For the FeIV intermediates, light was shown to disproportionate [{(corrole)FeIV}2-μ-oxo] to form the corresponding [(corrole)FeV(O)], hence increasing its reactivity. For the corresponding [FeIV(O)(MePy2tacn)]2+, the increase in reactivity was explained by the formation of an excited state via spin change. The possibility of rate enhancement in the reactivity of 2 due to the presence of light clearly exists and is currently being investigated.
Conclusions
In conclusion, we have demonstrated the first example of an iron-complex catalysed photocatalytic hydroxylation and epoxidation reaction, with a variety of substrates, using water as the oxygen source. The [{(bTAML)FeIV}2(μ-O)]2− (2) dimer, produced by oxidative activation of a water molecule, remains the active oxidant and results in hydroxylation or epoxidation with high selectivity. 18O-labelling experiments support the metal-based selective and controlled oxygenation of substrates with C–H and CC bonds. Although the reactivity of this photochemical system is lower compared to that of the FeIII-bTAML/NaOCl system and the use of [Co(NH3)5Cl]2+ as the electron acceptor is not optimal, we believe that conjugating complex 1 to a light harvesting system can increase its efficiency manyfold. Such work is being attempted currently in our laboratory.
Experimental section
General procedure for the photocatalytic hydroxylation reactions
The photocatalytic oxidation of alkanes was carried out under an argon atmosphere in an acetonitrile and phosphate buffer (pH 10, 10 mM) (3:2 v/v) mixed solvent. The reaction solution (1.0 mL) containing catalyst 1 (1.0 × 10−4 M) [Ru(bpy)3]Cl2·6H2O (2.0 × 10−3 M), [Co(NH3)5Cl]Cl2 (2.0 × 10−2 M) and substrate (3.0 × 10−3 M) was irradiated with a blue LED light source (3 W, 440 nm) and stirred for 40 min at room temperature. The temperature was kept constant using a water circulating system during the whole reaction. The final reaction mixture was extracted with dichloromethane (five times with 2 mL of dichloromethane each time) and dried over Na2SO4. After concentrating the reaction solution by purging with nitrogen gas, the product was identified and quantified by GC-MS. Control experiments were performed under the same conditions as mentioned above.
General procedure for the photocatalytic epoxidation reactions
The photocatalytic epoxidation of alkenes was carried out in an acetonitrile and phosphate buffer (pH 10, 10 mM) (3:2 v/v) mixed solvent. The reaction solution (1.0 mL) containing catalyst 1 (1.0 × 10−4 M), [Ru(bpy)3]Cl2·6H2O (2.0 × 10−3 M), [Co(NH3)5Cl]Cl2 (2.0 × 10−2 M) and substrate (5.0 × 10−3 M) was irradiated with a blue LED light source (3 W, 440 nm) and stirred for 40 min at room temperature (27 °C). The temperature was kept constant using a water circulating system during the whole reaction. After 40 min, the reaction mixture was extracted with dichloromethane (five times with 2 mL of dichloromethane each time) and dried over Na2SO4. After concentrating the reaction solution by purging with nitrogen gas, the product was identified and quantified using GC-MS.
18O-labelling experiment
18O-labelling experiments were carried out for the photochemical oxidation of styrene, adamantane, cis-1,2-dimethylcyclohexane and ambroxide. A mixture of acetonitrile and H218O (3:2 v/v) solution containing catalyst 1 (1.0 × 10−4 M), [Ru(bpy)3]Cl2·6H2O (2.0 × 10−3 M), [Co(NH3)5Cl]Cl2 (2.0 × 10−2 M) and substrates (styrene, adamantane, cis-1,2-dimethylcyclohexane and ambroxide) (5.0 × 10−3 M) was stirred and irradiated with light (3 W blue LED, 440 nm) for 40 min at room temperature. The resulting solution was extracted with dichloromethane and the products were analyzed by GC-MS.
UV-vis experiment
The photochemical generation of [{(bTAML)FeIV}2-μ-oxo]2− (2) was observed by UV-vis spectroscopy. A solution mixture of acetonitrile and phosphate buffer (3:2 v/v) containing catalyst (1.0 × 10−4 M), [Ru(bpy)3]Cl2·6H2O (2.0 × 10−5 M) and [Co(NH3)5Cl]Cl2 (6.0 × 10−4 M) was added to a 1.0 cm (path length) quartz cuvette and spectra were recorded at 0, 1 and 3 min of photoirradiation with a blue LED light source (3 W, 440 nm). The chemical formation of [{(bTAML)FeIV}2-μ-oxo]2− (2) was also examined from changes in the absorption spectra of the solution mixture of acetonitrile and phosphate buffer (3:2 v/v) containing catalyst (1) and NaOCl (0.5 equivalent) (Fig. S5†).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. S. G. acknowledges SERB, New Delhi (grant no. EMR/2014/00016) and IISER-Kolkata (“Start-up Grant”) for funding. K. K. S. and B. C. acknowledge UGC-New Delhi for fellowship.
Notes and references
-
(a) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed;
(b) A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hamarström, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
-
(a) B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS PubMed;
(b) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed;
(c) J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
-
(a) K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed;
(b) J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821–825 CrossRef CAS PubMed.
-
(a) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed;
(b) M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed;
(c) C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger Jr, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed;
(d) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed;
(e) L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed;
(f) L. Shu, J. C. Nesheim, K. Kauffmann, E. Munck, J. D. Lipscomb and L. Que Jr, Science, 1997, 275, 515–518 CrossRef CAS PubMed;
(g) M. Costas, J.-U. Rohde, A. Stubna, R. Y. N. Ho, L. Quaroni, E. Munck and L. Que Jr, J. Am. Chem. Soc., 2001, 123, 12931–12932 CrossRef CAS PubMed.
- H. Inoue, M. Sumitani, A. Sekita and M. Hida, J. Chem. Soc., Chem. Commun., 1987, 1681–1682 RSC.
-
(a) D. W. Low, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 1996, 118, 117–120 CrossRef CAS;
(b) J. Berglund, T. Pascher, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 1997, 119, 2464–2469 CrossRef CAS.
-
(a) S. Funyu, T. Isobe, S. Takagi, D. A. Tryk and H. Inoue, J. Am. Chem. Soc., 2003, 125, 5734–5740 CrossRef CAS PubMed;
(b) F. Li, M. Yu, Y. Jiang, F. Huang, Y. Li, B. Zhanga and L. Sun, Chem. Commun., 2011, 47, 8949–8951 RSC;
(c) D. Kalita, B. Radaram, B. Brooks, P. P. Kannam and X. Zhao, ChemCatChem, 2011, 3, 571–573 CrossRef CAS;
(d) P. Guillo, O. Hamelin, P. Batat, G. Jonusauskas, N. D. McClenaghan and S. Ménage, Inorg. Chem., 2012, 51, 2222–2230 CrossRef CAS PubMed;
(e) D. Chao and W. F. Fu, Chem. Commun., 2013, 49, 3872–3874 RSC;
(f) T. V. Pho, M. V. Sheridan, Z. A. Morseth, B. D. Sherman, T. J. Meyer, J. M. Papanikolas, K. S. Schanze and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2016, 8, 9125–9133 CrossRef CAS PubMed;
(g) L. Bai, F. Li, Y. Wang, H. Li, X. Jianga and L. Sun, Chem. Commun., 2016, 52, 9711–9714 RSC.
- S. Ohzu, T. Ishizuka, Y. Hirai, S. Fukuzumi and T. Kojima, Chem.–Eur. J., 2013, 19, 1563–1567 CrossRef CAS PubMed.
-
(a) S. Fukuzumi, T. Kishi, H. Kotani, Y.-M. Lee and W. Nam, Nat. Chem., 2011, 3, 38–41 CrossRef CAS PubMed;
(b) S. Fukuzumi, T. Mizuno and T. Ojiri, Chem.–Eur. J., 2012, 18, 15794–15804 CrossRef CAS PubMed.
-
(a) X. Wu, X. Yang, Y. M. Lee, W. Nam and L. Sun, Chem. Commun., 2015, 51, 4013–4016 RSC;
(b) D. Shen, C. Saracini, Y.-M. Lee, W. Sun, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 15857–15860 CrossRef CAS PubMed;
(c) G. Chen, L. Chen, L. Ma, H.-K. Kwong and T.-C. Lau, Chem. Commun., 2016, 52, 9271–9274 RSC.
-
(a) A. C. Rosenzweig, C. A. Frederick, S. J. Lippard and P. Nordlund, Nature, 1993, 336, 537–543 CrossRef PubMed;
(b) R. L. Lieberman and A. C. Rosenzweig, Nature, 2005, 434, 177–182 CrossRef CAS PubMed;
(c) I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed;
(d) J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- H. Kotani, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 3249–3251 CrossRef CAS PubMed.
- A. Company, G. Sabenya, M. González-Béjar, L. Gómez, M. Clémancey, G. Blondin, A. J. Jasniewski, M. Puri, W. R. Browne, J. Latour, L. Que Jr, M. Costas, J. Pérez-Prieto and J. Lloret-Fillol, J. Am. Chem. Soc., 2014, 136, 4624–4633 CrossRef CAS PubMed.
-
(a) S. Sen Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K.-W. Schraman and T. J. Collins, Science, 2002, 296, 326–328 CrossRef CAS PubMed;
(b) T. J. Collins, Acc. Chem. Res., 2002, 35, 782–790 CrossRef CAS PubMed;
(c) T. J. Collins, Acc. Chem. Res., 1994, 27, 279–285 CrossRef CAS;
(d) B. C. Ellis, C. T. Tran, M. A. Denardo, A. Fischer, A. D. Ryabov and T. J. Collins, J. Am. Chem. Soc., 2009, 131, 18052–18053 CrossRef PubMed;
(e) C. Panda, M. Ghosh, T. Panda, R. Banerjee and S. Sen Gupta, Chem. Commun., 2011, 47, 8016–8018 RSC.
-
(a) F. T. de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que Jr, E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835–838 CrossRef PubMed;
(b) M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. Sen Gupta, J. Am. Chem. Soc., 2014, 136, 9524–9527 CrossRef CAS PubMed;
(c) S. Pattanayak, A. Rana, A. J. Jasniewski, K. K. Singh, A. Weitz, M. Hendrich, L. Que Jr, A. Dey and S. Sen Gupta, Inorg. Chem., 2017, 56, 6352–6361 CrossRef CAS PubMed;
(d) S. Pattanayak, D. R. Chowduri, B. Garai, K. K. Singh, A. Paul, B. B. Dhar and S. Sen Gupta, Chem.–Eur. J., 2017, 23, 3414–3424 CrossRef CAS PubMed.
- C. Panda, J. Debgupta, D. Diaz Diaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS PubMed.
-
(a) K. K. Singh, M. k. Tiwari, M. Ghosh, C. Panda, A. Weitz, M. P. Hendrich, B. B. Dhar, K. Vanka and S. Sen Gupta, Inorg. Chem., 2015, 54, 1535–1542 CrossRef CAS PubMed;
(b) K. K. Singh, M. k. Tiwari, B. B. Dhar, K. Vanka and S. Sen Gupta, Inorg. Chem., 2015, 54, 6112–6121 CrossRef CAS PubMed.
- M. Ghosh, Y. L. K. Nikhil, B. B. Dhar and S. Sen Gupta, Inorg. Chem., 2015, 54, 11792–11798 CrossRef CAS PubMed.
- S. Jana, M. Ghosh, M. Ambule and S. Sen Gupta, Org. Lett., 2017, 19, 746–749 CrossRef CAS PubMed.
- K. Chen, A. Eschenmoser and P. S. Baran, Angew. Chem., Int. Ed., 2009, 48, 9705–9708 CrossRef CAS PubMed.
-
(a) J. Rosenthal, J. Bachman, J. L. Dempsey, A. J. Esswein, T. G. Gray, J. M. Hodgkiss, D. R. Manke, T. D. Luckett, B. J. Pistorio, A. S. Veige and D. G. Nocera, Coord. Chem. Rev., 2005, 249, 1316–1326 CrossRef CAS;
(b) D. N. Harischandra, G. Lowery, R. Zhang and M. Newcomb, Org. Lett., 2009, 11, 2089–2092 CrossRef CAS PubMed.
- K.-B. Cho, H. Hirao, S. Shaik and W. Nam, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, general instrumentation and additional figures. See DOI: 10.1039/c7sc02780j |
| ‡ Both authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*