
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNb2©Au6: a molecular wheel with a short Nb![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) Nb triple bond coordinated by an Au6 ring and reinforced by σ aromaticity†
Nb triple bond coordinated by an Au6 ring and reinforced by σ aromaticity†
Tian
Jian
,
Ling Fung
Cheung
 ,
Joseph
Czekner
,
Teng-Teng
Chen
,
Gary V.
Lopez
,
Wei-Li
Li
and
Lai-Sheng
Wang
*
,
Joseph
Czekner
,
Teng-Teng
Chen
,
Gary V.
Lopez
,
Wei-Li
Li
and
Lai-Sheng
Wang
*
Department of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: Lai-Sheng_Wang@brown.edu
First published on 15th September 2017
Abstract
We report a photoelectron spectroscopy and high-resolution photoelectron imaging study of a bimetallic Nb2Au6− cluster. Theoretical calculations, in conjunction with the experimental data, reveal that Nb2Au6−/0 possess high-symmetry D6h structures featuring a Nb–Nb axis coordinated equatorially by an Au6 ring. Chemical bonding analyses show that there are two π bonds and one σ bond in the Nb2 moiety in Nb2©Au6, as well as five totally delocalized σ bonds. The Nb![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Nb triple bond is strengthened significantly by the delocalized σ bonds, resulting in an extremely short Nb–Nb bond length comparable to the quintuple bond in gaseous Nb2. The totally delocalized σ bonding in Nb2©Au6 is reminiscent of σ aromaticity, representing a new bonding mode in metal–ligand systems. The unusually short Nb–Nb bond length in Nb2©Au6 shows that the Au6 ring can serve as a bridging ligand to facilitate multiple bonding in transition metal dimers via delocalized σ bonding.
Nb triple bond is strengthened significantly by the delocalized σ bonds, resulting in an extremely short Nb–Nb bond length comparable to the quintuple bond in gaseous Nb2. The totally delocalized σ bonding in Nb2©Au6 is reminiscent of σ aromaticity, representing a new bonding mode in metal–ligand systems. The unusually short Nb–Nb bond length in Nb2©Au6 shows that the Au6 ring can serve as a bridging ligand to facilitate multiple bonding in transition metal dimers via delocalized σ bonding.
1. Introduction
Due to the strong relativistic effects,1 negatively charged gold clusters can be planar up to a surprisingly large size.2 The Au6 cluster is very special with a huge energy gap between its highest occupied (HOMO) and lowest unoccupied molecular orbitals (LUMO), as revealed in the photoelectron spectra of Au6−.2c,3 On the basis of vibrationally-resolved photodetachment spectroscopy, Au6 was initially deduced to have a D6h ring structure,3b but was later confirmed to have a D3h triangular structure.2 The high electronic stability of Au6 is due to σ aromaticity from its delocalized σ electrons,4 as is also found in the isoelectronic Au5Zn+ cluster.5 Photoelectron spectroscopy (PES) of MAu6− (M = Ti, V, Cr) along with density functional theory (DFT) calculations found these doped-clusters to have planar structures featuring an Au6 ring with a central transition metal atom (M©Au6−) with variable magnetic moments.6 It was shown that, while the out-of-plane 3d orbitals of the transition-metal atoms do not participate in bonding with Au, the in-plane dxy and dx2−y2 orbitals are important to maintain the disk structure due to bonding interactions with the Au6 ring. Subsequent DFT calculations found that Au6 doped with 3d transition metals all have similar M©Au6 disk structures for M = Sc–Ni,7 whose electronic structures were also rationalized using an electron shell model.8 Thus, the Au6 ring provides a good host for a central transition metal atom with tunable magnetic properties.In the current study, we report the observation of Nb2©Au6−/0 molecular wheels, in which an Au6 ring is hosting a NbNb triple bond equatorially. The first report in 1965 by Cotton et al. of K2[Re2Cl8]·2H2O with a quadruple bond (σ2π4δ2) between the two Re atoms opened the field of metal–metal multiple bonds.9 The σ, π, and δ bonds between two transition metal atoms (M2) are mainly formed by the overlap of their d orbitals with the same symmetry. Employing bulky or bridging ligands, Cr2 complexes containing a quintuple bond (σ2π4δ4) have also been successfully synthesized.10 The quintuple bonded complexes synthesized so far are mainly related to group VI metals.11 Although gaseous Nb2 is characterized to possess a quintuple Nb–Nb bond with a 1π4u1σ2g2σ2g1δ2g electron configuration,12 synthesized diniobium complexes usually contain a Nb24+ core and maintain a Nb–Nb triple bond at the most.9b,13 To surpass the Nb–Nb triple bond, a Nb2 complex needs to free more d electrons from metal–ligand bonding to participate metal–metal δ bonding.
The Nb2Au6− cluster anions were produced by a laser vaporization supersonic cluster source and characterized by a magnetic-bottle PES analyzer,14 as well as high-resolution velocity-map imaging (VMI).15 Global minimum searches along with high-level quantum calculations reveal that the D6h Nb2Au6−/0 molecular wheels are the most stable structures. Chemical bonding analyses show that there are one σ bond and two π bonds in the Nb2 moiety, and five totally delocalized σ bonds in Nb2Au6. An extremely short Nb–Nb bond length is observed, showing that the Au6 ring can serve as a bridging ligand to facilitate multiple bonding in Nb2via delocalized σ bonding.
2. Experimental methods
The Nb2Au6− cluster was studied using two different PES apparatuses. The experiment was first carried out using a magnetic-bottle PES apparatus equipped with a laser vaporization supersonic cluster source, details of which has been published elsewhere.14 The second harmonic radiation (532 nm) from a Nd:YAG laser was used to vaporize a disk target compressed from niobium and gold powders. A helium carrier gas seeded with 5% argon was used to quench the plasma, initiating nucleation and cluster formation. Clusters formed in the nozzle were entrained in the carrier gas and underwent a supersonic expansion. The anionic clusters were extracted from the collimated cluster beam and analyzed by a time-of-flight (TOF) mass spectrometer. The Nb2Au6− anion of interest was mass-selected, decelerated and photodetached by a laser beam at 193 nm from an ArF excimer laser, and 266 nm from a Nd:YAG laser. Photoelectrons were collected at nearly 100% efficiency and analyzed in a 3.5 meter long electron flight tube. The photoelectron spectra were calibrated by the known spectra of Au− or Bi−. The energy resolution of the apparatus was ΔEk/Ek ≈ 2.5%, that is, ∼25 meV for 1 eV electrons.High-resolution PES experiment was then performed using a VMI apparatus, which has been previously described in detail.15 The cluster source and TOF mass spectrometer are similar to the magnetic-bottle PES apparatus. The Nb2Au6− cluster was mass selected before entering the imaging detector. A laser beam from a 355 nm pumped Deyang dye laser was used to detach electrons from the cluster anions. A voltage of −300 V was used on the repeller of the imaging lens.15 The electrons were focused onto a microchannel plate (MCP) coupled with a phosphorus screen and charged-coupled device (CCD) camera for detection. The experiments required between 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 200000 laser shots to achieve a satisfactory signal-to-noise ratio. Images were analyzed with the maximum entropy concept using the MEVELER program.16 The detector was calibrated using the well-known detachment energy of Au− at various photon energies.
000 and 200000 laser shots to achieve a satisfactory signal-to-noise ratio. Images were analyzed with the maximum entropy concept using the MEVELER program.16 The detector was calibrated using the well-known detachment energy of Au− at various photon energies.
3. Theoretical methods
Global minimum searches were carried out for Nb2Au6− using the simulated annealing algorithm17 coupled with density functional theory (DFT) geometric optimization. The PW91 (ref. 18) functional with the Los Alamos basis set and effective core potential (LANL2DZ)19 on both Au and Nb atom was used in the initial structure searches. Low-lying isomers from the initial searches were refined at PW91 and PBE0 (ref. 20) levels of theory using the Def2-TZVPPD basis set21 and the Stuttgart–Dresden (SDD) relativistic pseudopotentials (ECP28MDF for Nb and ECP60MDF for Au).22 Vibrational frequencies of the low-lying isomers were calculated to ensure they are true energy minima. All the DFT calculations were carried out using Gaussian 09.23 Single-point energies of the five lowest isomers were calculated using the coupled-cluster theory with single, double and perturbative triple excitations [CCSD(T)]24 implemented in Molpro 2015.1.25The first vertical detachment energy (VDE) was computed as the energy difference between the neutral and the anion at the optimized geometry of the anion. The first adiabatic detachment energy (ADE) was computed as the energy difference between the optimized anion and the neutral. Further VDEs were approximated by adding excitation energies from the time-dependent density functional theory (TDDFT)26 calculations to the first vertical detachment energy. The simulated spectrum was obtained by fitting a unit-area Gaussian function of 0.03 eV width to each VDE. The ezSpectrum program was employed to calculate Franck–Condon factors, which were fitted with a unit-area Gaussian function of 2 meV width (FWHM) to simulate the vibrational features.27 Natural population analyses (NPA) were carried out to illustrate the charge distribution on the neutral Nb2Au6 cluster.28 A charge decomposition analysis (CDA)29 was done using the Multiwfn program30 to describe the interaction between the Au6 ring and the Nb2 axis. Chemical bonding analyses were done using the adaptive natural density partitioning (AdNDP)31 method at the PBE0/Def2-TZVP level of theory. The structural and chemical bonding pictures were visualized by GaussView32 and Molekel.33
4. Experimental results
Fig. 1 displays the photoelectron spectra of Nb2Au6− at 266 nm and 193 nm from the magnetic-bottle PES apparatus, which are compared with the simulated spectrum. The PES bands are labeled as X, A, B… which denote detachment transitions from the ground electronic state of the anion to the ground (X) and excited electronic states (A, B…) of neutral Nb2Au6. The VDEs of the electronic transitions are measured from the peak maxima and summarized in Table 1, where they are compared with theoretical calculations. The high-resolution spectra shown in Fig. 2 and S1† are taken from the VMI apparatus. The peaks labeled in lower case letters (a, b, c…) represent the vibrational levels resolved for the ground electronic state of neutral Nb2Au6. The binding energies and the corresponding assignments for the vibrational features are summarized in Table 2.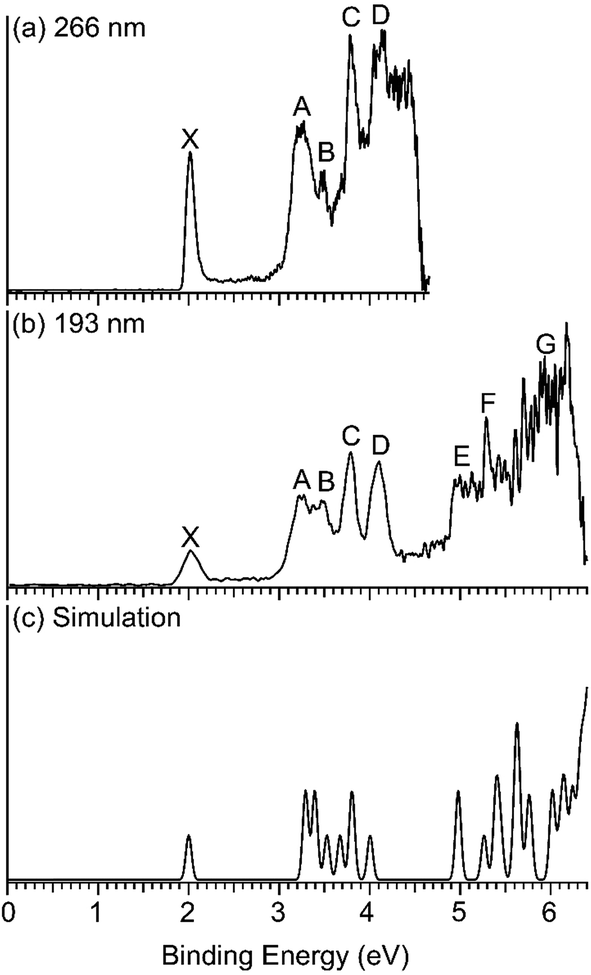 | ||
| Fig. 1 Photoelectron spectra of Nb2Au6− at (a) 266 nm (4.661 eV) and (b) 193 nm (6.424 eV) obtained from the magnetic-bottle PES apparatus, and (c) comparison with the simulated spectrum of the D6h global minimum at PBE0/Def2-TZVPPD. The theoretical first VDE is shifted to align with the experimental first VDE, while the higher theoretical VDEs are shifted accordingly (see Table 1). | ||
| VDE(exp)a | Final states and electronic configurationb | PBE0c | |
|---|---|---|---|
| a Numbers in the parentheses are the uncertainty in the last digit. b The orbitals shown in bold face indicate the major electron detachment channels. c The first VDE was computed to be 1.79 eV (see Table S2), but shifted to align with the experimental value. All higher VDEs are shifted accordingly to better compare with the experimental data. | |||
| X | 1.9969(4) | 1A1g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a21g5e41u2a02u | 2.00 |
| A | 3.25(3) | 3E1g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a21g5e31u2a12u | 3.29 |
| 3A2u⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a11g5e41u2a12u | 3.39 | ||
| B | 3.48(3) | 1E1g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a21g5e31u2a12u | 3.53 |
| C | 3.79(3) | 1A2u⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a11g5e41u2a12u | 3.67 |
| 3E2u⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e32g4a21g5e41u2a12u | 3.80 | ||
| D | 4.11(5) | 1E2u⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e32g4a21g5e41u2a12u | 4.00 |
| E | ∼5.0 | 3E1g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e31u4e42g4a21g5e41u2a12u | 4.98 |
| F | ∼5.3 | 1E1g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e31u4e42g4a21g5e41u2a12u | 5.26 |
| 3A1u⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a12g4e41u4e42g4a21g5e41u2a12u | 5.40 | ||
| 1A1u⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a12g4e41u4e42g4a21g5e41u2a12u | 5.45 | ||
| 3B2g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b11u1a22g4e41u4e42g4a21g5e41u2a12u | 5.61 | ||
| 3A2g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a11u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 5.64 | ||
| 1A2g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a11u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 5.75 | ||
| 1B2g⋯2e42u3e41u3e42g1b21u1b22g2e41g1a21u2b11u1a22g4e41u4e42g4a21g5e41u2a12u | 5.77 | ||
| G | ∼5.9 | 3E1u⋯2e42u3e41u3e42g1b21u1b22g2e31g1a21u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 6.02 |
| 1E1u⋯2e42u3e41u3e42g1b21u1b22g2e31g1a21u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 6.10 | ||
| 3E2u⋯2e42u3e41u3e32g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 6.15 | ||
| 3B1u⋯2e42u3e41u3e42g1b21u1b12g2e41g1a21u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 6.24 | ||
| 3B2g⋯2e42u3e41u3e42g1b11u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 6.32 | ||
| 3E2g⋯2e32u3e41u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 6.36 | ||
| 3E1g⋯2e42u3e31u3e42g1b21u1b22g2e41g1a21u2b21u1a22g4e41u4e42g4a21g5e41u2a12u | 6.40 | ||
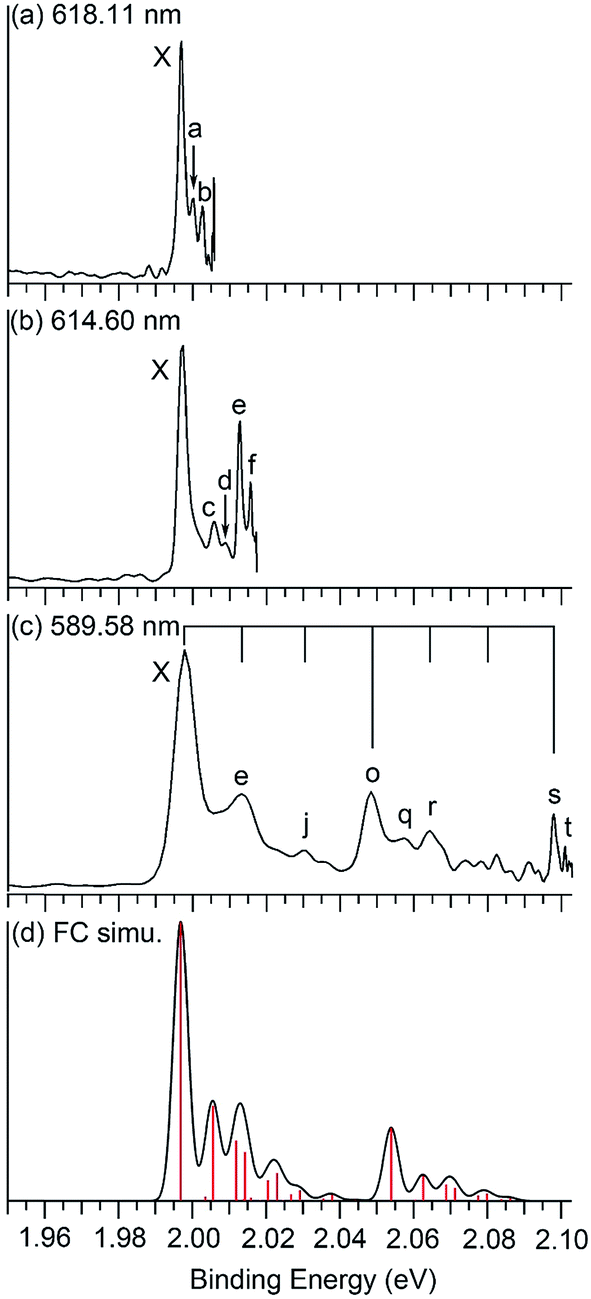 | ||
| Fig. 2 Photoelectron spectra of Nb2Au6− at (a) 618.11 nm (2.0059 eV), (b) 614.60 nm (2.0173 eV), and (c) 589.58 nm (2.1029 eV) obtained from the VMI PES apparatus, and (d) the Franck–Condon simulation. The black vertical lines in (c) indicate the vibrational progressions of the two totally symmetric modes, ν8 (short lines) and ν12 (long lines) (see Fig. 4 and Table 2). The red vertical lines in (d) represent the calculated Franck–Condon factors. | ||
| Peak | BE (eV) | ΔE (cm−1) | Assignment | Symmetry | Calculateda |
|---|---|---|---|---|---|
| a The values without and with parentheses are from the PW91/Def2-TZVPPD and PBE0/Def2-TZVPPD calculations, respectively. | |||||
| X | 1.9969 ± 0.0004 | 0.0 | |||
| a | 2.0001 ± 0.0006 | 26 ± 5 | ν 1 | E2u | 24 (26) |
| b | 2.0027 ± 0.0003 | 47 ± 2 | ν 3 | E1g | 48 (68) |
| c | 2.0067 ± 0.0003 | 79 ± 2 | 2ν 2 | A 1g | 64 (70) |
| d | 2.0092 ± 0.0002 | 99 ± 2 | ν 7 | E1u | 81 (83) |
| e | 2.0128 ± 0.0003 | 128 ± 2 | ν 8 | A 1g | 121 (121) |
| f | 2.0157 ± 0.0002 | 152 ± 2 | 4ν 2 | A 1g | 128 (140) |
| g | 2.0190 ± 0.0005 | 178 ± 3 | ν 8 + ν3 | E1g | 169 (189) |
| h | 2.0215 ± 0.0013 | 198 ± 5 | ν 8 + 2ν 2 | A 1g | 185 (191) |
| i | 2.0250 ± 0.0008 | 227 ± 3 | 6ν 2 | A 1g | 192 (210) |
| j | 2.0285 ± 0.0004 | 255 ± 2 | 2ν 8 | A 1g | 242 (242) |
| k | 2.0349 ± 0.0003 | 306 ± 2 | ν 8 + 4ν 2 | A 1g | 249 (261) |
| l | 2.0375 ± 0.0004 | 328 ± 2 | 2ν 8 + 2ν 2 | A 1g | 306 (312) |
| m | 2.0391 ± 0.0004 | 340 ± 2 | ν 8 + 6ν 2 | A 1g | 313 (331) |
| n | 2.0444 ± 0.0004 | 383 ± 2 | 2ν 8 + 4ν 2 | A 1g | 370 (382) |
| o | 2.0475 ± 0.0003 | 408 ± 2 | ν 12 | A 1g | 423 (460) |
| p | 2.0509 ± 0.0002 | 436 ± 2 | ν 12 + ν1 | E2u | 447 (486) |
| q | 2.0575 ± 0.0014 | 489 ± 6 | ν 12 + 2ν 2 | A 1g | 487 (530) |
| r | 2.0643 ± 0.0011 | 544 ± 5 | ν 12 + ν 8 | A 1g | 544 (581) |
| s | 2.0979 ± 0.0005 | 815 ± 3 | 2ν 12 | A 1g | 846 (920) |
| t | 2.1010 ± 0.0002 | 840 ± 2 | 2ν12 + ν1 | E2u | 870 (946) |
The 266 nm spectrum (Fig. 1a) displays a sharp ground-state band X around 2.0 eV. Following a large energy gap of ∼1.25 eV, a relatively broad band A at 3.25 eV is observed, as well as three more bands (B, C, D) at VDEs of 3.48, 3.79, and 4.11 eV, respectively. Following another energy gap after band D, more congested features are observed above 5 eV in the 193 nm spectrum (Fig. 1b). The labels E, F, and G are given tentatively for the sake of discussion. The large gap between the X and A bands suggests that neutral Nb2Au6 is closed shell with a large HOMO–LUMO gap, indication of a very stable electronic system. A series of high-resolution spectra were obtained using the VMI PES apparatus to resolve vibrational structures for the ground-state transition. Three are shown in Fig. 2 and three more are given in Fig. S1.† The 589.58 nm spectrum (Fig. 2c) yields two main vibrational progressions with frequencies of 408 and 128 cm−1. The short vibrational progressions suggest a minimal geometry change between the anionic and neutral ground states and high symmetries for Nb2Au6−/0. More weak vibrational features are resolved in the spectra at even lower photon energies (Fig. 2a and b). The 618.11 nm spectrum (Fig. 2a) defines the most accurate 0–0 transition at 1.9969 ± 0.0004 eV, which represents the first VDE and ADE, as well as the electron affinity (EA) of neutral Nb2Au6. The peak d resolved in Fig. S1a† has a line width of 2 cm−1, which is the narrowest peak resolved in PE imaging for any molecular systems.34
5. Theoretical results
The optimized low-lying isomers of Nb2Au6− within 1.5 eV of the global minimum are given in Fig. S2.† All the low-lying isomers were found to have the six Au atoms arranged around a Nb2 moiety. Structures with two separated Nb atoms were all much higher in energy. At the PW91/Def2-TZVPPD level, the top five isomers are within 0.1 eV of each other with isomer II (D3d, 2A2u) being the lowest in energy. Isomer I (D6h, 2A2u) is a close-lying isomer, but shows a small imaginary frequency (16.5i cm−1) at PW91/Def2-TZVPPD level. Distortion along this mode of imaginary frequency led to the D3d isomer II. At the PBE0/Def2-TZVPPD level, isomer I was found to be the most stable structure, while isomers II and III optimized to isomer I. The energy difference between isomers I and II was also calculated using different density functionals, which are summarized in Table S1.† Isomer II was calculated to be slightly more stable at PW91, PBE0, BP86, B3PB6, and TPSS levels, while isomer I was calculated to be more stable at PBE0, TPSSh, and LC-ωPBE levels.35 This discrepancy in relative energies and imaginary frequency suggests that the choice of density functional is important to describe the potential energy surface correctly. To establish a better relative energy order, we calculated single-point energies at the CCSD/Def2-TZVPPD and CCSD(T)/Def2-TZVPPD levels for isomers I–V at the PW91/Def2-TZVPPD structures. At the CCSD and CCSD(T) levels, we found isomer I was most stable, while the second lowest-lying isomer II was 0.029 eV and 0.066 eV higher in energy, respectively. The imaginary frequency of isomer I at PW91/Def2-TZVPPD level is likely to be a result of computational artifact. The optimized neutral structures are shown in Fig. S3,† where the D6h structure is found to be appreciably more stable at both the PW91/Def2-TZVPPD and PBE0/Def2-TZVPPD levels of theory. The optimized structures of Nb2Au6− and Nb2Au6 are nearly identical with only minor bond length changes, as shown in Fig. 3.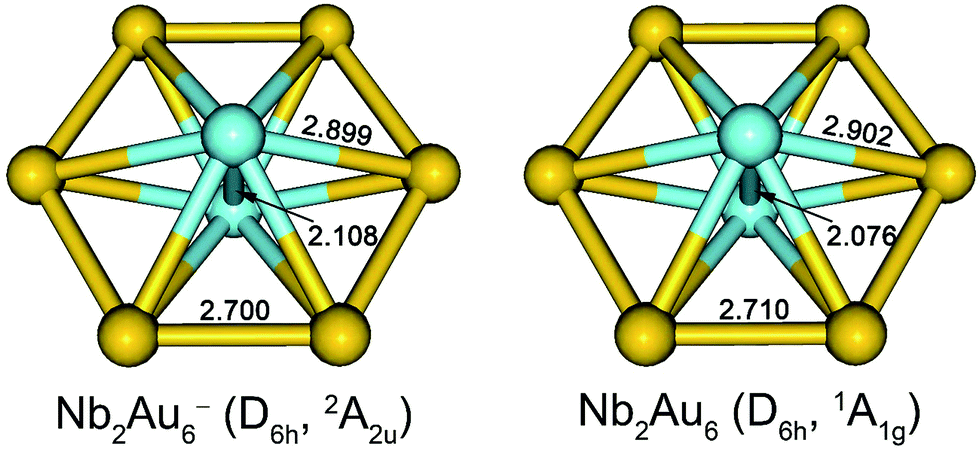 | ||
| Fig. 3 The optimized structures for the global minimum D6h Nb2Au6− and Nb2Au6 at the PBE0/Def2-TZVPPD level. Bond lengths are given in Å. | ||
6. Comparison between experiment and theory
To compare with the experimental data, we computed the first ADE and VDE of isomers I–V at the PW91 and PBE0 levels, as given in Table S2.† It was shown previously that the PW91 functional was excellent in predicting the first ADE and VDE for the M©Au6− clusters.6a Indeed, we found that the calculated first ADE (2.01 eV) and VDE (2.02 eV) of isomer I at PW91 are in good agreement with the experimental ADE and VDE of 1.9969 eV, whereas the first VDEs computed for isomers II–V at PW91 are much larger. Furthermore, the large differences between the first ADE and VDE predicted for isomers II–V at PW91 are also inconsistent with the observed sharp band X. The first calculated VDE (1.79 eV) and ADE (1.77 eV) at the PBE0 level for isomer I are slightly lower than the experimental values. These results are consistent with previous observations that the PBE0 functional tends to underestimate the first VDEs of gold clusters by 0.1–0.3 eV.36 Since isomers II–IV are not stable at the PBE0 level (Fig. S2†), their first VDE and ADE could not be computed at this level of theory. The calculated first ADE and VDE of isomer V are much larger than the experimental value at the PBE0 level. At both PW91 and PBE0 levels, only the calculated fist VDE and ADE of isomer I are consistent with the observed band X.We further computed the vibrational frequencies for the neutral D6h global minimum at the PW91 and PBE0 levels, as given in Table S3.† There are only two totally symmetric fundamental modes (Fig. 4) for the D6h structure, the ring breathing (ν8: 121 cm−1 at both PW91 and PBE0) and the Nb–Nb stretching (ν12: 423 cm−1 at PW91 and 460 cm−1 at PBE0). To better compare with the vibrationally-resolved spectra in Fig. 2 and S1,† we also calculated the Franck–Condon factors for the detachment transition from the ground state of the D6h anion (2A2u) to that of the D6h neutral (1A1g) using output files at the PBE0 level.27 The Franck–Condon simulated spectrum of isomer I is shown in Fig. 2d. Apart from the two totally symmetric fundamental modes, the totally symmetric overtone of the out-of-plane bending mode (ν2: 32 cm−1 at PW91 and 35 cm−1 at PBE0) also gives a significant Franck–Condon factor. Most of the observed vibrational features can be assigned to the combination/overtone of these three modes (2ν2, ν8, and ν12), as shown in Table 2. Several low frequency modes involving ring distortions are also observed (Fig. 2 and S1†) and the observed frequencies for these weak vibrational features are also in good agreement with the calculated frequencies (Table 2). The Franck–Condon factors of the transition from the ground state of the D3d anion (2A2u) to that of the D6h neutral (1A1g) are negligible, ruling out the possibility of contributions from isomer II to the observed vibrational features. The excellent agreement between the computed first detachment energies and Franck–Condon factors for isomer I and the experimental data confirms firmly the D6h global minimum for both Nb2Au6− and Nb2Au6. The observation of the weak vibrational features involving the ring distortion modes indicate that the Au6 ring is somewhat floppy in the anion.
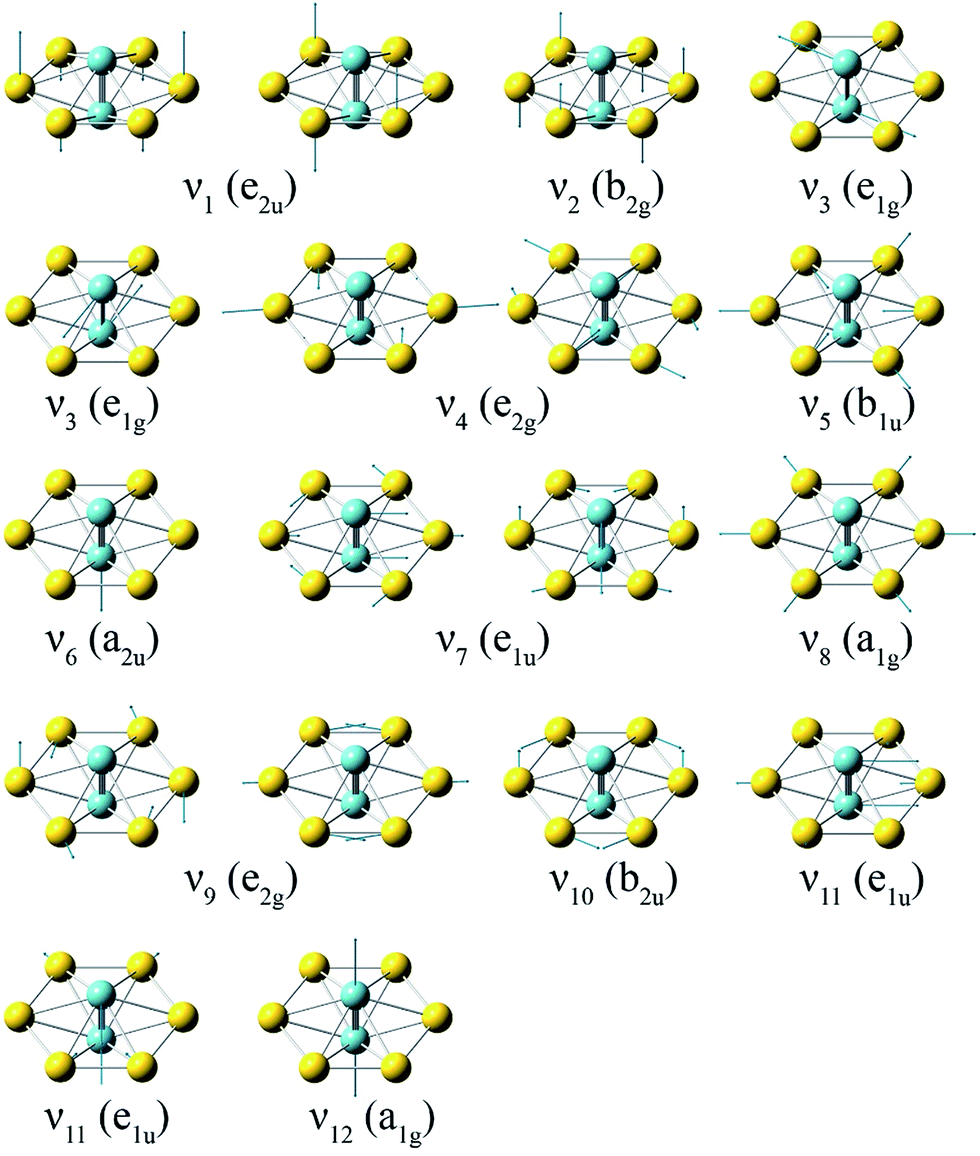 | ||
| Fig. 4 The normal modes and their symmetries for the D6h Nb2Au6 cluster at the PBE0/Def2-TZVPPD level. | ||
The valence canonical molecular orbital (CMO) contours of isomer I are shown in Fig. S4.† The first PES band X corresponds to electron detachment from the singly occupied molecular orbital (SOMO) (a2u), which involves weak antibonding interactions between the two Nb atoms and weak bonding interactions between Nb and Au. Thus, the removal of the SOMO electron should lead to a slight decrease of the Nb–Nb bond length and a slight increase of the Nb–Au bond length in the Nb2Au6 neutral, consistent with the optimized structures (Fig. 3) and the Franck–Condon activities observed for the Au6 ring breathing and Nb–Nb stretching modes (Fig. 2).
To help assign the PE spectra at the high photon energies (Fig. 1), we calculated higher VDEs of the D6h global minimum of Nb2Au6− using TDDFT at the PBE0/Def2-TZVPPD level, which are compared with the experimental VDEs in Table 1 and plotted in Fig. 1c by fitting each VDE with a unit-area Gaussian function of 0.03 eV width. The calculated VDEs from deeper orbitals are consistent with the observed bands at the higher binding energy side. We also performed TDDFT calculations for isomers I and II using different functionals and plotted the simulated spectra in the ESI (Fig. S5–S7†). Overall, only the simulated spectral pattern of isomer I is in good agreement with the experimental PE spectra (Fig. 1), lending further credence to the established D6h global minimum for Nb2Au6−.
7. Discussion
7.1. The extremely short Nb–Nb bond length in Nb2Au6
The optimized structures for the global minima of Nb2Au6− and Nb2Au6 at the PBE0/Def2-TZVPPD level are shown in Fig. 3. Both Nb2Au6− and Nb2Au6 possess D6h symmetry featuring an Au6 ring building around the Nb–Nb axis. The slight bond length variations between Nb2Au6− and Nb2Au6 are consistent with the nature of the SOMO of the anion (or the LUMO of the neutral), as discussed above. Interestingly, the size of the D6h Au6 ring in Nb2Au6− is almost identical to that in the M©Au6− (M = Ti, V, Cr) clusters,6a suggesting the Au6 ring is a robust host to accommodate a dopant from a single atom to a dimer. The Nb–Nb bond lengths in Nb2Au6− and Nb2Au6 are 2.108 Å and 2.076 Å, respectively. These short Nb–Nb bond lengths are similar to the observed bond length of 2.078 Å in gaseous Nb2, which possesses a 3Σg− ground state with a 1π4u1σ2g2σ2g1δ2g electron configuration.12 With two unpaired weakly bonding electrons in the two degenerate δ orbitals, the bare Nb2 dimer can be approximately viewed to possess a quintuple bond. The Nb–Nb bond lengths in Nb2Au6−/0 are much shorter than the Nb–Nb triple bond length (2.32 Å) from Pyykkö's self-consistent covalent radii.37 Indeed, the calculated Mayer bond orders38 between Nb–Nb in Nb2 and Nb2Au6 are 5.07 and 3.61 at the PBE0/Def2-TZVP level, respectively. This implies that the Nb–Nb bond strength in Nb2Au6 is beyond a triple bond. How does the Au6 ring accommodate the Nb2 dimer and facilitate the multiple bonding of Nb2 in Nb2Au6?7.2. Chemical bonding in Nb2Au6
The binding energy between Nb2 and Au6 is calculated to be 8.71 eV at the PBE0/Def2-TZVPPD level based on Nb2Au6 (D6h, 1A1g) → Nb2 (D∞h, 3Σg−) + Au6 (D6h, 1A1g), indicating strong guest–host interactions. The charge distribution on each atom in Nb2Au6 is shown in Table S4.† Although the Au atom is more electronegative than the Nb atom, the charge distribution shows each Nb atom actually draws electron (NPA charge: −0.195|e|; Mulliken charge: −0.457|e|) from the Au atoms in Nb2Au6. Using the results from the CDA analysis, we made an orbital correlation diagram about the interactions between the Nb2 and the Au6 fragments in Nb2Au6, as shown in Fig. 5. The HOMO (e1u) and HOMO-1 (a1g) of Nb2Au6 are mainly from the Nb2 fragment, representing two π bonds and one σ bond formed by the overlap of 4d orbitals. The HOMO-2 (e2g) of Nb2Au6 results from symmetric overlap between the SOMO (1δg) of Nb2 and the LUMO (e2g) of the Au6 ring, providing the key factor to stabilize the ring structure and facilitate the multiple bonding of Nb2 in Nb2Au6.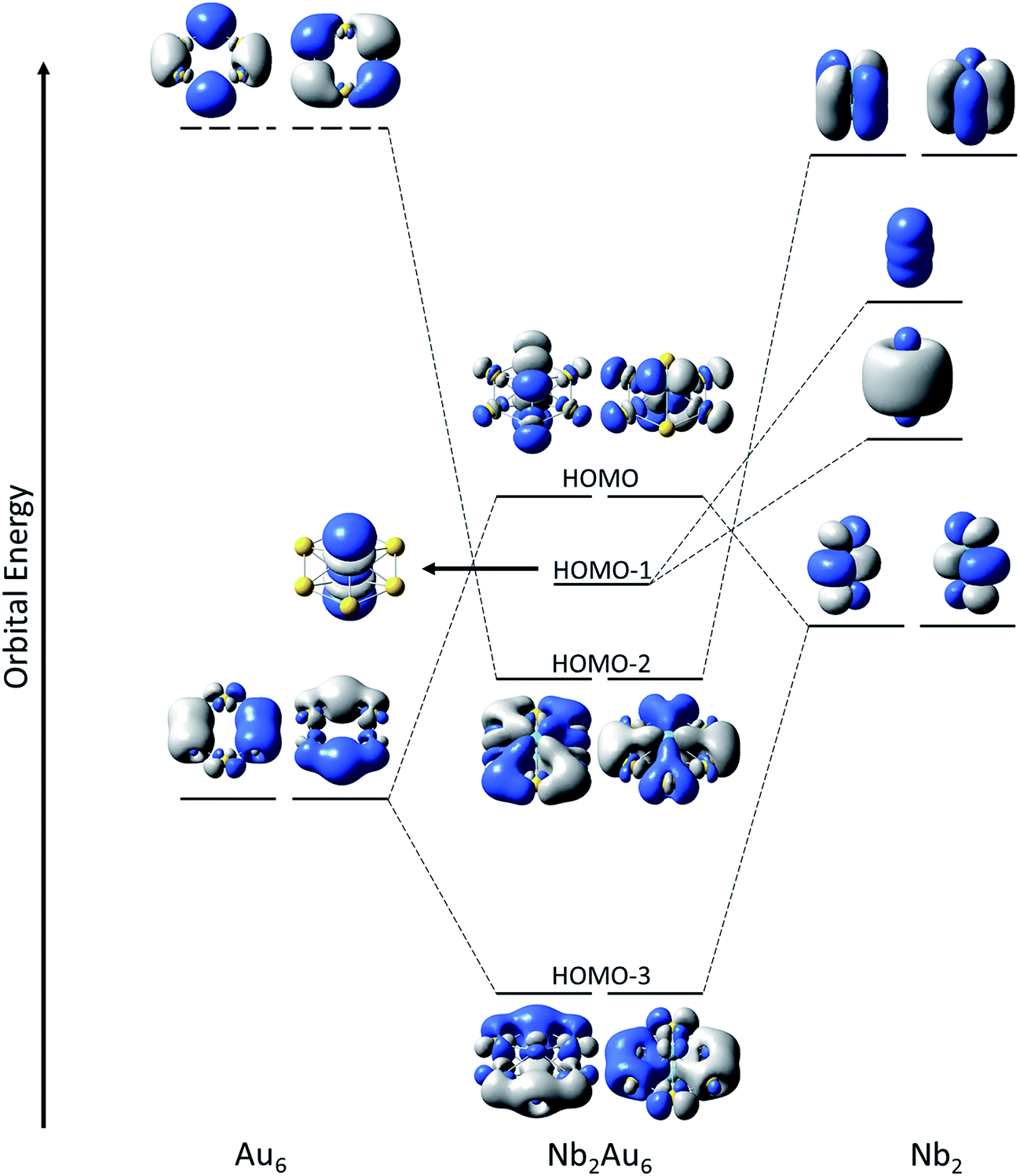 | ||
| Fig. 5 Qualitative orbital correlation diagram of the interactions between the D∞h Nb2 fragment and the D6h Au6 fragment in the D6h Nb2Au6. The horizontal dashed and solid lines represent virtual and occupied molecular orbitals, respectively. | ||
However, it is difficult to establish a clear bonding picture between Nb2 and Au6 in the deeper CMOs due to strong hybridization. We further analyzed the bonding in Nb2, Au6, and Nb2Au6 using the AdNDP method at the PBE0/Def2-TZVPPD level. The AdNDP analysis can yield a chemically intuitive bonding picture, displaying not only localized but also delocalized bonds in complicated molecular and cluster systems.31,39 The results of the AdNDP analyses are shown in Fig. 6. For the Au6 ring, there are three totally delocalized σ bonds mainly from the 6s orbitals. For the isolated Nb2, there are one 4dσ bond, two 4dπ bonds, one 5sσ bond, and two two-center one-electron (2c-1e) 5dδ bonds. In Nb2Au6, the one 4dσ bond and two 4dπ bonds are preserved, defining a triple bond in the Nb2 moiety. In addition, there are five totally delocalized σ bonds, describing bonding within the Au6 ring and the Nb2–Au6 guest–host bonding. These five delocalized 8c-2e bonds are remarkable, reminiscent of σ-aromaticity. The first three 8c-2e σ bonds in Nb2Au6 are similar to those in the Au6 ring but contain extra contributions from 5s/5p orbitals of Nb. The first and following two 8c-2e σ bonds show the overlap between the bonding orbitals of Au6 with the 5sσ orbital and the 5pπ orbitals of Nb2, respectively. The last two 8c-2e bonds mainly represent interactions between the δ MOs of Nb2 and the antibonding orbitals of Au6. The totally delocalized bonding between Nb2 and Au6 represents a new mode of guest–host interactions, reinforcing the NbNb triple bond and giving rise to the extraordinarily short Nb–Nb bond length.
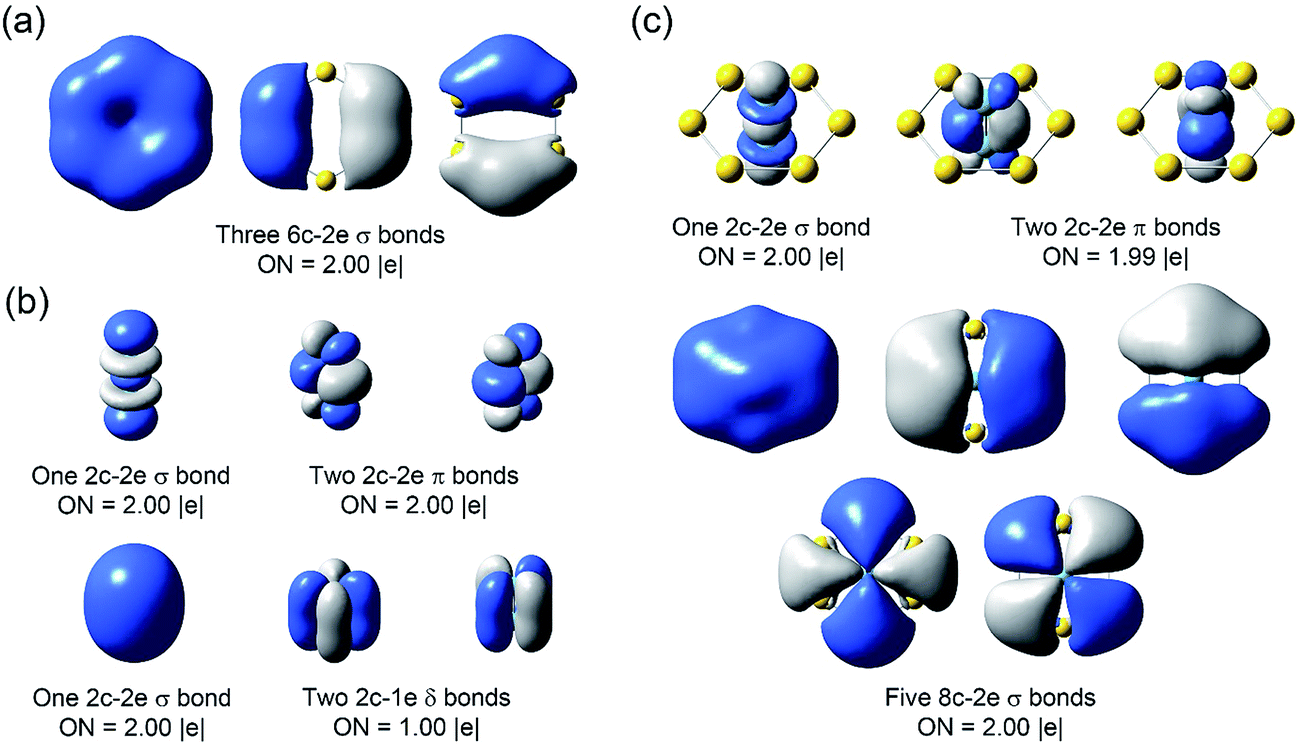 | ||
| Fig. 6 The AdNDP chemical bonding analyses of (a) D6h Au6, (b) D∞h Nb2, and (c) D6h Nb2Au6 at the PBE0/Def2-TZVP level. ON stands for occupation number. | ||
7.3. The Au6 ring as a bridging ligand to facilitate metal–metal multiple bonding
It is a well-established strategy to use bridging ligands to facilitate metal–metal multiple bonding.9b In fact, using 1,3,4,6,7,8-hexahydropyrimido[1,2-a]pyrimidinate (hpp) as bridging ligands, Cotton et al. synthesized the Nb2(hpp)4 complex, which contains the shortest Nb–Nb triple bond (σ2π4) with a bond length of 2.204 Å.13 This kind of complexes require four electrons of Nb2 for metal–ligand bonding, restricting the Nb–Nb bond order to be no more than three. Note that the Nb–Nb bond length in Nb2Au6 is significantly shorter than that in Nb2(hpp)4. The δ orbitals in Nb2 can interact with the antibonding orbitals of the equatorial Au6 ring by symmetry, providing the key mechanism for reinforcing the NbNb triple bond. Thus, the Au6 ring acts as a new type of bridging ligand and provides a new strategy to facilitate stronger multiple bonding in transition metal dimers.
8. Conclusions
In summary, we report the observation of highly symmetric Nb2©Au6−/0 molecular wheels, in which an extremely short NbNb triple bond is coordinated equatorially by an Au6 ring and stabilized by σ aromaticity. A large HOMO–LUMO gap is observed in the photoelectron spectra, suggesting the neutral Nb2Au6 cluster is a highly stable electronic system. High-resolution photoelectron spectra from velocity map imaging indicate that neutral Nb2Au6 is highly symmetric. Global minimum searches coupled with high-level quantum calculations show that the most stable structures of Nb2Au6−/0 consist of a D6h Au6 ring building around a short Nb2 axis. Chemical bonding analyses reveal one σ bond and two π bonds in the Nb2 axis, and five totally delocalized σ bonds in Nb2Au6. The antibonding orbitals of the Au6 ring can interact with the δ orbitals in Nb2 by symmetry, which is critical for strengthening the NbNb triple bond. The Au6 ring acts as a novel bridging ligand, providing a new strategy to facilitate stronger multiple bonding in transition metal dimers. A whole class of M2©Au6 type molecular wheels containing a strengthened M–M multiple bond may be possible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE-1263745). We are indebted to Prof. Xueming Yang and Dr Tao Wang for supplying us the Deyang ( ) dye laser system, which is critical for the high-resolution PE imaging experiment. The calculations were performed using resources at the Center for Computation and Visualization (CCV) of Brown University. T. J. wishes to thank the Chemistry Department of Brown University for the Vince Wernig Fellowship.
) dye laser system, which is critical for the high-resolution PE imaging experiment. The calculations were performed using resources at the Center for Computation and Visualization (CCV) of Brown University. T. J. wishes to thank the Chemistry Department of Brown University for the Vince Wernig Fellowship.
References
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- (a) F. Furche, R. Ahlrichs, P. Weis, C. Jacob, S. Gilb, T. Bierweiler and M. M. Kappes, J. Chem. Phys., 2002, 117, 6982–6990 CrossRef CAS; (b) H. Hakkinen, M. Moseler and U. Landman, Phys. Rev. Lett., 2002, 89, 033401 CrossRef PubMed; (c) H. Hakkinen, B. Yoon, U. Landman, X. Li, H. J. Zhai and L. S. Wang, J. Phys. Chem. A, 2003, 107, 6168–6175 CrossRef; (d) M. P. Johansson, A. Lechtken, D. Schooss, M. M. Kappes and F. Furche, Phys. Rev. A, 2008, 77, 053202 CrossRef; (e) W. Huang and L. S. Wang, Phys. Rev. Lett., 2009, 102, 153401 CrossRef PubMed.
- (a) K. Taylor, C. L. Pettiette-Hall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 92, 3319–3329 CrossRef; (b) K. Taylor, C. Jin, J. Conceicao, L. S. Wang, O. Cheshnovsky, B. Johnson, P. Nordlander and R. E. Smalley, J. Chem. Phys., 1990, 93, 7515–7518 CrossRef CAS.
- I. A. Popov, A. A. Starikova, D. V. Steglenko and A. I. Boldyrev, Chem.–Eur. J., 2017 DOI:10.1002/chem.201702035.
- H. Tanaka, S. Neukermans, E. Janssens, R. E. Silverans and P. Lievens, J. Am. Chem. Soc., 2003, 125, 2862–2863 CrossRef CAS PubMed.
- (a) X. Li, B. Kiran, L. F. Cui and L. S. Wang, Phys. Rev. Lett., 2005, 95, 253401 CrossRef PubMed; (b) The © sign was first proposed to designate a disk or wheel structure with a central atom in a boron ring. See C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2011, 50, 9334–9337 CrossRef CAS PubMed.
- M. Zhang, L. M. He, L. X. Zhao, X. J. Feng and Y. H. Luo, J. Phys. Chem. C, 2009, 113, 6491–6496 CAS.
- (a) W. D. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Chou and M. L. Cohen, Phys. Rev. Lett., 1984, 52, 2141 CrossRef CAS; (b) T. Höltzl, P. Lievens, T. Veszprémi and M. T. Nguyen, J. Phys. Chem. C, 2009, 113, 21016–21018 CrossRef.
- (a) F. A. Cotton, N. F. Curtis, C. B. Harris, B. F. G. Johnson, S. J. Lippard, J. T. Mague, W. R. Robinson and J. S. Wood, Science, 1964, 145, 1305–1307 CAS; (b) F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds between Metal Atoms, Springer, New York, 2005 Search PubMed.
- (a) T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef CAS PubMed; (b) K. A. Kreisel, G. P. Yap, O. Dmitrenko, C. R. Landis and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 14162–14163 CrossRef CAS PubMed; (c) G. Frenking, Science, 2005, 310, 796–797 CrossRef CAS PubMed.
- N. V. S. Harisomayajula, A. K. Nair and Y. C. Tsai, Chem. Commun., 2014, 50, 3391–3412 RSC.
- A. M. James, P. Kowalczyk, R. Fournier and B. Simard, J. Chem. Phys., 1993, 99, 8504–8518 CrossRef CAS.
- F. A. Cotton, J. H. Matonic and C. A. Murillo, J. Am. Chem. Soc., 1997, 119, 7889–7890 CrossRef CAS.
- (a) L. S. Wang, H. S. Cheng and J. Fan, J. Chem. Phys., 1995, 102, 9480–9493 CrossRef CAS; (b) L. S. Wang, Int. Rev. Phys. Chem., 2016, 35, 69–142 CrossRef CAS.
- I. León, Z. Yang, H. T. Liu and L. S. Wang, Rev. Sci. Instrum., 2014, 85, 083106 CrossRef PubMed.
- B. Dick, Phys. Chem. Chem. Phys., 2014, 16, 570–580 RSC.
- S. Kirkpatrick, C. D. Gelatt and M. P. Vecchi, Science, 1983, 220, 671–680 CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 12947 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (b) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- (a) D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS; (b) K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009.
- (a) G. D. Purvis III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef; (b) G. E. Scuseria, C. L. Janssen and H. F. Schaefer III, J. Chem. Phys., 1988, 89, 7382–7387 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schutz, et al., MOLPRO, a package of ab initio programs, see http://www.molpro.net Search PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- V. A. Mozhayskiy and A. I. Krylov, ezSpectrum, http://iopenshell.usc.edu/downloads Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- S. Dapprich and G. Frenking, J. Phys. Chem., 1995, 99, 9352 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- (a) D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC; (b) D. Y. Zubarev and A. I. Boldyrev, J. Org. Chem., 2008, 73, 9251–9258 CrossRef CAS PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, version 4.1, Semichem, Inc, Shawnee Mission, KS, 2007 Search PubMed.
- U. Varetto, Molekel 5.4.0.8, Swiss National Supercomputing Center, Manno, Switzerland, 2009 Search PubMed.
- (a) I. Leon, Z. Yang and L. S. Wang, J. Chem. Phys., 2013, 138, 184304 CrossRef PubMed; (b) C. Hock, J. B. Kim, M. L. Weichman, T. I. Yacovitch and D. M. Neumark, J. Chem. Phys., 2012, 137, 244201 CrossRef PubMed.
- (a) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (c) J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed; (d) Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed; (e) O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109–234117 CrossRef PubMed; (f) O. A. Vydrov, J. Heyd, A. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 074106–074114 CrossRef PubMed; (g) O. A. Vydrov, G. E. Scuseria and J. P. Perdew, J. Chem. Phys., 2007, 126, 154109–154117 CrossRef PubMed.
- (a) L. M. Wang, R. Pal, W. Huang, X. C. Zeng and L. S. Wang, J. Chem. Phys., 2010, 132, 114306 CrossRef PubMed; (b) N. Shao, W. Huang, Y. Gao, L. M. Wang, X. Li, L. S. Wang and X. C. Zeng, J. Am. Chem. Soc., 2010, 132, 6596–6605 CrossRef CAS PubMed.
- P. Pyykkö, J. Phys. Chem. A, 2014, 119, 2326–2337 CrossRef PubMed.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- A. I. Boldyrev and L. S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 11589–11605 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Photoelectron spectra of Nb2Au6− at (a) 616.86 nm, (b) 610.55 nm, (c) 603.38 nm, and (d) 589.58 nm; relative energies of low-lying isomers of Nb2Au6− within 1.5 eV at the PW91, PBE0 (in square brackets), CCSD (in braces) and CCSD(T) (in angle brackets) levels; the four lowest-lying isomers of Nb2Au6 at the levels of PW91 and PBE0; valence canonical molecular orbital contours for the D6h Nb2Au6− at the PBE0 level; comparison between the 193 nm spectrum of Nb2Au6− with the simulated spectra of isomers I and II at the PW91/Def2-TZVPPD and BP86/Def2-TZVPPD levels; comparison between the 193 nm spectrum of Nb2Au6− with the simulated spectra of isomer I at PBE0/Def2-TZVPPD and TPSSh/Def2-TZVPPD levels; the energy difference between isomers I and II of Nb2Au6− calculated at various levels of theory; the first VDEs of isomers I, II, III, IV and V of Nb2Au6− computed at the PW91 and PBE0 levels; calculated vibrational frequencies for the D6h global minimum of Nb2Au6 at PW91 and PBE0 levels; calculated bond orders and charges of D∞h Nb2, D6h Au6, and D6h Nb2Au6 at the PBE0/Def2-TZVP level. See DOI: 10.1039/c7sc02881d |
| This journal is © The Royal Society of Chemistry 2017 |