
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective catalytic β-amination through proton-coupled electron transfer followed by stereocontrolled radical–radical coupling†
Zijun
Zhou
a,
Yanjun
Li
a,
Bowen
Han
a,
Lei
Gong
 *a and
Eric
Meggers
*ab
*a and
Eric
Meggers
*ab
aCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: gongl@xmu.edu.cn
bFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse 4, 35043 Marburg, Germany. E-mail: meggers@chemie.uni-marburg.de
First published on 15th June 2017
Abstract
A new mechanistic approach for the catalytic, enantioselective conjugate addition of nitrogen-based nucleophiles to acceptor-substituted alkenes is reported, which is based on a visible light induced and phosphate base promoted transfer of a single electron from a nitrogen nucleophile to a catalyst-bound acceptor-substituted alkene, followed by a stereocontrolled C–N bond formation through stereocontrolled radical–radical coupling. Specifically, N-aryl carbamates are added to the β-position of α,β-unsaturated 2-acyl imidazoles using a visible light activated photoredox mediator in combination with a chiral-at-rhodium Lewis acid catalyst and a weak phosphate base, affording new C–N bonds in a highly enantioselective fashion with enantioselectivities reaching up to 99% ee and >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr for a menthol-derived carbamate. As an application, the straightforward synthesis of a chiral β-amino acid ester derivative is demonstrated.
:1 dr for a menthol-derived carbamate. As an application, the straightforward synthesis of a chiral β-amino acid ester derivative is demonstrated.
Chiral amines featuring a stereogenic carbon connected to a nitrogen are important structural motifs in pharmaceuticals and natural products.1 The development of methodology for the catalytic, asymmetric construction of C–N bonds is therefore of high relevance.2 In this respect, the conjugate amination of readily available α,β-unsaturated carbonyl compounds is a principal strategy which provides useful β-amino carbonyl building blocks and exploits the intrinsic nucleophilicity of nitrogen lone pairs, with or without a prior deprotonation of the N–H group (Fig. 1).3,4 However, the high nucleophilicity of such nitrogen reagents needs to be controlled carefully in order to prevent side reactions, catalyst deactivation, and uncatalyzed background reactions reducing the enantioselectivity.1
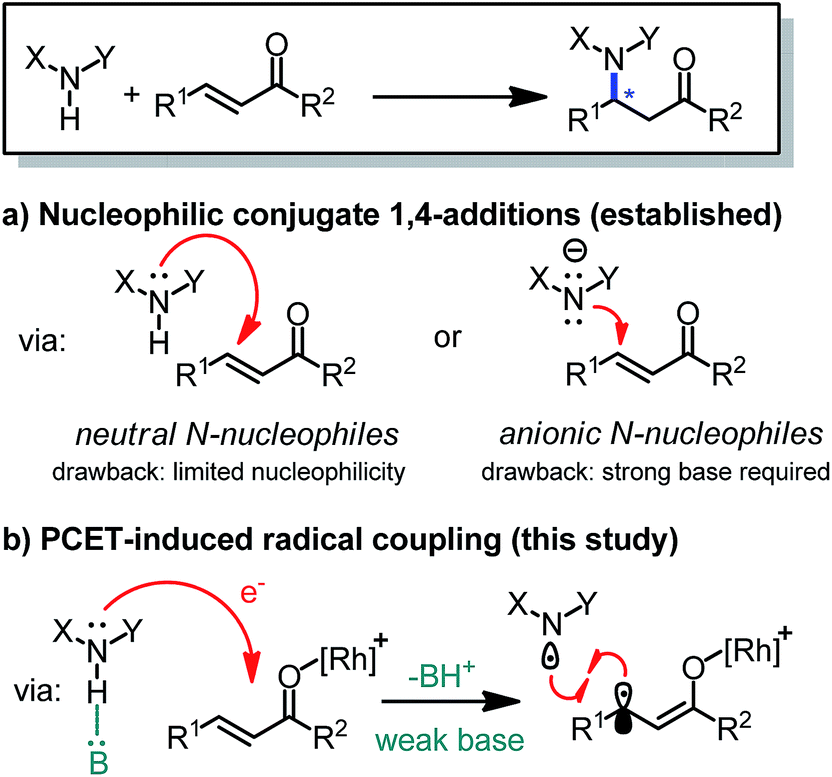 | ||
| Fig. 1 Formation of C–N bonds by β-amination of readily available α,β-unsaturated carbonyl compounds. Scope limitations for the PCET-induced radical coupling reported in this study: X = aryl, Y = CO2R, R1 = aryl, R2 = 2-imidazolyl. | ||
Recently, Knowles introduced an elegant strategy making use of proton-coupled electron transfer (PCET) to convert N–H groups into nitrogen-centered radicals, whose high reactivities can then be exploited for radical additions to electron-rich double bonds or hydrogen atom transfer reactions.5–8 The activation of N–H containing compounds by PCET is attractive for its mild reaction conditions: it requires only a weak base, such as a phosphate, and the single electron transfer can be induced by visible light using a photoredox mediator. Unfortunately, the electron-deficient nature of nitrogen-centered radicals renders their application to the direct radical β-amination of unsaturated carbonyl compounds unfavorable, at least for intermolecular reactions.9–11
Herein, we introduce a novel strategy to interface photoinduced PCET with the catalytic, enantioselective conjugate amination of α,β-unsaturated carbonyl compounds. The mild and visible light induced enantioselective C–N bond formation is catalyzed by a previously developed chiral-at-rhodium Lewis acid12 and couples N-aryl carbamates with α,β-unsaturated 2-acyl imidazoles with high yields and excellent enantioselectivities.
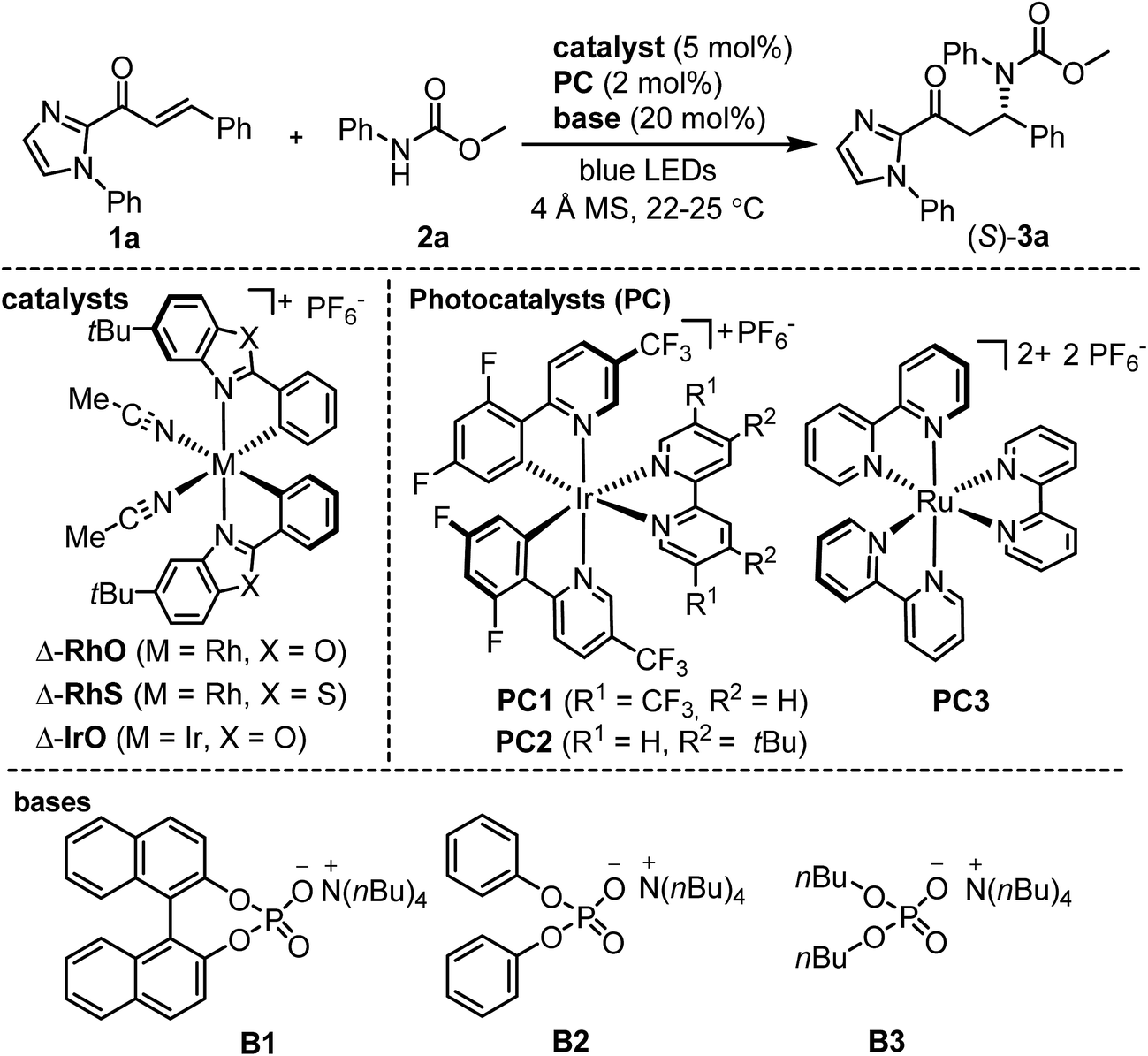
We commenced our study by investigating the reaction of the α,β-unsaturated 2-acyl imidazole 1a with N-phenyl carbamic acid methyl ester 2a under PCET conditions8 using the iridium-based photoredox mediator PC113 and the weak phosphate base B1, and combined this with the chiral-at-rhodium Lewis acid Δ-RhO14 (Table 1). Encouragingly, upon irradiation with blue LEDs at room temperature for 16 hours we observed a conversion of 43% to the C–N bond formation product (S)-3a with 96% ee (entry 1). Changing the solvent from CHCl3 to CH2Cl2 and slightly elongating the reaction time provided full conversion while retaining the high enantioselectivity (entry 2). The enantiomeric excess could be further improved to 98% ee upon optimization of the phosphate base (B2 and B3, entries 3 and 4). Examining other photoredox mediators provided inferior results (entries 5 and 6) and it is also worth noting that the related chiral-at-metal catalysts Δ-RhS15 (entry 7) and Δ-IrO16 (entry 8) are not suitable for this method. Meanwhile, control experiments verified that Δ-RhO, a phosphate base (B2), and blue light are all essential for product formation (entries 9–11). Interestingly, product is formed with high enantioselectivity even in the absence of the photoredox mediator, but the conversion is more sluggish and cannot be brought to full completion even after elongated reaction times (entry 12).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | PC | hν | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: α,β-unsaturated acyl imidazole 1a (0.10 mmol), carbamate 2a (0.12 mmol), metal catalyst (0.005 mmol), PC1-3 (0.002 mmol), B1-3 (0.02 mmol), 4 Å molecular sieves (MS) (10 mg), and solvent (1.0 mL). The reactions were irradiated with blue LEDs (24 + 36 W), except for entry 11. b Yields determined by crude 1H NMR analysis. c Ee values determined by HPLC analysis on a chiral stationary phase. d Not determined. e Cyclobutane side product formed. f Not applicable. | |||||||
| 1 | Δ-RhO | B1 | PC1 | Light | CHCl3 | 43 | 96 |
| 2 | Δ-RhO | B1 | PC1 | Light | CH2Cl2 | Quant. | 96 |
| 3 | Δ-RhO | B2 | PC1 | Light | CH2Cl2 | Quant. | 98 |
| 4 | Δ-RhO | B3 | PC1 | Light | CH2Cl2 | 24 | 74 |
| 5 | Δ-RhO | B2 | PC2 | Light | CH2Cl2 | 41 | 97 |
| 6 | Δ-RhO | B2 | PC3 | Light | CH2Cl2 | 34 | 98 |
| 7 | Δ-RhS | B2 | PC1 | Light | CH2Cl2 | <10 | n.d.d |
| 8 | Δ-IrO | B2 | PC1 | Light | CH2Cl2 | Trace | n.d.d |
| 9 | None | B2 | PC1 | Light | CH2Cl2 | 0e | n.a.f |
| 10 | Δ-RhO | None | PC1 | Light | CH2Cl2 | 8 | 93 |
| 11 | Δ-RhO | B2 | PC1 | Dark | CH2Cl2 | 0 | n.a.f |
| 12 | Δ-RhO | B2 | None | Light | CH2Cl2 | 39 | 98 |
The proposed mechanism is shown in Fig. 2. The photoactivated photoredox mediator (PC*) initiates a proton-coupled electron transfer (PCET) involving the Brønsted-base-activated carbamate to generate a nitrogen-centered radical and a reduced photoredox mediator (PC˙−), which in turn transfers an electron to the Rh-coordinated substrate (I → II). Thus, the photoredox mediator serves as a single electron shuttle from the carbamate to the Rh-bound α,β-unsaturated 2-acyl imidazole, providing an electron deficient carbamoyl N-radical and the persistent electron rich rhodium enolate radical intermediate II,17,18 which subsequently recombines to the rhodium enolate intermediate III.19 After protonation by the protonated phosphate base (III → IV), product release, and coordination of new substrate (IV → I), a new catalytic cycle can be initiated. The function of the rhodium catalyst in this mechanism is twofold. First, the established N,O-bidentate coordination12 to the unsaturated 2-acyl imidazole facilitates the reduction of the Rh-bound substrate. Secondly, the chiral Lewis acid controls the stereochemistry of the radical–radical coupling by providing a high asymmetric induction. Note that in this radical–radical coupling step the stereocontrol occurs through controlling the reaction of a catalyst-bound prochiral carbon-centered radical.20
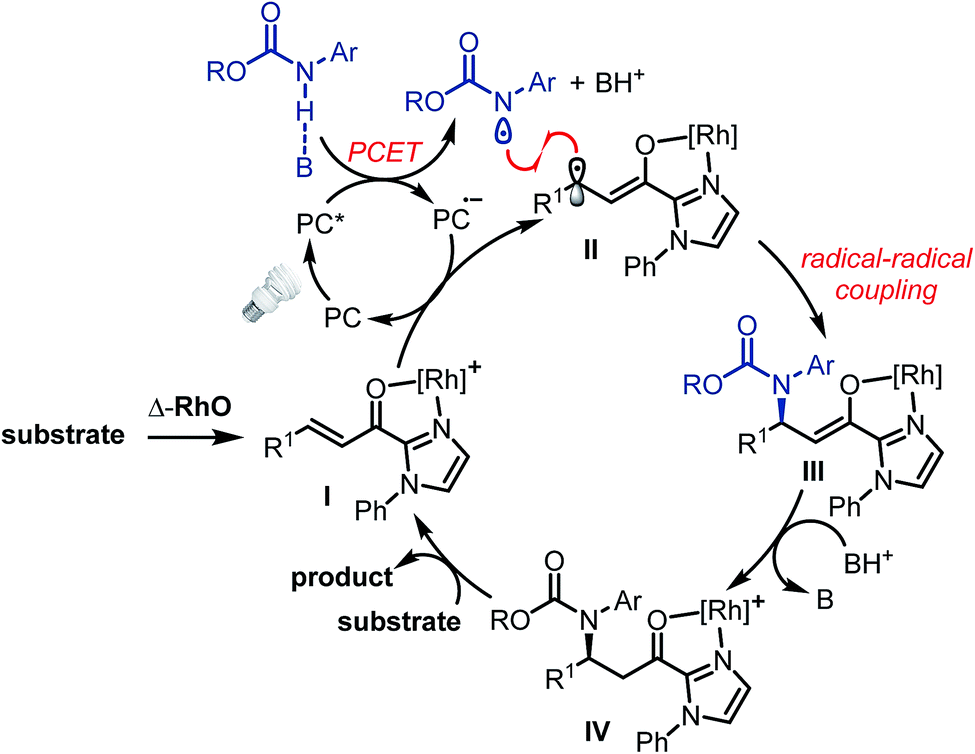 | ||
| Fig. 2 Proposed mechanism. | ||
A number of control experiments back this mechanism. First, visible light, rhodium catalyst, and phosphate base are all required for product formation. Second, intermediate N-centered radical formation was verified by trapping the nitrogen-centered radical by an intramolecular radical addition to an alkene (2b) followed by reaction of the formed carbon-centered radical with the unsaturated 2-acyl imidazole 1a, affording the product 4 with 89% yield and 97% ee (Fig. 3a). Third, in the absence of any Rh-catalyst, the cyclobutane complex 5 is formed as the major product in 36% yield. This reaction is analogous to Yoon's reported photoredox-mediated [2 + 2] cycloaddition involving unsaturated 2-acyl imidazoles which was proposed to proceed through the single electron reduction of the unsaturated 2-acyl imidazoles and supports the involvement of the Rh-enolate radical intermediate II in our pathway.18,21 Finally, when we added one equivalent of TEMPO to the reaction, no product was formed which is indicative of a radical mechanism.
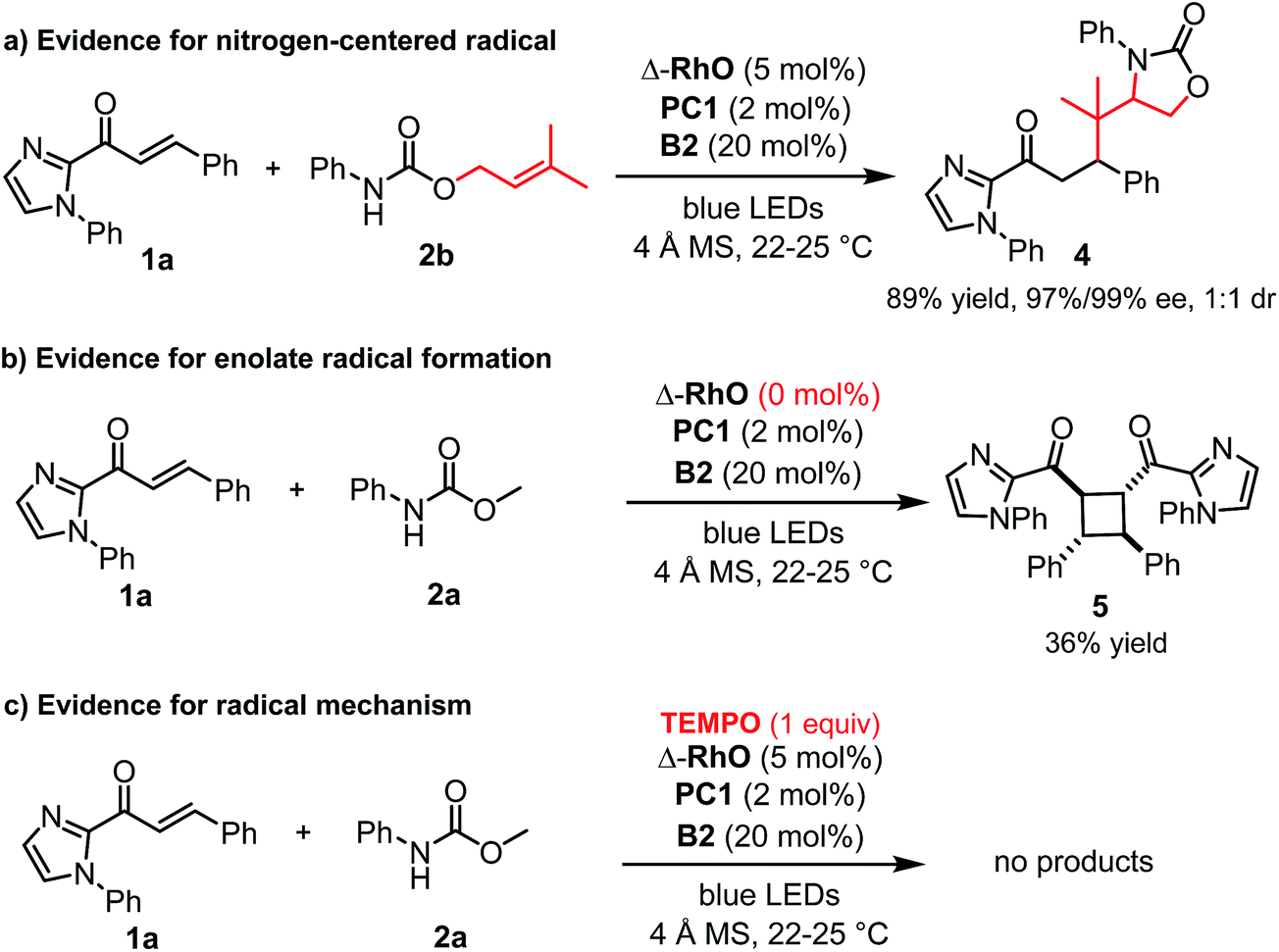 | ||
| Fig. 3 Control experiments. | ||
Alternative mechanistic scenarios through the addition of N-carbamoyl radicals or carbamate anions to the rhodium-coordinated α,β-unsaturated 2-acyl imidazoles are unlikely and are briefly discussed. N-centered carbamoyl and amidyl radicals are classified as electrophilic radicals which prefer to react with electron rich alkenes.11,22,23 Rhodium coordination to the α,β-unsaturated 2-acyl imidazole substrates would further decrease the radical addition rate and is therefore not consistent with the observed high enantioselectivity under rhodium catalysis. On the other hand, a mechanism through the conjugate addition of carbamate anions is unlikely due to the requirement for light activation and the experimental evidence for the intermediate formation of a carbamoyl radical (Fig. 3a). As an additional control experiment we performed the standard reaction in the presence of the strong base sodium ethoxide instead of the weak phosphate base but no product was formed in the absence of light (see ESI for details†). Furthermore, the reduction of an intermediate carbamoyl radical to the anion (by the reduced photoredox mediator) followed by a conjugate addition is unlikely because the reaction also proceeds in the absence of an additional photoredox mediator with reduced yields but identical enantioselectivity (Table 1, entry 12).
This proposed mechanism resembles some conceptional similarity with Melchiorre's recently reported visible-light-excited enantioselective β-alkylation of α,β-unsaturated aldehydes catalyzed by a chiral secondary amine.20g A photoactivated conjugated iminium ion intermediate oxidizes the alkylsilane co-substrate by single electron transfer followed by a radical–radical recombination. The iminium ion in Melchiorre's system resembles our rhodium-coordinated α,β-unsaturated 2-acyl imidazole (intermediate I), both of which serve as in situ assembled chiral single electron acceptors, accepting the single electron from a cosubstrate, followed by a stereocontrolled radical–radical recombination. Other photoactivated catalytic asymmetric reactions in which single electrons are transferred between a substrate-bound catalyst and a co-substrate have been reported.20
Next, after having investigated the mechanism of this novel catalytic, enantioselective C–N bond formation, we evaluated the scope (Fig. 4). The visible-light-induced enantioselective β-amination of 2a to electron-deficient alkenes provided the C–N bond formation products (3b–g) with 82–97% yields and 94–99% ee. Different substituted aromatic moieties with respect to steric and electronic effects are well tolerated. With an electron-donating methoxy substituted aromatic moiety (product 3i), the reaction time needed to be extended to 72 hours in order to achieve a high yield (87%) and high enantioselectivity (97% ee). However, when we applied this β-amination to a substrate with an ortho-methylated phenyl group, only traces of the product 3h were detected. Apparently, the methyl group prevents a proper conjugation of the entire π-system, which suppresses the entire PCET mechanism. This also explains the requirement for an aromatic moiety in β-position of the α,β-unsaturated 2-acyl imidazole substrates as the formation of the product 3j failed.
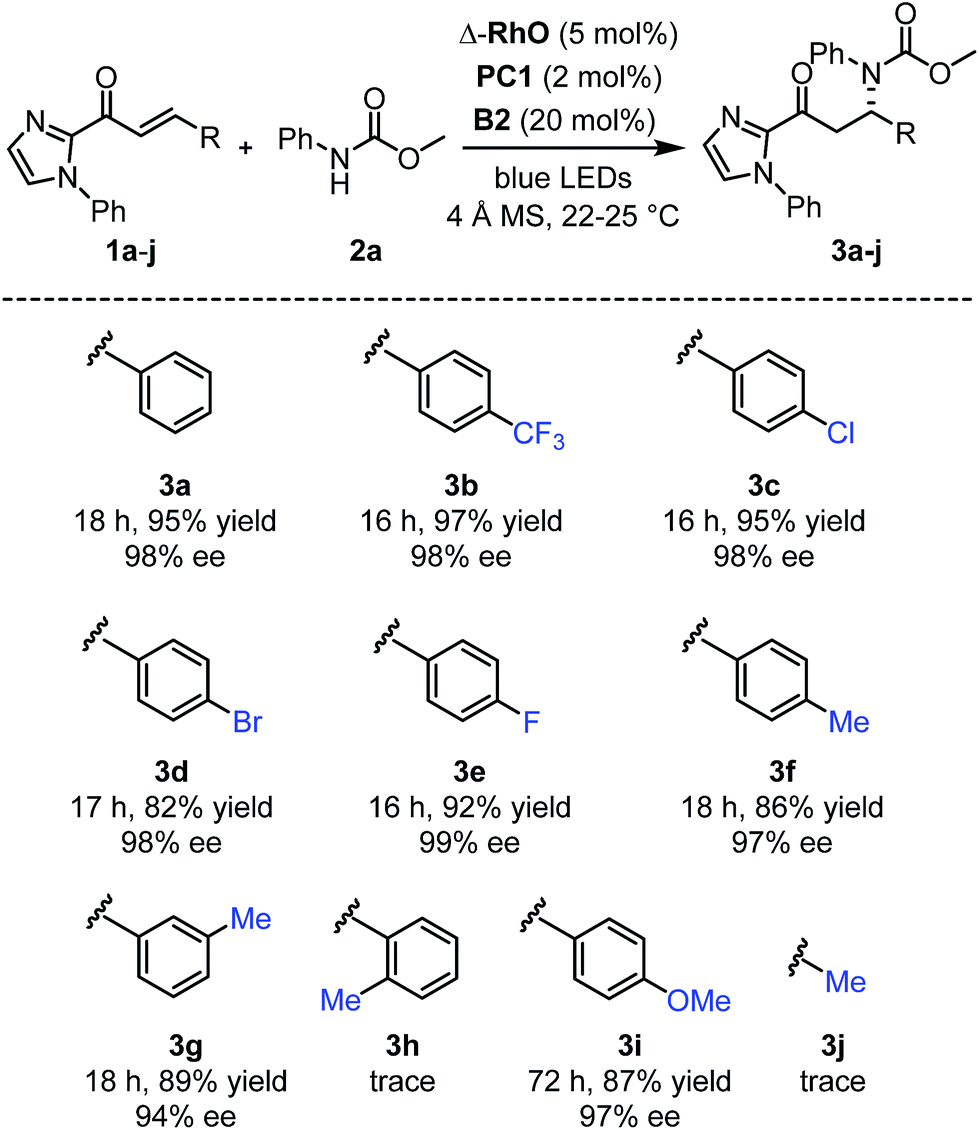 | ||
| Fig. 4 Reaction scope with regards to 2-acyl imidazoles. | ||
The scope of this reaction with respect to substituted carbamates is outlined in Fig. 5. Different O-substituents such as ethyl, isopropyl, tert-butyl, and isobutyl (products 3k–n) in the ester protected group are tolerated and provide high yields (87–98%) with excellent enantioselectivities (94–98% ee). Carbamates with O-benzyl or an O-phenethyl group (products 3o, p) reacted more slowly. The N-para-methoxyphenyl (PMP) protection group was not tolerated and did not provide the β-amination product 3q but instead a dimer of the 2-acyl imidazole substrate. However, methyl and halogen substituents at the N-phenyl moiety (products 3r–v) are well tolerated with β-amination yields of 84–92% and 96–99% ee. Also, a menthol-derived carbamate provided the β-amination product 3w in 85% yield with >99:1 d.r. As a limitation, the carbamate relies on N-aryl substituents (3x), and amides do not or almost not provide the desired products (3y, z) but instead the cyclobutane complex 5 as a side product.
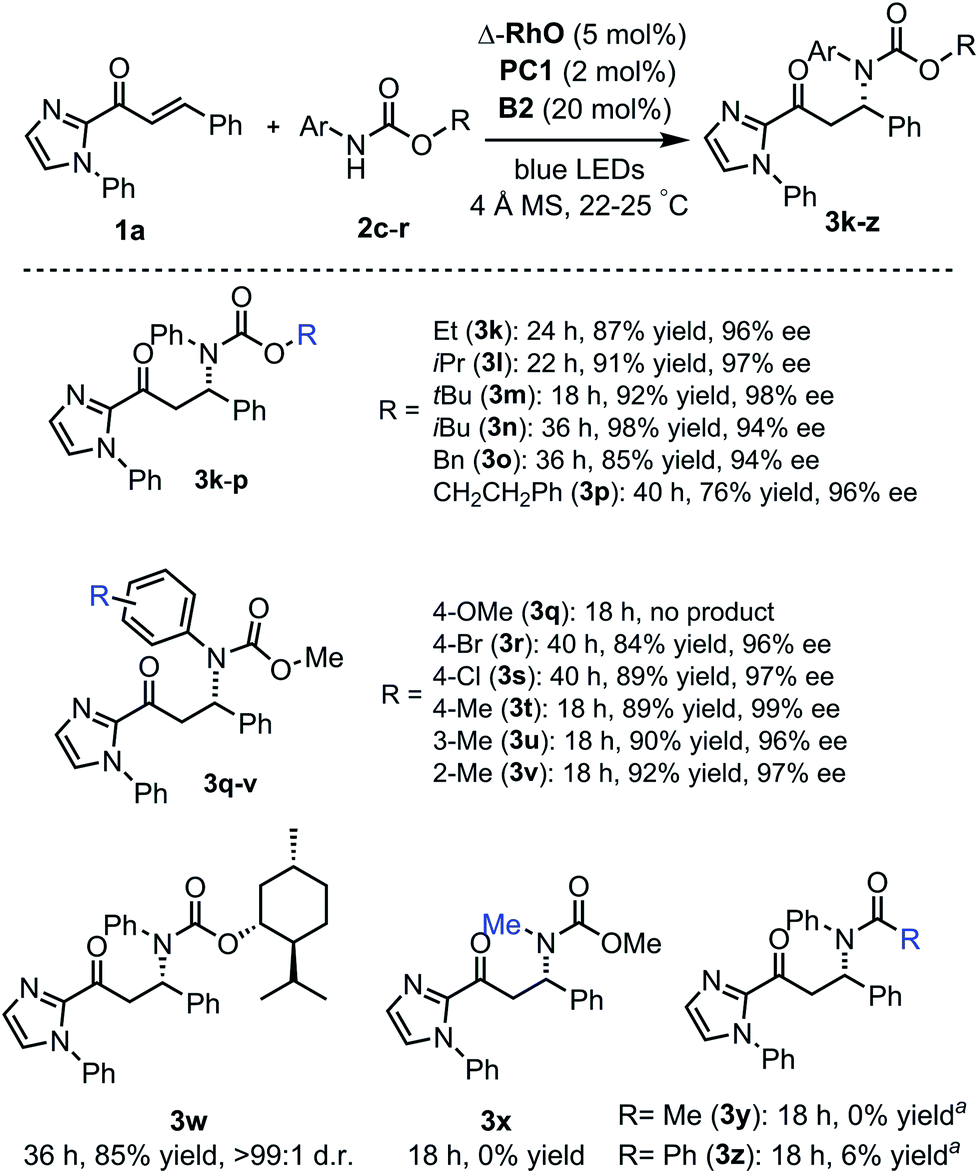 | ||
| Fig. 5 Reaction scope with respect to carbamates. aNo desired product formed but instead a cyclobutane side product. | ||
Finally, the conversion of the typical product 3m to a useful synthetic building block is illustrated in Fig. 6. Removal of the N-phenylimidazole moiety smoothly proceeded to provide the intermediate ester following an established procedure.24 Then, it was dissolved in CH2Cl2 and trifluoroacetic acid (10 equivalents) was added to cleave the Boc-protection group and subsequently form 1,2-aminoester derivative (S)-3m′, which was also used to assign the absolute configuration of the formed products in this study.25
 | ||
| Fig. 6 Follow-up chemistry with a cross-coupling reaction product 3m. | ||
In conclusion, we have introduced a novel strategy to establish a C–N bond in β-position of an α,β-unsaturated carbonyl compound in a catalytic, enantioselective fashion via photoinduced proton-coupled electron transfer, followed by a highly stereoselective radical–radical recombination controlled by a chiral rhodium-enolate radical intermediate. This method permits the catalytic, enantioselective synthesis of chiral amines without the necessity for a strong base or a highly nucleophilic nitrogen reagent. Although there are clear restrictions with respect to the substrate scope, within these limitations the obtained yields and enantioselectivities are excellent. Future work will need to address the currently limited scope.
Experimental section
Representative asymmetric catalysis: to a solution of α,β-unsaturated 2-acyl imidazole 1a (32.90 mg, 0.12 mmol) in fresh distilled anhydrous CH2Cl2 (1.0 mL) was added rhodium catalyst Δ-RhO (4.15 mg, 0.005 mmol), PC1 (2.29 mg, 0.002 mmol), B2 (9.83 mg, 0.02 mmol), 4 Å MS (10.0 mg) in a 10 mL Schlenk tube and the resulting solution was stirred at room temperature for 15 min. Then, tert-butyl N-phenylcarbamate (19.30 mg, 0.10 mmol) was added and the resulting solution was purged for 15 min with argon. The reaction was stirred under argon in a thermostatic cabinet under irradiation with blue LEDs (24 + 36 W), providing a reaction temperature of 22–25 °C. The Schlenk tube was kept at a distance of approximately 3 cm from the light source. The reaction was monitored by TLC analysis. After 18 hours full conversion was reached and the mixture was diluted with CH2Cl2 and purified by flash chromatography on silica gel (EtOAc/n-hexane = 1/5 to 1/3) to afford the product (S)-3m (43 mg, yield: 92%) as a white solid. The enantiomeric excess of 98% ee was established by HPLC analysis using a Daicel Chiralpak IC column (250 × 4.6 mm) under the following conditions: UV-detection at 254 nm, mobile phase n-hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, column temperature of 25 °C. Retention times: tr(minor) = 17.44 min, tr(major) = 26.98 min. 1H NMR (500 MHz, CD2Cl2): δ (ppm) 7.47–7.37 (m, 3H), 7.30–7.14 (m, 12H), 6.89–6.76 (m, 2H), 6.10 (t, J = 7.5 Hz, 1H), 3.86 (dd, J = 15.6, 6.7 Hz, 1H), 3.55 (dd, J = 15.6, 8.3 Hz, 1H), 1.30 (s, 9H). 13C NMR (126 MHz, CD2Cl2): δ (ppm) 188.2, 154.5, 142.7, 140.2, 138.2, 129.7, 129.3, 128.6, 128.3, 128.0, 127.9, 127.8, 127.2, 127.0, 126.5, 125.8, 125.6, 79.8, 56.8, 41.6, 27.7. IR (film): ν (cm−1) 2963, 2920, 2859, 1692, 1404, 1260, 1093, 1020, 829, 699, 559. HRMS (ESI, m/z) calcd for C29H29N3NaO3 (M + Na)+: 490.2101, found: 490.2110.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of P. R. China (grant no. 21572184 and 21472154) and the Fundamental Research Funds for the Central Universities (grant no. 20720160027).References
- For reviews on different aspects of bioactive chiral amines, see: (a) H.-J. Federsel, M. Hedberg, F. R. Qvarnstroam, M. P. T. Sjoegren and W. Tian, Acc. Chem. Res., 2007, 40, 1377–1384 CrossRef CAS PubMed; (b) H.-J. Federsel, Drug News Perspect., 2008, 21, 193–199 CrossRef CAS PubMed; (c) R. Edupuganti and F. A. Davis, Org. Biomol. Chem., 2012, 10, 5021–5031 RSC; (d) S. Jones and C. J. A. Warner, Org. Biomol. Chem., 2012, 10, 2189–2200 RSC; (e) M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS PubMed; (f) A. K. Mailyan, J. A. Eickhoff, A. S. Minakova, Z.-H. Gu, P. Lu and A. Zakarian, Chem. Rev., 2016, 116, 4441–4557 CrossRef CAS PubMed.
- Chiral Amine Synthesis: Methods, Developments and Applications, ed. T. C. Nugent, Wiley-VCH, Weinheim, 2010 Search PubMed.
- For reviews on catalytic asymmetric aza-Michael additions, see: (a) L.-W. Xu and C.-G. Xia, Eur. J. Org. Chem., 2005, 633–639 CrossRef CAS; (b) P. R. Krishna, A. Sreeshailam and R. Srinivas, Tetrahedron, 2009, 65, 9657–9672 CrossRef CAS; (c) D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058–11076 CrossRef CAS PubMed; (d) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626–2704 CrossRef CAS PubMed; (e) M. Sánchez-Roselló, J. L. Aceña, A. Simón-Fuentes and C. del Pozo, Chem. Soc. Rev., 2014, 43, 7430–7453 RSC; (f) J. Gmach, Ł. Joachimiak and K. M. Błażewska, Synthesis, 2016, 48, 2681–2704 CrossRef CAS.
- For selected reviews and examples of biologically active chiral β-amino carbonyl compounds, see: (a) G. Cardillo and C. Tomassini, Chem. Soc. Rev., 1996, 25, 117–128 RSC; (b) E. Juaristi and H. Lopez-Ruiz, Curr. Med. Chem., 1996, 6, 983–1004 Search PubMed; (c) W. J. Drury, D. Ferraris, C. Cox, B. Young and T. Leckta, J. Am. Chem. Soc., 1998, 120, 11006–11007 CrossRef CAS; (d) D. L. Steer, R. A. Lew, P. Perlmutter, A. I. Smith and M.-I. Aguilar, Curr. Med. Chem., 2002, 9, 811–822 CrossRef CAS PubMed; (e) A. Kuhl, M. G. Hahn, M. Dumic and J. Mittendorf, Amino Acids, 2005, 29, 89–100 CrossRef CAS PubMed; (f) V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793 CrossRef CAS PubMed; (g) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS PubMed; (h) D. H. Paull, C. J. Abraham, M. T. Scerba, E. A. Danforth and T. Lectka, Acc. Chem. Res., 2008, 41, 655–663 CrossRef CAS PubMed; (i) M. Bartók, Chem. Rev., 2010, 110, 1663–1705 CrossRef PubMed; (j) L. Kiss and F. Fulop, Chem. Rev., 2014, 114, 1116–1169 CrossRef CAS PubMed; (k) C. Cabrele, T. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed; (l) B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed; (m) M. Kaur, M. Singh, N. Chadha and O. Silakari, Eur. J. Med. Chem., 2016, 123, 858–894 CrossRef CAS PubMed.
- (a) R. Knowles and H. Yayla, Synlett, 2014, 25, 2819–2826 CrossRef; (b) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed; (c) L. Q. Nguyen and R. R. Knowles, ACS Catal., 2016, 6, 2894–2903 CrossRef CAS.
- For recent reviews and accounts on PCET, see: (a) O. S. Wenger, Acc. Chem. Res., 2013, 46, 1517–1526 CrossRef CAS PubMed; (b) M. T. M. Koper, Chem. Sci., 2013, 4, 2710–2723 RSC; (c) B. A. Barry, Nat. Chem., 2014, 6, 376–377 CrossRef CAS PubMed; (d) B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53, 6427–6443 CrossRef CAS PubMed; (e) A. Migliore, N. F. Polizzi, M. J. Therien and D. N. Beratan, Chem. Rev., 2014, 114, 3381–3465 CrossRef CAS PubMed; (f) B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53, 6427–6443 CrossRef CAS PubMed; (g) I. Siewert, Chem.–Eur. J., 2015, 21, 15078–15091 CrossRef CAS PubMed; (h) S. Hammes-Schiffer, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef CAS PubMed; (i) H. Kisch, Acc. Chem. Res., 2017, 50, 1002–1010 CrossRef CAS PubMed; (j) N. Hoffmann, Eur. J. Org. Chem., 2017, 1982–1992 CrossRef CAS.
- For the electrochemical generation of nitrogen-centered radicals, see: (a) H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2008, 130, 13542–13543 CrossRef CAS PubMed; (b) H.-C. Xu, J. M. Campbell and K. D. Moeller, J. Org. Chem., 2014, 79, 379–391 CrossRef CAS PubMed; (c) F. Xu, L. Zhu, S. B. Zhu, X. M. Yan and H.-C. Xu, Chem.–Eur. J., 2014, 20, 12740–12744 CrossRef CAS PubMed; (d) L. Zhu, P. Xiong, Z. Y. Mao, Y. H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226–2229 CrossRef CAS PubMed; (e) H.-B. Zhao, Z.-W. Hou, Z.-J. Liu, Z.-F. Zhou, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 587–590 CrossRef CAS PubMed; (f) P. Xiong, H.-H. Xu and H.-C. Xu, J. Am. Chem. Soc., 2017, 139, 2956–2959 CrossRef CAS PubMed.
- For visible-light-induced PCET for the generation of nitrogen-centered radicals, see: (a) G. J. Choi and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 9226–9229 CrossRef CAS PubMed; (b) D. C. Miller, G. J. Choi, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 13492–13495 CrossRef CAS PubMed; (c) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed; (d) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed.
- For reviews on different aspects of nitrogen-centered radicals, see: (a) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603–1618 RSC; (b) M. Minozzi, D. Nanni and P. Spagnolo, Chem.–Eur. J., 2009, 15, 7830–7840 CrossRef CAS PubMed; (c) M. Heinrich and S. Höfling, Synthesis, 2010, 173–189 Search PubMed; (d) B. Quiclet-Sire and S. Z. Zard, Beilstein J. Org. Chem., 2013, 9, 557–576 CrossRef CAS PubMed; (e) J. C. Walton, Acc. Chem. Res., 2014, 47, 1406–1416 CrossRef CAS PubMed; (f) J. Hioe, D. Šakic, V. Vrček and H. Zipse, Org. Biomol. Chem., 2015, 13, 157–169 RSC; (g) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC; (h) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC.
- For recent examples on using photoredox chemistry to generate nitrogen-centered radicals, see: (a) L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607–5610 CrossRef CAS PubMed; (b) X.-Q. Hu, J.-R. Chen, Q. Wei, F.-L. Liu, Q.-H. Deng, A. M. Beauchemin and W.-J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 12163–12167 CrossRef CAS; (c) T. W. Greulich, C. G. Daniliuc and A. Studer, Org. Lett., 2015, 17, 254–257 CrossRef CAS PubMed; (d) K. Miyazawa, T. Koike and M. Akita, Chem.–Eur. J., 2015, 21, 11677–11680 CrossRef CAS PubMed; (e) J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017–14021 CrossRef CAS PubMed; (f) Q.-Q. Zhao, X.-Q. Hu, M.-N. Yang, J.-R. Chen and W.-J. Xiao, Chem. Commun., 2016, 52, 12749–12752 RSC; (g) J. Davies, T. D. Svejstrup, D. F. Reina, N. S. Sheikh and D. Leonori, J. Am. Chem. Soc., 2016, 138, 8092–8095 CrossRef CAS PubMed; (h) X.-Q. Hu, J. Chen, J.-R. Chen, D.-M. Yan and W.-J. Xiao, Chem.–Eur. J., 2016, 22, 14141–14146 CrossRef CAS PubMed; (i) K. Tong, X. Liu, Y. Zhang and S. Yu, Chem.–Eur. J., 2016, 22, 15669–15673 CrossRef CAS PubMed; (j) X.-Q. Hu, X. Qi, J.-R. Chen, Q.-Q. Zhao, Q. Wei, Y. Lan and W.-J. Xiao, Nat. Commun., 2016, 7, 11188 CrossRef PubMed; (k) Y. Zhao, B. Huang, C. Yang, B. Li, B. Gou and W. Xia, ACS Catal., 2017, 7, 2446–2451 CrossRef CAS; (l) D. F. Reina, E. M. Dauncey, S. P. Morcillo, T. D. Svejstrup, M. V. Popescu, J. J. Douglas, N. S. Sheikh and D. Leonori, Eur. J. Org. Chem., 2017, 2108–2111 CrossRef CAS.
- Nitrogen-centered radicals containing additional electron-withdrawing substituents prefer to react with electron-rich alkenes in intermolecular reactions. For recent examples, see: (a) G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521–11524 CrossRef CAS PubMed; (b) X. Shen, K. Harms, M. Marsch and E. Meggers, Chem.–Eur. J., 2016, 22, 9102–9105 CrossRef CAS PubMed; (c) X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636–12642 CrossRef CAS PubMed.
- L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed.
- For selected reviews and accounts on photoredox catalysis with transition metal based photoredox catalysts, see: (a) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (c) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed; (d) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295–2306 CrossRef CAS PubMed; (e) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C. Wang, L.-A. Chen, H. Huo, X. Shen, K. Harms, L. Gong and E. Meggers, Chem. Sci., 2015, 6, 1094–1100 RSC.
- J. Ma, X. Shen, K. Harms and E. Meggers, Dalton Trans., 2016, 45, 8320–8323 RSC.
- H. Huo, C. Fu, K. Harms and E. Meggers, J. Am. Chem. Soc., 2014, 136, 2990–2993 CrossRef CAS PubMed.
- For the persistent radical effect, see: A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS.
- The stabilization of enolate radical anions derived from α,β-unsaturated 2-acyl imidazoles through Lewis acid coordination has been reported. See: E. L. Tyson, E. P. Farney and T. P. Yoon, Org. Lett., 2012, 14, 1110–1113 CrossRef CAS PubMed.
- In the absence of a photoredox mediator, apparently the photoexcited Rh-substrate complex intermediate I is directly initiating the electron transfer, albeit less efficiently (see Table 1, entry 12).
- For the stereocontrolled chemistry of photochemically generated prochiral radicals, see: (a) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed; (b) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS PubMed; (c) J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed; (d) D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768–13771 CrossRef CAS PubMed; (e) C. Wang, J. Qin, X. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685–688 CrossRef CAS PubMed; (f) T. Kizu, D. Uraguchi and T. Ooi, J. Org. Chem., 2016, 81, 6953–6958 CrossRef CAS PubMed; (g) M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017 DOI:10.1038/nchem.2748.
- (a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed; (b) J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604–14605 CrossRef CAS PubMed; (c) M. A. Ischay, M. S. Ament and T. P. Yoon, Chem. Sci., 2012, 3, 2807–2811 RSC . See also ref. 18 and 20c.
- J. H. Horner, O. M. Musa, A. Bouvier and M. Newcomb, J. Am. Chem. Soc., 1998, 120, 7738–7748 CrossRef CAS.
- For a failed intramolecular addition of carbamoyl nitrogen-centered radical to an acceptor-substituted alkene, see for example: K. C. Nicolaou, P. S. Baran, R. Kranich, Y.-L. Zhong, K. Sugita and N. Zou, Angew. Chem., Int. Ed., 2001, 40, 202–206 CrossRef CAS PubMed.
- (a) D. A. Evans, K. R. Fandrick and H.-J. Song, J. Am. Chem. Soc., 2005, 127, 8942–8945 CrossRef CAS PubMed; (b) D. A. Evans and K. R. Fandrick, Org. Lett., 2006, 8, 2249–2252 CrossRef CAS PubMed; (c) D. A. Evans, H.-J. Song and K. R. Fandrick, Org. Lett., 2006, 8, 3351–3354 CrossRef CAS PubMed; (d) D. A. Evans, K. R. Fandrick, H.-J. Song, K. A. Scheidt and R. Xu, J. Am. Chem. Soc., 2007, 129, 10029–10032 CrossRef CAS PubMed.
- H.-J. Zheng, W.-B. Chen, Z.-J. Wu, J.-G. Deng, W.-Q. Lin, W.-C. Yuan and X.-M. Zhang, Chem.–Eur. J., 2008, 14, 9864–9867 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02031g |
| This journal is © The Royal Society of Chemistry 2017 |