
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMono-N-protected amino acid ligands stabilize dimeric palladium(II) complexes of importance to C–H functionalization†
Joseph J.
Gair
 a,
Brandon E.
Haines
b,
Alexander S.
Filatov
a,
Djamaladdin G.
Musaev
*b and
Jared C.
Lewis
*a
a,
Brandon E.
Haines
b,
Alexander S.
Filatov
a,
Djamaladdin G.
Musaev
*b and
Jared C.
Lewis
*a
aDepartment of Chemistry, The University of Chicago, Chicago, Illinois 60637, USA. E-mail: jaredlewis@uchicago.edu
bCherry L. Emerson Center for Scientific Computation, Emory University, 1515 Dickey Drive, Atlanta, Georgia 30322, USA. E-mail: dmusaev@emory.edu
First published on 16th June 2017
Abstract
Mono-protected amino acid (MPAA) ligands are used in a number of Pd-catalyzed C–H functionalization reactions. MPAAs have been proposed to bind to Pd(II) via κ2-(N,O) coordination, but such binding has not yet been experimentally validated. Herein, we report the synthesis and detailed characterization of a series of MPAA complexes prepared via cyclopalladation of dimethylbenzylamine in the presence of MPAAs. The isolated complexes exist as μ-carboxylato (MPAA) bridged dimers and feature potential M–M cooperativity and secondary sphere hydrogen bonding. Selective MPAA coordination and relay of stereochemistry, previously suggested to uniquely result from κ2-(N,O) MPAA coordination, are both observed. The isolated MPAA complexes undergo C–C and C–X (X = Cl, Br, I) bond formation when treated with electrophiles used for catalytic C–H functionalization. Stoichiometric iodination of MPAA palladacycles was found to proceed via a dinuclear palladium species with one equivalent of iodine in the rate limiting transition structure, and the isolated complexes also served as viable precatalysts for catalytic C–H functionalization. Together, these results provide a number of insights into the reactivity of Pd-MPAA complexes relevant to C–H bond functionalization.
Introduction
One of the foremost challenges in transition metal catalyzed C–H functionalization has been the development of catalysts that selectively modify one C–H bond in the presence of others with similar steric and electronic properties.1–6 Palladium catalyzed transformations that exploit the ability of Lewis basic functional groups within a substrate to coordinate to the metal catalyst and thus confer site selectivity to C–H bond cleavage (activation) have long proven particularly useful in this regard.7–9 In combination with this approach, mono-N-protected amino acid (MPAA) ligands have been found to accelerate10,11 and impart enantioselectivity12–17 to several Pd(II)-catalyzed C–H functionalization reactions.18Numerous approaches, including DFT calculations,19–23 mass spectrometry,24,25 and steady state kinetics10,26 have been used to investigate the mechanism(s) by which MPAA ligands affect Pd(II)-catalyzed C–H functionalization. These studies have concluded that MPAA-promoted, Pd(II)-catalyzed C–H bond functionalization proceeds via a N–H cleavage and subsequent C–H activation mechanism.21 The proposed active catalyst is generated by κ2-(N,O) coordination of MPAA to Pd(II) (Chart 1) followed by deprotonation of the N-protected amide. Concerted metallation-deprotonation (CMD)27–31 of the substrate C–H bond by either the N-protecting group25 of the MPAA ligand or an external base19 could then occur. κ2-(N,O) binding of the MPAA ligand is thought to enforce a rigid structure capable of relaying chirality from the MPAA to a prochiral substrate in the CMD transition state.24 With the development of this mechanistic paradigm, it has become widely accepted that the active catalysts are monomeric κ2-(N,O) Pd(II)MPAA complexes.
 | ||
| Chart 1 Three potential modes for coordination of the MPAA N-acetylglycine to Pd(II). | ||
Notably, however, no Pd(II)MPAA intermediates in these reactions have been rigorously characterized in solution or by X-ray diffraction. To shed light on the nature of MPAA coordination to Pd(II) and to understand the impact that MPAA coordination has on the nuclearity and stoichiometric reactivity of these complexes, we sought to isolate and characterize Pd(II)MPAA complexes analogous to those invoked for catalysis. Such complexes would also serve as models to specifically probe and understand individual steps of proposed catalytic cycles and guide the design of new and more effective ligands for C–H functionalization catalysis.
Results and discussion
Synthesis of MPAA complexes via ligand exchange
Putative κ2-(N,O) binding of MPAA ligands to Pd(II) during catalytic C–H functionalization was first proposed17 in the literature based on the previously reported32 synthesis of 1, Pd(dmba)(NAc-Gly) (Fig. 1a, dmba = N,N-dimethylbenzylamine, NAc-Gly = N-acetyl-glycine). Indeed, we obtained a good yield (71%) of a complex consistent with the 1H and 13C NMR data reported for 1 by following this procedure. The κ2-(N,O) MPAA coordination in 1 was originally assigned based on analogy to the corresponding unprotected glycine complex.32,33 To gain further confidence in this assignment, we determined the structure of the isolated product by single-crystal X-ray analysis. The structure observed in the solid state is the μ2-(O,O) MPAA-bridged dimer complex 2 (Fig. 1c) instead of the reported κ2-(N,O) complex 1. | ||
| Fig. 1 (a) Previously reported synthesis of 1, which is herein reassigned as complex 2. (b) Relative calculated energies (ΔG/ΔH) of 1, 2, and 3 with observed NOEs noted on 2; (c) ORTEP diagram of 2 with 50% ellipsoids; in structures throughout yellow = Pd, red = O, blue = N, grey = C, white = H, and green = halogen; (d) portion of 1H NOESY spectrum with diagnostic cross peaks; (e) overlaid 1H NMR spectra of MPAA complex 2 with acetate complex 4 offset by 0.1 ppm for clarity. | ||
To address the possibility that this dimer may only exist in the solid state, a solution of 2 was analyzed by 1H NMR, NOESY, and ESI-MS—all of which are consistent with a dimeric MPAA-bridged palladium complex. An initial indication of a dimeric complex in solution is the large upfield shift in one of the inequivalent benzylic resonances (Ha) and N-methyl resonances (Mea) relative to the precursors [Pd(dmba)(Cl)]2 and Pd(dmba) (acac) (acac = acetylacetonate) (Fig. 1b and e). This large upfield shift is consistent with proximity to the ring current of an aromatic π-system—as one would expect for the carboxylate bridged dimer 2—and is strikingly similar to the 1H NMR of known acetate bridged dimer 4 [Pd(dmba)(OAc)]2 (Fig. 1e).34–36 The most direct evidence for the existence of 2 as a dimer in solution is the presence of an NOE between the aromatic C–H ortho to palladium with the upfield NMea and while the downfield resonance NMeb has no such NOE (Fig. 1d).
DFT calculations37 were conducted to gain further insight into the stability of the dimeric structure of 2. Good agreement between the calculated and crystal structures of 2 is observed; for example, the Pd–Pd distances in the two structures are 2.95 and 2.99 Å, respectively (see ESI† for full structural analysis). Moreover, the free energy/enthalpy of dimerization (ΔGdimer/ΔHdimer) for 2 (i.e. energy of the reaction 2(1) → 2) is calculated to be −21.6/−32.3 kcal mol−1 (Fig. 1b). This finding shows that the dimeric structure 2 is significantly more stable than that of two putative monomers (1). Calculations also indicate that the κ2-(N,O) coordination mode of NAc-Gly in 1 is less stable than the κ2-(O,O) binding mode (3) by 2.0 kcal mol−1, suggesting that the proposed κ2-(N,O) coordination mode may not be the most stable monomer complex. We also located a μ-(N,O) dimer structure with dmba and NAc-Gly, 2-μ-(N,O), that is higher in energy than 2 by 16.0 kcal mol−1. These results show, for the first time, that widely used MPAA ligands can stabilize dimeric, μ-(O,O) carboxylate-bridged Pd(II)MPAA complexes directly analogous to widely-studied acetate-bridged palladacycles.38–43
Synthesis of MPAA complexes via C–H activation
To determine if 2 could also be prepared via cyclopalladation of dmba in the presence of MPAA, an equimolar mixture of palladium acetate, dmba, and NAc-Gly was stirred at room temperature in methanol. Analysis of the crude reaction mixture revealed 2 as the major product despite the presence of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 excess of acetate:MPAA, indicating selective coordination of MPAA over acetate. Complete conversion of dmba was observed overnight, and a 57% isolated yield of 2 was obtained after crystallization. These conditions were used to prepare MPAA-bridged complexes 6a–d (NAc-Gly, NAc-Ala, NAc-Leu, NAc-Ile) from trifluoromethyl dmba derivative 5a (F3C-dmba) in 62–91% yield by 19F NMR (Scheme 1).
:1 excess of acetate:MPAA, indicating selective coordination of MPAA over acetate. Complete conversion of dmba was observed overnight, and a 57% isolated yield of 2 was obtained after crystallization. These conditions were used to prepare MPAA-bridged complexes 6a–d (NAc-Gly, NAc-Ala, NAc-Leu, NAc-Ile) from trifluoromethyl dmba derivative 5a (F3C-dmba) in 62–91% yield by 19F NMR (Scheme 1).
 | ||
| Scheme 1 Synthesis and 19F NMR yields of carboxylate bridged F3C-dmba dimers 6a–d. | ||
DFT calculations performed for 6a–d and their acetate-bridged analogue, 9, are consistent with these experimental findings. In all cases, the formation of carboxylate-bridged dimers, [(dmba)Pd(MPAA)]2, from separated monomers is calculated to be highly exergonic: ΔGdimer/ΔHdimer = −18.5/−35.9, −18.5/−36.7, −21.0/−39.0, −21.5/−39.6 and −17.3/−33.1 kcal mol−1 for 6a, 6b, 6c, 6d and 9, respectively. These computational data demonstrate that all studied MPAAs form more stable dimeric complexes than their acetate analogs. Furthermore, the stability of [(dmba)Pd(MPAA)]2 relative to the corresponding monomers increases with increasing size of the MPAA ligand side chain (i.e., 6c/6d > 6a/6b). Similar to our previous findings for acetate-bridged Pd(II) complexes, however, it is expected that the stability of MPAA-bridged complexes will depend on the nature of substrate directing group, MPAA, and environment (solvent, additives, etc.).44
Characterization of dimeric Pd(II)MPAA complexes
Single crystal X-ray diffraction studies of 6b–d confirm that each of these complexes exists as a carboxylate-bridged dimer in the solid state. However, detailed mechanistic studies are not possible without an understanding of structure in solution: though solid state structures can provide insight, solution structures and aggregation states must be determined independently.45,46 Pulse gradient spin-echo (PGSE) experiments were conducted to assess the nuclearity of the MPAA complexes in solution.47,48 The molecular volume of the monomeric acac complex 7 was benchmarked at 1.0 and the dimeric acetate and chloride bridged complexes (8 and 9) gave molecular volumes of 1.9 and 2.4 relative to 7, indicating the ability of PGSE experiments to differentiate monomeric from dimeric Pd(dmba) complexes (Chart 2). Under the same conditions, the MPAA complexes 6a–d all gave relative molecular radii greater than the dimeric control complexes (3.9–4.8) consistent with dimeric MPAA complexes in solution. | ||
| Chart 2 Relative molecular volumes approximated from diffusion coefficients obtained by pulse gradient spin-echo NMR. | ||
The dimeric nature of complexes 6a–d in solution is further supported by their 1H NMR and NOESY spectra, which display the same diagnostic features as the acetate bridged complexes 4 and 9 (Fig. 1e and Chart 2, respectively). Specifically, upfield shifts due to magnetic anisotropy and NOE cross peaks with upfield-shifted resonances were observed. Full characterization for 6a–d is provided in the ESI† (1H, 13C{1H}, 19F, HMQC, COSY, and NOESY).
A noteworthy feature of carboxylate bridged palladacycles is the chirality of the stacked, square planar coordination spheres (Chart 3). As expected for an achiral ligand, all of the NAc-Gly complexes crystallize as racemic mixtures with both enantiomers present in the unit cell. When an additional stereogenic unit is introduced to the MPAA (6b–d), the chirality of the complex gives rise to two diastereomers, which are present in an approximately equal ratio for 6b–d and are differentiable by NMR as illustrated in Fig. 2. It is interesting to note that, despite a nearly 10 Å separation of the CF3 substituents and the stereogenic centers of the MPAA ligands, the extent of diastereotopic differentiation of 19F NMR appears to be correlated with the size of the MPAA side chain (Δδ = 4.5, 15, and 26 Hz for R = Me (6b), iso-Bu (6c), and sec-Bu (6d)).
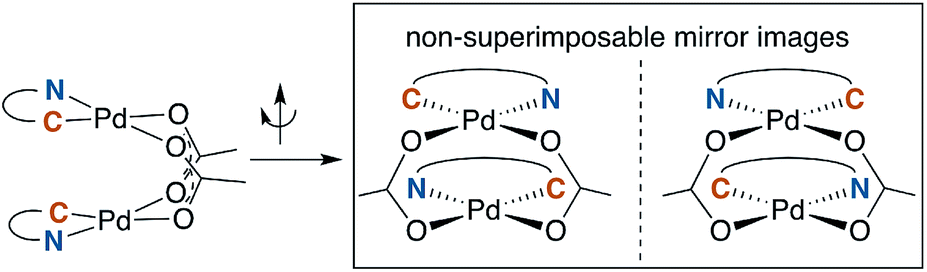 | ||
| Chart 3 Chirality of carboxylated bridged palladacycles. | ||
 | ||
| Fig. 2 19F NMR of 6a–d show correlation between diastereotopic differentiation and size of MPAA side chain. | ||
Assessing monomer dimer equilibrium
To address the possibility that other species (e.g., κ2-(N,O) monomer) may be in equilibrium with the observed MPAA-bridged dimers, a crossover experiment was conducted which revealed rapid equilibration of homodimers to a statistical mixture of hetero and homodimers (Scheme 2 and ESI S1.5†).43 Rapid crossover at room temperature is consistent with a small equilibrium concentration of monomeric MPAA complex capable of rapidly recombining with other free monomers in solution. This rapid equilibrium was further assessed by 19F EXSY experiments which revealed ΔG‡exchange = 19.2 kcal mol−1 at 21 °C.49 The observed ΔG‡exchange is comparable to the calculated ΔGdimer of 6a (18.5 kcal mol−1) and thus compatible with an exchange pathway proceeding through high energy monomeric intermediates (see ESI S1.5† for 19F EXSY). | ||
| Scheme 2 Crossover experiment with MPAA bridged dimers 2 and 6a reveals rapid exchange. | ||
The rapid exchange of homodimers to equilibrium mixtures of homo and heterodimers enabled analysis by the method of continuous variation (MCV) which has proven especially powerful for characterizing the structure and aggregation state of complex mixtures in solution.45,46,50 This approach confirmed the dimeric nature of 2 and 6a in solution by revealing a characteristic statistical ensemble of dimers (see ESI S1.5† for Job plot).50 Complexes 6a and 6b were further characterized by variable temperature 1H NMR (233–353 K), variable concentration 19F NMR (3–39 mM), and variable concentration UV-vis (0.02–0.5 mM) to further assess the presence of the putative monomeric species involved in the equilibrium shown in Scheme 2. Across this range of conditions, there was no observable monomer or any other species in equilibrium with the characterized dimeric (MPAA)Pd-complexes. Attempts to observe monomeric (MPAA)Pd-complexes by trapping with excess of amine substrate revealed no change in the 1H and 19F spectra of 6a with up to 100 equivalents of 5a (ESI S1.4†). Collectively, these results as well as the DFT-calculated dimerization energies are consistent with the existence of high energy monomer complexes in rapid equilibrium with the observed MPAA-bridged dimer complexes.
Effect of MPAA on rate of cyclopalladation
As noted above, C–H activation of dmba with palladium acetate gives selective formation of MPAA-bridged over acetate-bridged di-palladium products, the later of which have been extensively characterized in stoichiometric studies and as catalytic intermediates.38–43This selectivity could result from more favorable binding of MPAA relative to acetate or faster cyclometallation by a Pd/MPAA species relative to Pd/OAc. Cyclopalladation of CF3-dmba to afford discrete MPAA complexes (6a–d) provides a unique model system to study the impact of MPAA on cyclopalladation rates. Initial rates for the reaction shown in Scheme 3 were similar in the presence and absence of NAc-Ala and sodium acetate (NaOAc). These results suggest that kinetic effects are not responsible for the selective coordination of MPAA over acetate in compounds 6a–d. Previous studies have suggested that observed rate enhancements in MPAA catalyzed C–H functionalization are the result of MPAA ligands lowering the barrier to C–H cleavage. However, MPAA accelerated C–H cleavage has not yet been directly confirmed in stoichiometric reactions, and in the system studied here, a rate effect does not account for selective MPAA coordination.
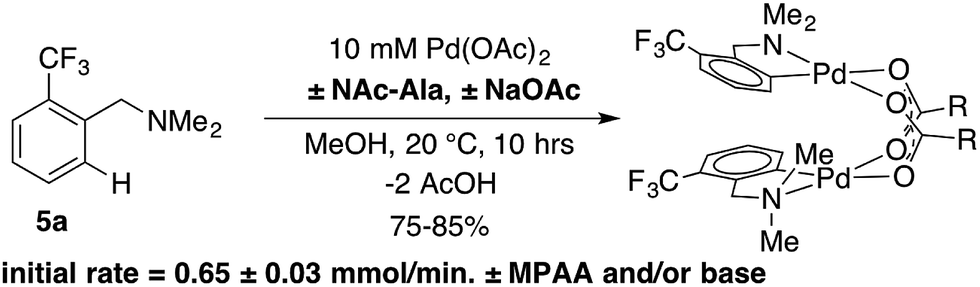 | ||
| Scheme 3 MPAA and carboxylate bases showed no effect on the rate of cyclopalladation of 5a. | ||
Pd(II)-MPAA binding affinity
To determine whether thermodynamic factors are responsible for selective MPAA coordination during the formation of 6a–d, carboxylate exchange reactions were conducted using AcOH, NAc-Gly, NAc-Ala, and NAc-Ile (Scheme 4). In forward and reverse reactions involving NAc-Gly, an equilibrium mixture containing approximately 5% acetate-bridged complex 9 was observed.51 In contrast, when 9 was equilibrated with NAc-Ala and NAc-Ile, the bulkier MPAA's drove the equilibrium to <1% complex 9. Poor solubility of NAc-Gly and overlapping chemical shifts of NAc-Ala and NAc-Ile complexes precluded quantitative evaluation of the binding affinities of MPAAs and acetate. Qualitatively however, our experiments show that NAc-Gly, NAc-Ala, and NAc-Ile selectively displace acetate and suggest that NAc-Ala and NAc-Ile have a greater binding affinity than NAc-Gly (see ESI S1.8† for 1H and 19F NMR of MPAA/OAc equilibrium mixtures). Thus, while these are structurally similar to acetate bridged dimers, differential binding strength leads to selective coordination of MPAAs, which can carry stereochemical information. | ||
| Scheme 4 Competitive carboxylate binding equilibria. | ||
To better understand bridging carboxylate binding affinity, we extended our DFT studies to examine ligand exchange equilibria (ΔGL/ΔHL) between acetate and MPAA ligands as defined by the equation: 9 + 2 MPAA → 6a/6b/6d + 2 AcOH (Scheme 4).52 The ligand exchange equilibrium was found to favor coordination of the MPAA ligands in the dimer complex, where ΔGL/ΔHL = −4.0/−6.5, −7.5/−7.7 and −9.8/−11.0 kcal mol−1 for 6a, 6b, and 6d, respectively. The thermodynamic preference for MPAA binding relative to acetate is consistent with the results of a previous study on carboxylate-bridged palladium(II) dimers, in which we found that electron-withdrawing groups on bridging carboxylates stabilize the dimer relative to separated monomers by increasing interaction energy between the Pd-center and ligands (i.e. via the Pd–OCO bonding motif) (see Table S2.3 in the ESI† for charge analysis).44 Thus, both experimental and computational results suggest bridging carboxylate binding affinity follows the trend acetate < NAc-Gly < NAc-Ala, NAc-Ile.
Substrate scope of C–H activation
To assess whether substrates used in C–H functionalization reactions catalyzed by Pd(II)/MPAA mixtures can form dimeric complexes analogous to 6a–d, 1 mM methanol solutions of Pd(OAc)2, NAc-Ala, and substrate were analyzed by ESI-MS after 30 minutes of mixing (Fig. 3a).17,53,54 The substrate dmba (in 11) was studied to confirm that reaction mixtures afforded similar mass spectra to the isolated complex 2. The substrates 2-pyridyl-diphenylmethane17 (in 12) and dimethylamino-ferrocenyl-methane53,54 (in 13) were also evaluated because both have been used in enantioselective Pd(II)/MPAA catalyzed C–H functionalization. Moreover, the former was the subject of computational,21 synthetic,21 and mass spectrometry24 experiments aimed at understanding the impact of MPAA ligands on catalysis, and the latter was used in stoichiometric enantioselective cyclopalladation reactions with MPAA additives.55–58 Thus, while these substrates cannot, of course, represent the full scope Pd(II)/MPAA catalyzed C–H functionalization due to the sensitivity of these reactions to different directing groups,59 they are structurally similar to dmba, which we have studied in depth, and both have been used for stoichiometric model studies21,24,55–58 and catalysis.17,53,54 | ||
| Fig. 3 (a) Reaction conditions to generate MPAA complexes for analysis by ESI-MS; (b) ions for di-palladium MPAA complexes (1 MPAA) (c) observed isotope patterns for ions 11–13 (black) and overlaid theoretical isotope patterns (green) from ESI-MS analysis of reactions in (a).62 | ||
As expected, the di-palladium ion 11 (with dmba substrate) was observed. While ions consistent with monomeric cyclopalladated (MPAA)Pd-complexes were previously reported for 2-pyridyl-diphenylmethane,24 we observe ions consistent with both monomeric and dimeric Pd species (12) for this substrate17,60 and for dimethylamino-ferrocenyl-methane53,54,61 (13) (Fig. 3). The observed di-palladium ions are analogous to those observed in the MS of isolated, characterized carboxylate-bridged dimers 2, 4, 6a–d and 9. Moreover, di-palladium ions 11–13 had a higher intensity than those corresponding MPAA to monomeric complexes. For all three substrates, the palladium-containing ion with the greatest intensity is the cyclopalladated substrate with no ancillary ligands (or a fragment thereof) (see Sections S4.1, S4.2 and S4.3 in the Section S3 in the ESI† for full spectra of 11, 12, and 13 respectively).
Given the observation of ions consistent with di-palladium substrate complexes by MS, we sought to determine whether di-palladium complexes are also present in the absence of substrate.63 While a previous report25 noted the monomeric species Pd(MPAA)+ under such conditions, we found that 2 mM acetonitrile solutions of Pd(OAc)2 and MPAA (NAc-Gly, NAc-Ala, and NAc-Leu) contain an ion corresponding to the dimeric complex Pd2(MPAA)2+ in addition to Pd(MPAA)+ in ratios dependent on fragmentor voltage (see Sections 4.4–4.6 in the ESI† for labeled spectra from 50–230 V). These results suggest that MPAA-bridged di-palladium species can form both in the absence and presence of multiple substrates used in enantioselective C–H functionalization under conditions similar to those used in the preparation of dimeric MPAA-bridged palladacycles 6a–d.
We have also calculated the ΔGdimer/ΔHdimer values for cyclopalladated carboxylate bridged Pd/MPAA species with 2-pyridyl-diphenylmethane60 and 2-benzylpyridine64 substrates, as well as with acetonitrile (i.e., no substrate). In all cases, we find that the dimer is favored over the separated two monomers (see Section S2.4 in the ESI† for full details). As in our previous study, these calculations show that the nature of substrate can have a large impact on the thermodynamic stability of dimeric complexes.44 For example, calculations show that the additional phenyl substituent of 2-pyridyl-diphenylmethane stabilizes the corresponding cyclopalladated dimer complex relative to that derived from 2-benzylpyridine. The presented calculations are consistent with the experimental data showing that MPAA-bridged dimeric complexes can form with a wide range of substrates relevant for C–H functionalization.
Reactivity of MPAA complexes toward electrophiles
The facile formation of dimeric Pd(II)MPAA complexes 2 and 6a–d, along with the observation by MS and the calculated energies of analogous complexes with substrates used in catalysis, suggests that MPAA-bridged complexes could form under conditions relevant to catalytic C–H functionalization. The reactivity of 2 and 6b toward electrophiles used in MPAA-facilitated C–H functionalization was therefore assessed. Both complexes underwent reaction with Michael acceptors,10,65 hypervalent iodine oxidants,66 and elemental halogens12,67 to give C–C and C–X (X = Cl, Br, I) bond formation as illustrated in Scheme 5.68 | ||
| Scheme 5 Representative reactions of cyclopalladated MPAA complexes with electrophiles. | ||
To determine whether 6b reacts with electrophiles as a dimer or perhaps as a putative monomeric species, a kinetic analysis was performed of the reaction of 6b with I2 (though 6b may react differently with other electrophiles). Initial control experiments revealed that iodination of 6b in the presence of increasing, catalytic amounts of exogenous iodide (0.033–0.53 equiv., Bu4NI) exhibits saturation behavior (Fig. 4b). Thus, to avoid interference from iodide that may be generated over the course of the iodination reaction, the order with respect to iodine was determined in the presence of a saturating concentration of Bu4NI (53 mol%). Under these conditions, initial rate analysis indicated that the reaction was first order in iodine from 3–39 mM (Fig. 4c). Under pseudo first order conditions, the disappearance of 6b could be fit using a single exponential function indicating first order kinetics (Fig. 4d). These results, in the presence of catalytic exogenous iodide, are consistent with a reaction of iodine with the dimer 6b in the rate limiting transition structure, rather than reaction at a monomeric intermediate. Additional data and discussion of iodination in the absence of exogenous iodide are presented in the ESI† (see Section S1.14 for Experimental data and S2.6† for computational analysis).
 | ||
| Fig. 4 (a) Iodination of cyclopalladated MPAA complex 6b; (b) plot of initial rate of iodination of 6bversus equivalents of exogenous iodide; (c) plot of initial rate of iodination of 6bversus initial concentration of I2 showing first order kinetics in I2 under saturating Bu4NI conditions; (d) overlaid reaction profiles of iodination of 6b from 3–39 mM I2 with exponential fits for pseudo-first order reactions; (e) overlaid reaction profiles of iodination of 6b in the presence and absence of Bu4NI with an exponential fit in the presence of Bu4NI and a linear fit for zero order portion in the absence of Bu4NI. | ||
Observation of C–H activation from MPAA palladacycles
While studying the reactivity of Pd(II)/MPAA complexes toward electrophiles, we found that cyclopalladated substrates with a C–H bond ortho to the directing group can undergo subsequent C–H activation chemistry. This reactivity affords a unique opportunity to study C–H activation in MPAA complexes with well-defined, di-palladium reactants and products. For example, combining 2 and iodobenzene dichloride led to the formation of chlorinated Pd(II)MPAA complexes 15a and 15b (Fig. 5a). This constitutes the first report of an MPAA-ligated palladacycle reacting to give discrete Pd(II)MPAA products.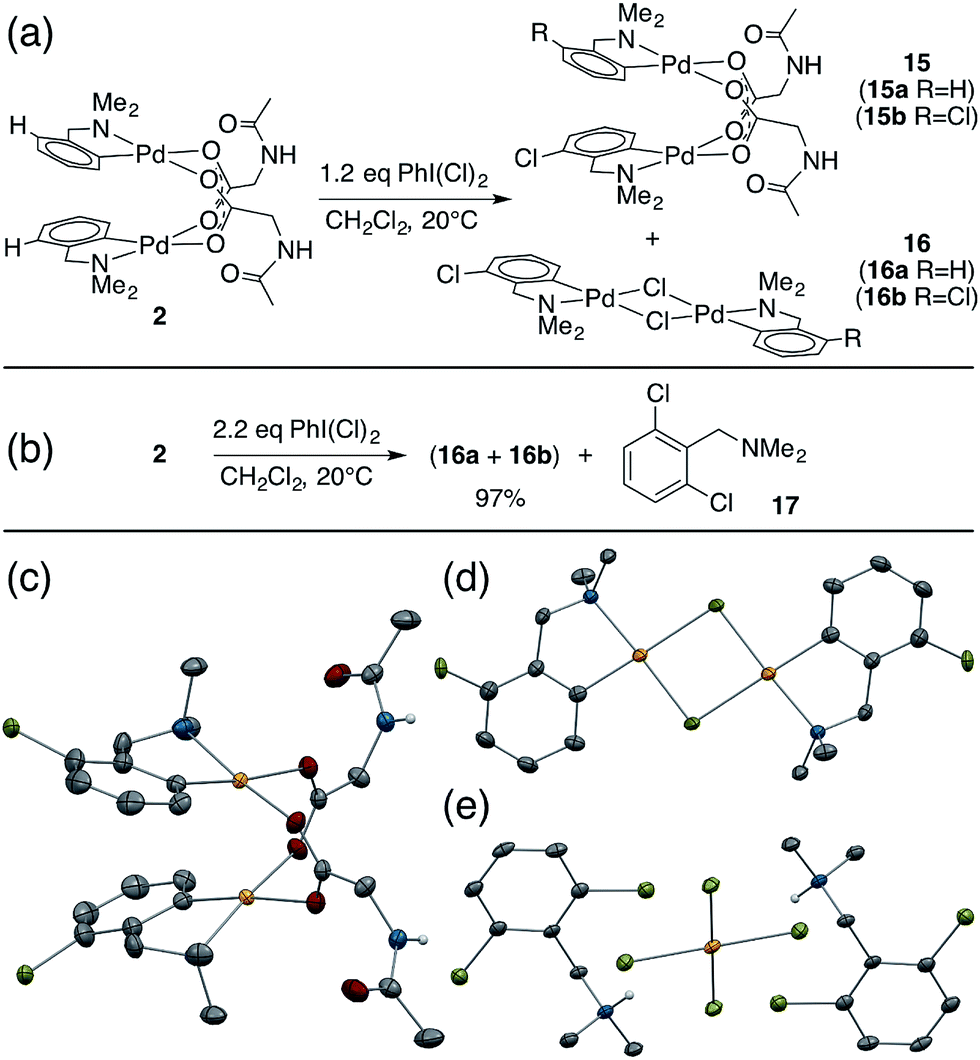 | ||
| Fig. 5 (a) Sequential chlorination and C–H activation of 2; (b) chlorination of 2 with excess iodobenzene dichloride; (c) ORTEP of co-crystallized C–H activation products 15a and 15b: chlorines are disordered over both positions in dimer and were refined at 50% chemical occupancy; (d) ORTEP of co-crystallized C–H activation products 16a and 16b: chlorines are disordered over both positions in dimer and were refined at 20% chemical occupancy; (e) ORTEP of product of dmba dichlorination (17) co-crystallized with chloro-palladate. | ||
Further investigation into the reaction of 2 with iodobenzene dichloride revealed the presence of chloride bridged dimers 16a, and 16b, which could result from carboxylate/chloride metathesis between 15a–b and HCl generated in situ (Fig. 5a). The MPAA- and chloride-bridged di-palladium products crystallized as needles and blocks, respectively, and could be separated manually. The mono- and di-chlorinated dimers co-crystallized, however, leading to partial occupancy of the chlorinated positions in the structures shown in Fig. 5c and d. Treating 2 with excess iodobenzene dichloride led to chlorination and C–H activation of both dmba ligands and complete metathesis with chloride to give 75% 16b and 22% 16a, which reacted further with excess oxidant to give partial conversion to dichlorinated dmba 17 (Fig. 5b and e). The dichlorinated compound (17) was protonated by HCl generated in situ and co-crystallized as a chloro-palladate salt (Fig. 5e).
The observation of stoichiometric C–H cleavage and Pd–C functionalization starting from dimeric MPAA-bridged Pd(II) complexes raises the possibility that both C–H activation and C–H functionalization could proceed via dimeric complexes. We envisioned that DFT calculations could provide further insight into potential reaction mechanisms, the nuclearity of potential intermediates (Pd(II)MPAA dimer or monomer), and factors controlling cyclopalladation and Pd–C bond functionalization for the F3C-dmba model complexes examined.
Computational analysis of cyclopalladation of dimeric and monomeric Pd(II)/MPAA complexes
Several computational studies have established cyclopalladation pathways for monomeric Pd(II)MPAA complexes,19–25 and we previously studied the effect of acetate-bridged dimeric Pd(II) complexes on cyclopalladation,44 but a similar study on dimeric MPAA-bridged Pd(II) complexes has not yet been reported. Here, we investigate the mechanisms of cyclopalladation of [Pd(II) (κ-N–F3C-dmba) (κ-OAc) (μ-NAc-Gly)]2 (18-D)69 and its monomer (21-M) complex (where -D and -M signify dimeric and monomeric species, respectively). The free energy surface for cyclopalladation of the lowest energy (F3C-dmba)Pd(II)/NAc-Gly dimer and monomer systems are shown in Fig. 6 (see Fig. S2.1 and S2.2† for full energy surfaces). Prior to C–H activation, the monomer–dimer equilibrium for the (F3C-dmba)Pd(II)/NAc-Gly system (i.e., 2 (21-M) → 18-D) is ΔGdimer/ΔHdimer = −3.3/−13.5 kcal mol−1, which is much smaller than that calculated for the cyclopalladated products 6a–d. This is attributed to the strong hydrogen bonding interaction between NAc-Gly and OAc in 21-M. As shown in Fig. 6, the overall free energy barrier (relative to 18-D) for the dimer pathway (ΔG‡/ΔH‡ = 23.2/20.3 kcal mol−1) is significantly lower than that for the monomer pathway (ΔG‡/ΔH‡ = 41.7/52.1 kcal mol−1). In addition, the dimer pathway has the larger driving force (again relative to the lowest reactant, 18-D) of ΔG/ΔH = −15.6/−15.5 kcal mol−1 compared to the monomer pathway, which is unfavorable by ΔG/ΔH = 7.2/23.9 kcal mol−1. | ||
| Fig. 6 Energy surfaces (ΔG/ΔH) for mononuclear and dinuclear C–H cleavage: carboxylate oxygens of NAc-Gly shown in red relevant acetate for CMD highlighted in green, and all complexes except 6a truncated for clarity, see above 6a for key. | ||
In both pathways, the C–H activation transition state requires an open coordination site on Pd to form the new Pd–C bond. Dissociation of one of the bridging MPAA ligands from 18-D, which is thermodynamically unfavorable by ΔG/ΔH = 20.3/18.7 kcal mol−1, leads to formation of an intermediate (19-D) that undergoes C–H activation with a small intrinsic barrier of 2.9 kcal mol−1 (TS1-D).70 Therefore, for the dimer pathway, cyclopalladation occurs via two sequential concerted-metallation-deprotonation (CMD) steps (at the transition states TS1-D and TS2-D) with ΔG‡/ΔH‡ = 23.2/20.3 and 19.0/18.3 kcal mol−1 energy barriers, respectively. Thus, the first cyclopalladation facilitates the second step of the reaction. On the other hand, formation of 22-M on the monomer pathway is unfavorable by ΔG/ΔH = 25.2/38.7 kcal mol−1 and the intrinsic barrier for C–H activation is 16.5 kcal mol−1. These results show that the dimer stabilizes the C–H activation reactant and transition state relative to the monomer. It is unlikely that only one factor is responsible for such a large difference in the dimer and monomer barriers (ΔΔG‡ = 13.6 kcal mol−1). Instead, we attribute the acceleration in the dimer to several additive or cooperative effects including decreased trans effect from the bridging carboxylate to the formed Pd–C bond, intramolecular hydrogen bonding, as well as Pd–Pd and dispersive interactions between the monomer fragments.
In summary, cyclopalladation of the (F3C-dmba)Pd(II)/MPAA dimer is calculated to be kinetically and thermodynamically favored over the monomer, indicating that the calculations generally support C–H cleavage/cyclopalladation via the dimer pathway. Notably, previous DFT studies of MPAA promoted C–H cleavage have evaluated energies of monomeric transitions states relative to monomeric MPAA reactants without commenting on possible dimeric pathways to C–H cleavage.19–25 When the full potential energy surface is analyzed, as in Fig. 6, it is apparent that assessing only the intrinsic barrier to C–H cleavage (i.e., 22-M → TS-M, where ΔG‡ = 16.5 kcal mol−1) neglects the fact the reactant in this step (22-M) is higher in energy than any of the intermediates or transition states on the dimer pathway. This illustrates the importance of assessing the energies of transformations involving monomeric and dimeric pathways relative to a common energetic reference when evaluating possible mechanisms for MPAA induced rate acceleration in C–H functionalization.
MPAA complexes as pre-catalysts
Based on our extensive characterization of stoichiometric C–H activation and Pd–C functionalization involving the dimeric, MPAA-bridged Pd(II) complexes outlined above, we sought to determine whether these complexes are also viable precatalysts for catalytic C–H functionalization reactions. Indeed, alkenylation of dmba was catalyzed with the same rate whether 2 or Pd(II)/NAc-Gly was used as the precatalyst (Scheme 6). However, similar to the stoichiometric C–H activations discussed above—in which selective MPAA coordination modified the complex's secondary coordination sphere, but did not afford rate enhancements—MPAA additives also did not accelerate catalytic C–H functionalization relative to palladium(II) acetate (see Fig. S1.3–S1.5 in the ESI†). These experiments thus confirm that MPAA bridged Pd dimer complexes can generate catalysts active for C–H functionalization, but they do not shed light the nuclearity or coordination mode of the active catalyst. | ||
| Scheme 6 Pre-catalyst 2 catalyzes olefination of dmba at the same rate as mixtures of Pd(OAc)2/NAc-Gly. | ||
On the other hand, examining the catalytic competence of isolated, structurally characterized Pd(II)MPAA complexes provides a concrete starting point for determining the nuclearity and coordination mode of intermediates in catalytic C–H functionalization reactions. Given the range of substrates and conditions used in reactions catalyzed by Pd(II)/MPAA mixtures and the potential impact of these differences on the coordination chemistry of complexes involved in catalysis, a detailed understanding of different coordination modes, such as those reported in this work, is essential. Importantly, however, characterizing the structure and reactivity of Pd(II)MPAA complexes with substrates relevant to catalysis is only a prerequisite to the loftier goal of understanding the molecular origins the rate acceleration and enantioselectivity afforded by MPAA ligands. The initial experiments summarized above using model complex 2 do not specifically implicate dimeric MPAA complexes in catalysis. On the other hand, further kinetic analysis of catalytic C–H functionalization reactions, informed by the structural characterization herein, provides a means to probe this possibility.
Summary and conclusion
Since the discovery that MPAA ligands can accelerate and confer enantioselectivity to C–H functionalization reactions, experimental and computational studies have been conducted to understand the molecular origins of these ligand effects. Prior to our work, the structure and reactivity of discrete Pd(II)MPAA complexes remained poorly characterized. The extensive synthetic, spectroscopic, and computational studies reported herein reveal—for the first time—an unprecedented class of dimeric MPAA-bridged palladacycles that are analogous to catalytically relevant acetate bridged dimers and that form selectively from mixtures of Pd(II), MPAA, and dmba derivatives. While our work focused on dmba model complexes, MS and computational analysis of analogous complexes generated from substrates used in catalysis suggests that dimeric, MPAA-bridged palladacycles could be relevant beyond dmba substrates.Our combined experimental and computational results show that structural features of putative Pd(II)MPAA complexes inferred from catalytic outcomes, including selective MPAA coordination and relay of stereochemistry, are observed in dmba model complexes. This finding indicates that such features do not, by themselves, necessitate κ2-(N,O) MPAA coordination to Pd(II) during catalysis. Selective formation of dimeric, MPAA-bridged complexes from MPAA/acetate mixtures results from greater MPAA binding affinity to Pd(II) relative to acetate. Diastereotopicity in bound dmba ligands results from relay of stereochemistry from distal MPAA side chains, but further experiments using substrates with prochiral C–H bonds will be required to determine if this phenomenon is relevant to enantioselective catalysis.
In addition to these observations, C–H activation of dmba in dimeric, MPAA-bridged complexes is calculated to be kinetically and thermodynamically favored over a monomeric pathway. Coordination of MPAA to Pd(II) in a κ2-(N,O) fashion is also not predicted to provide access to lower barrier pathways to C–H cleavage in dmba, consistent with our experimental observation that MPAA does not influence the rate of dmba cyclopalladation. On the other hand, calculations indicate that the relative stability of different Pd(II)MPAA dimer complexes is substrate and ligand specific, such that different complexes and reaction pathways could dominate for different reactions or under different conditions.
Despite their stability with respect to carboxylate exchange and calculated monomer–dimer equilibrium, the dmba model complexes studied undergo both Pd–C functionalization and subsequent C–H cleavage reactions relevant to C–H functionalization chemistry. A detailed kinetic analysis of stoichiometric iodination revealed that MPAA-bridged palladacycles react as dimers rather than as putative monomeric intermediates. This analysis is consistent with previous calculations showing a lower barrier to iodination at a dimeric palladacycle than the corresponding monomeric complex.71
In addition to their stoichiometric reactivity, dimeric MPAA-bridged palladacycles are competent precatalysts in palladium catalyzed C–H functionalization. Thus, the structure and reactivity of the complexes reported herein provide a starting point for understanding the role of MPAA ligands in catalysis. For example, MPAA bridged palladacycles project hydrogen bond donors and acceptors into the secondary coordination sphere of the Pd centers and create the potential for cooperative metal–metal interactions. Given reports72–76 of these features in catalytic mechanisms, these structural studies open the possibility of a range of additional mechanisms by which MPAA ligands might impact C–H functionalization. Alternatively, MPAA complexes could serve as stable, off-cycle catalyst reservoirs. Ongoing studies by our groups will focus on delineating possible roles of MPAA-bridged palladacycles in C–H functionalization catalysis. These fundamental studies will, in turn, provide a framework for designing improved ligands and discrete catalysts for more advanced applications.
Funding sources
This work was supported by the NSF under the CCI Center for Selective C–H Functionalization (CCHF, CHE-1205646) and by The University of Chicago Louis Block Fund for Basic Research and Advanced Study. JJG was supported by an NSF predoctoral fellowship (DGE-1144082) and by the University of Chicago Department of Chemistry. The authors gratefully acknowledge NSF MRI-R2 grant (CHE-0958205) and the use of the resources of the Cherry Emerson Center for Scientific Computation. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06-CH11357. ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1346572.Acknowledgements
We would like to thank CCHF members, including Jin-Quan Yu and Huw M. L. Davies, for many helpful discussions. We would also like to thank Alexander Rago for synthesizing 5a and 9, Alan M. Swartz for assistance acquiring mass spectra, and Zekai Lin for a preliminary solution to the structure of 15.References
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45(6), 936 CrossRef CAS PubMed.
- R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451(7177), 417 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40(4), 1857 RSC.
- J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2(5), 281 CrossRef CAS PubMed.
- J. F. Hartwig, J. Am. Chem. Soc., 2016, 138(1), 2 CrossRef CAS PubMed.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45(6), 788 CrossRef CAS PubMed.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48(28), 5094 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110(2), 1147 CrossRef CAS PubMed.
- K. M. Engle, D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2010, 132(40), 14137 CrossRef CAS PubMed.
- R. D. Baxter, D. Sale, K. M. Engle, J. Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134(10), 4600 CrossRef CAS PubMed.
- L. Chu, K. J. Xiao and J. Q. Yu, Science, 2014, 346(6208), 451 CrossRef CAS PubMed.
- X.-F. Cheng, Y. Li, Y.-M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X.-S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135(4), 1236 CrossRef CAS PubMed.
- L. Chu, X.-C. Wang, C. E. Moore, A. L. Rheingold and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135(44), 16344 CrossRef CAS PubMed.
- K.-J. Xiao, L. Chu and J.-Q. Yu, Angew. Chem., 2016, 128(8), 2906 CrossRef.
- B. N. Laforteza, K. S. L. Chan and J.-Q. Yu, Angew. Chem., 2015, 127(38), 11295 CrossRef.
- B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47(26), 4882 CrossRef CAS PubMed.
- K. M. Engle, Pure Appl. Chem., 2016, 88(1–2), 1 Search PubMed.
- B. E. Haines and D. G. Musaev, ACS Catal., 2015, 5(2), 830 CrossRef CAS.
- D. G. Musaev, T. M. Figg and A. L. Kaledin, Chem. Soc. Rev., 2014, 43(14), 5009 RSC.
- D. G. Musaev, A. Kaledin, B. F. Shi and J. Q. Yu, J. Am. Chem. Soc., 2012, 134(3), 1690 CrossRef CAS PubMed.
- X. M. Zhong, G. J. Cheng, P. Chen, X. H. Zhang and Y. D. Wu, Org. Lett., 2016, 18(20), 5240 CrossRef CAS PubMed.
- X. F. Cong, H. R. Tang, C. Wu and H. M. Zeng, Organometallics, 2013, 32(21), 6565 CrossRef CAS.
- G. J. Cheng, P. Chen, T. Y. Sun, X. H. Zhang, J. Q. Yu and Y. D. Wu, Chem.–Eur. J., 2015, 21(31), 11180 CrossRef CAS PubMed.
- G.-J. Cheng, Y.-F. Yang, P. Liu, P. Chen, T.-Y. Sun, G. Li, X. Zhang, K. N. Houk, J.-Q. Yu and Y.-D. Wu, J. Am. Chem. Soc., 2014, 136(3), 894 CrossRef CAS PubMed.
- R. D. Baxter, D. Sale, K. M. Engle, J.-Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134(10), 4600 CrossRef CAS PubMed.
- B. Biswas, M. Sugimoto and S. Sakaki, Organometallics, 2000, 19(19), 3895 CrossRef CAS.
- D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127(40), 13754 CrossRef CAS PubMed.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130(33), 10848 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- J. C. Gaunt and B. L. Shaw, J. Organomet. Chem., 1975, 102(4), 511 CrossRef CAS.
- M. D. Diaz-de-Villegas, J. Organomet. Chem., 1995, 490, 35 CrossRef.
- K. Severin, R. Bergs and W. Beck, Angew. Chem., Int. Ed., 1998, 37, 1634 CrossRef.
- A. D. Ryabov, I. K. Sakodinskaya and A. K. Yatsimirsky, J. Chem. Soc., Dalton Trans., 1985, 12, 2629 RSC.
- J. M. Thompson and R. F. Heck, J. Org. Chem., 1975, 4(18), 2667 CrossRef.
- A. Van Der Ploeg, G. Van Koten and K. Vrieze, J. Organomet. Chem., 1981, 222(1), 155 CrossRef CAS.
- Geometry optimizations and frequency calculations were performed at the B3LYP-D3BJ/[6-31G(d, p) + Lanl2dz (Pd, I)] level of theory (B3LYP-D3BJ/BS1) with a polarizable continuum model (PCM) using dichloromethane to account for solvent effects. The reported energies are calculated at the B3LYP-D3BJ/[6-311+G(2d, p) + SDD (Pd, I)] level with Gibbs free energy corrections to a solution standard state of 1 M at 298.15 K with either the PCM model for methanol or dichloromethane. (See ESI† for full description of computational methodology).
- N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131(31), 11234 CrossRef CAS PubMed.
- A. K. Cook and M. S. Sanford, J. Am. Chem. Soc., 2015, 137(8), 3109 CrossRef CAS PubMed.
- D. C. Powers, E. Lee, A. Ariafard, M. S. Sanford, B. F. Yates, A. J. Canty and T. Ritter, J. Am. Chem. Soc., 2012, 134(29), 12002 CrossRef CAS PubMed.
- D. C. Powers, D. Y. Xiao, M. A. L. Geibel and T. Ritter, J. Am. Chem. Soc., 2010, 132(41), 14530 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Nat. Chem., 2009, 1(4), 302 CrossRef CAS PubMed.
- D. C. Powers, D. Benitez, E. Tkatchouk, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132(40), 14092 CrossRef CAS PubMed.
- B. E. Haines, J. F. Berry, J.-Q. Yu and D. G. Musaev, ACS Catal., 2016, 6(2), 829 CrossRef CAS.
- L. R. Liou, A. J. McNeil, A. Ramirez, G. E. S. Toombes, J. M. Gruver and D. B. Collum, J. Am. Chem. Soc., 2008, 130(14), 4859 CrossRef CAS PubMed.
- L. R. Liou, A. J. McNeil, G. E. S. Toombes and D. B. Collum, J. Am. Chem. Soc., 2008, 130(51), 17334 CrossRef CAS PubMed.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42(1), 288 CrossRef CAS.
- P. S. Pregosin, P. G. A. Kumar and I. Fernández, Chem. Rev., 2005, 105(8), 2977 CrossRef CAS PubMed.
- C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90(6), 935 CrossRef CAS.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52(46), 11998 CrossRef CAS PubMed.
- Mixed MPAA/OAc complex 10 is also observed in the equilibrium mixture and is shown in the ESI in Section S1.8.†.
- We also calculated the mixed intermediate species in the exchange equilibrium with Gly. The overall equilibrium is 2e + 2 AcNH-Gly-OH → 2h + AcOH + AcNH-Gly-OH → 2a + 2 AcOH with ΔG = −2.3 and −1.7 kcal mol−1 for each step. These energy values are consistent with a product distribution of 2a/2h/2e = 89.7%/10.2%/0.1%. Thus, the DFT calculations slightly overestimate the ligand exchange equilibrium, but the trend is consistent with the experimental results.
- D.-W. Gao, Y.-C. Shi, Q. Gu, Z.-L. Zhao and S.-L. You, J. Am. Chem. Soc., 2013, 135(1), 86 CrossRef CAS PubMed.
- C. Pi, Y. Li, X. Cui, H. Zhang, Y. Han and Y. Wu, Chem. Sci., 2013, 4(6), 2675 RSC.
- V. I. Sokolov, L. L. Troitskaya and O. A. Reutov, J. Organomet. Chem., 1979, 182(4), 537 CrossRef CAS.
- V. I. Sokolov, L. L. Troitskaya and N. S. Khrushchova, J. Organomet. Chem., 1983, 250(1), 439 CrossRef CAS.
- N. Dendele, F. Bisaro, A.-C. Gaumont, S. Perrio and C. J. Richards, Chem. Commun., 2012, 48(14), 1991 RSC.
- M. E. Günay and C. J. Richards, Organometallics, 2009, 28(19), 5833 CrossRef.
- L. V. Desai, K. J. Stowers and M. S. Sanford, J. Am. Chem. Soc., 2008, 130(40), 13285 CrossRef CAS PubMed.
- A. J. Canty, N. J. Minchin, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1986, 10, 2205 RSC.
- T. K. Hollis and L. E. Overman, Tetrahedron Lett., 1997, 38(51), 8837 CrossRef CAS.
- The observed ions are represented as carboxylate bridged dimers based on our characterization of 1a and 2a–e, as well as reported acetate and trifluoroacetate bridged analogues of 3c and 3b, though further structural work would be required to rigorously establish the proposed connectivity.
- I. Y. Yakushev, A. E. Gekhman and A. P. Klyagina, Russ. J. Coord. Chem., 2016, 42(9), 604 CrossRef CAS.
- Y. Fuchita, Inorg. Chem., 1982, 20(12), 4316 Search PubMed.
- B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132(2), 460 CrossRef CAS PubMed.
- R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507(7491), 215 CrossRef CAS PubMed.
- X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135(28), 10326 CrossRef CAS PubMed.
- Bromination of 6b proceeds orders of magnitude faster than iodination, consistent with calculated barriers to palladacycle halogenation: see Fig. S1.2 in the ESI.†.
- J. Vicente, I. Saura-Llamas, M. G. Palin, P. G. Jones and M. C. Ramírez de Arellano, Organometallics, 1997, 16(5), 826 CrossRef CAS.
- It should be noted that the lowest energy dimer pathway for C–H activation calculated here is different than the pathway reported by us in ref. 44. See Fig. S2.1† for a comparison of these pathways.
- B. E. Haines, H. Xu, P. Verma, X.-C. Wang, J.-Q. Yu and D. G. Musaev, J. Am. Chem. Soc., 2015, 137(28), 9022 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45(6), 840 CrossRef CAS PubMed.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137(39), 12486 CrossRef CAS PubMed.
- R. B. Siedschlag, V. Bernales, K. D. Vogiatzis, N. Planas, L. J. Clouston, E. Bill, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2015, 150323142749002 Search PubMed.
- A. S. Borovik, Acc. Chem. Res., 2005, 38(1), 54 CrossRef CAS PubMed.
- S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48(8), 2407 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical and spectral characterization data for all new compounds, and crystallographic information files (CIF) for compounds 2, 6a–c, 15, 16, 17, S1.4, and S5.1. CCDC 1544213–1544221. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01674c |
| This journal is © The Royal Society of Chemistry 2017 |