
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of MR probes for targeted-reporter imaging†
Kirti Dhingra
Verma‡
a,
Justin O.
Massing‡
b,
Sarah G.
Kamper
b,
Christiane E.
Carney
b,
Keith W.
MacRenaris
b,
James P.
Basilion
*a and
Thomas J.
Meade
 *b
*b
aDepartment of Biomedical Engineering, Case Center for Imaging Research, The NFCR Center for Molecular Imaging, Case Western Reserve University, Cleveland, Ohio 44106-7207, USA
bDepartment of Chemistry, Molecular Biosciences, Neurobiology, Biomedical Engineering, and Radiology, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208-3113, USA
First published on 13th June 2017
Abstract
Visualizing disease heterogeneity remains a challenging task since most imaging agents are targeted to a single receptor. We describe the development of an MR platform able to report on multiple molecular events. Enzyme activation and enhanced cellular uptake of this modular probe make it suitable for subsequent targeted-reporter imaging applications.
Extracellular receptors enable cells to interact with and adapt to their surrounding environment through a series of interwoven signalling cascades.1 Receptor expression levels therefore provide valuable insight with regard to cellular health. For instance, given the increased nutrient requirements necessary for uncontrolled cellular proliferation, cancerous cells are often characterized by the concerted overexpression of multiple cell surface receptors.2 Specifically, overexpression and amplification of epidermal growth factor receptor (EGFR) has been reported as the primary gain-of-function mutation observed in brain,3,4 breast,5 lung,6 ovarian,7 and skin cancers.8 Moreover, transferrin receptor (TfR) has been implicated in several cancers given the involvement of the iron-dependent enzyme ribonucleotide reductase during rapid DNA synthesis.9,10 Thus, targeted molecular imaging strategies able to report on the status of multiple biomarkers have the potential to function as innovative diagnostic agents during drug development.
The Basilion lab has demonstrated the ability to visualize multiple biomarkers using complementing β-galactosidase (β-gal) fragments targeted to EGFR and TfR.11 This approach relied on colocalization of complementing enzyme fragments at extracellular surfaces overexpressing the target receptors to restore enzyme activity, thus permitting their detection following colorimetric staining with X-gal in vitro. While this approach represents a first attempt at multimarker visualization, the colorimetric output is restricted to translucent specimens, thereby precluding longitudinal studies in vivo. More recently, Grimm and coworkers achieved in vivo imaging of multiple biomarkers by utilizing Cerenkov luminescence produced by targeted positron-emitting radionuclides to excite nearby bioactivatable fluorescent nanoparticles.12 However, the use of radionuclides make this imaging platform unsuitable for longitudinal studies given prolonged exposure to ionizing radiation. To overcome these limitations, we designed a complementary approach employing magnetic resonance (MR) imaging.
MR imaging has greatly impacted diagnostic medicine due to its high spatiotemporal resolution and unlimited penetration depth.13,14 To improve image contrast, paramagnetic agents are frequently administered to shorten the longitudinal (T1) or transverse (T2) relaxation times of nearby water protons. This increase in the observed water relaxation rate (1/Ti,obs, i = 1, 2) yields a change in image contrast commensurate with the rate in the presence (1/Ti,p) and absence (1/Ti,d) of the paramagnetic species. With seven unpaired electrons and high magnetic moment, Gd(III) chelates are the most commonly employed T1 agents. The efficiency with which these agents shorten T1 is termed relaxivity (r1), and is influenced by the number of bound waters (q), their residency lifetime (τm), and the rotational correlation time of the complex (τR). Modulating any one of the above variables upon interaction with a stimulus generates responsive MR probes.15–23
Here, we present a modular platform for the noninvasive visualization of enzyme activity and receptor expression. To achieve this aim we designed a Gd(III) chelate that incorporates a proximal glycoside and a distal biotin that allow signal transduction and receptor targeting, respectively. The corresponding galactose residue is covalently tethered to the Gd(III) core through a self-immolative linker capable of modulating the coordination chemistry about the metal center in a manner similar to that previously reported by our group.24 Biotinylation of the probe enables facile complexation with avidin, a tetrameric protein capable of binding four biotin molecules (Kd = 10−15 M).25 Complexation of three MR agents to one avidin was predicted to enable facile targeting to the desired receptor through subsequent complexation between the remaining subunit and the desired biotinylated ligand (e.g., Tf). β-gal could then similarly be targeted to a separate receptor with the appropriate biotinylated ligand (e.g., EGF). Overexpression of both receptors is therefore expected to result in colocalization, receptor-mediated internalization, and subsequent activation through targeted-reporter complex formation (Fig. 1).
 | ||
| Fig. 1 Formation of a targeted-reporter complex is achieved by conjugating a targeting ligand to an enzyme or its corresponding substrate. Thus, multiple cell surface receptors may be targeted simply by varying the ligands used. Cells overexpressing the receptors of interest enable positioning of the targeted enzyme and substrate close to one another, resulting in activation of the MR probe via a self-immolative cascade. | ||
Briefly, 1,4,7,10-tetraazacyclododecane was protected at the 1 and 7 positions with Cbz-Cl26 prior to alkylation with ethyl bromoacetate; subsequent deprotection of the Cbz groups via catalytic hydrogenation yielded N-trans symmetrical material (Scheme S1†). Alkylation of this material with half an equivalent of 1-(tert-butyl)6-ethyl 2-bromohexanedioate27 (3) followed by acid-catalyzed cleavage of the t-butyl ester gave trisubstituted macrocycle in 34% over five steps. Reaction with 11, followed by global deprotection and subsequent metalation yielded untargeted complex Gd14 in 5% after preparative HPLC. Peptide coupling with norbiotinamine (15)28 and 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 gave the desired biotinylated MR agents Gd2 and Gd1, respectively (Table 1 and Scheme S3†). The corresponding Tb(III) complexes were similarly generated to quantify q following luminescence lifetime decay measurements.
29 gave the desired biotinylated MR agents Gd2 and Gd1, respectively (Table 1 and Scheme S3†). The corresponding Tb(III) complexes were similarly generated to quantify q following luminescence lifetime decay measurements.
|
|
||||
|---|---|---|---|---|
| Relaxivity at 1.41 T (60 MHz) | q | |||
| (−) Avidin (mM−1 s−1) | (+) Avidin (mM−1 s−1) | (−) Avidin | (+) Avidin | |
| Gd1 | 7.8 | 7.8 | 0.95 | 0.14 |
| Gd1 + β-gal | 4.2 | 17.2 | 1.3 | 1 |
| Gd2 | 8.6 | 19.3 | 1.0 | 0.9 |
| Gd2 + β-gal | 4.2 | n.a. | 1.2 | n.a. |
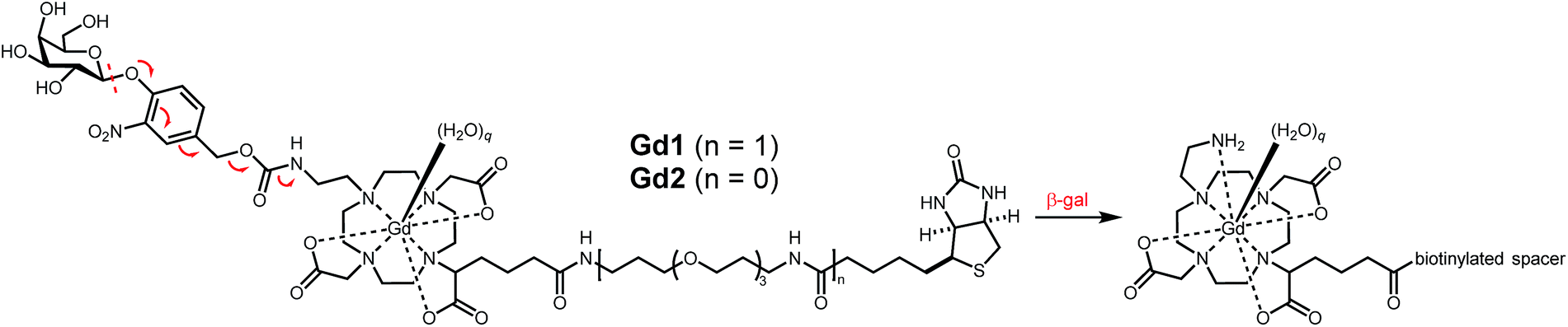
Gd1 and Gd2 were assessed regarding their ability to influence the 1/T1 of nearby water protons at pH 7.4 in 100 mM MOPS buffer. T1 measurements made at 1.5 T and 37 °C yielded r1 values for Gd1 and Gd2 of 7.8 and 8.6 mM−1 s−1, respectively. Exposing each agent to β-gal afforded a measurable change in absorbance and 1/T1 (Fig. S19†). The apparent decrease in the latter is consistent with findings previously reported by our lab,24 and is likely due to the decrease in molecular weight upon enzyme activation.30 Both agents exhibited complete activation within three hours.
We next examined the ability of Gd1 and Gd2 to form host–guest complexes with avidin. This was confirmed by changes in 1/T1 and 1/T2 of nearby water protons upon successive additions of avidin. Measurements were performed identically (vide supra), and converted to r1 and r2 values following ICP-MS. As anticipated, Gd2 exhibited an increase in both r1 (149%) and r2 (382%) upon complexation with avidin (Fig. S20†). This finding is expected given an increase in τR following complexation with biomacromolecules. Moreover, r1 and r2 values appeared to saturate upon binding of approximately three Gd2 molecules to a single avidin tetramer, as has been seen before.31 While Gd1 produced a similar increase in r2 (40%) that became saturated at the desired 4:1 stoichiometry, r1 remained largely unchanged throughout the experiment (Fig. 2). This response is important for subsequent targeted-reporter imaging and was later attributed to a concurrent decrease in q (vide infra). We therefore aimed to further establish Gd1 as a viable candidate for such imaging applications by demonstrating its ability to drastically alter relaxation rates of nearby water protons upon activation while complexed with avidin.
 | ||
| Fig. 2 r 1 and r2 as a function of avidin equivalents for Gd1 (0.15 mM). | ||
Attempts to activate Gd2 complexed to avidin failed to alter r1, suggesting the proximity of avidin to the glycoside was sufficient to obviate β-gal recognition, and hence MR activation. However, Gd1 readily underwent enzyme activation while complexed with avidin owing to the increased linker length. However, unlike before, activation of this macromolecular complex by β-gal afforded a desirable increase in r1 from 7.8 to 17.2 mM−1 s−1.
To understand these changes in r1 prior to, and following complexation with avidin and/or β-gal activation, luminescence lifetime measurements were performed with Tb1 and Tb2 (Table 1). As noted above, the constant r1 value for Gd1 may be rationalized given a concurrent decrease in q and increase in τR, the latter of which dominates at clinical field strengths.13 These findings indicate that avidin, like carbonate,24,27,32 is capable of efficiently masking the open coordination sites in Gd1 to afford an MR agent exhibiting a desirable increase in r1 (130%) following activation by β-gal.
We determined the binding stoichiometry between this agent and avidin via a colorimetric assay using 2-(4-hydroxyphenylazo)benzoic acid (HABA). To a reconstituted HABA/avidin solution was titrated in a solution of Gd1 and the change in HABA absorption at 500 nm monitored. Plotting the difference in absorption between each addition against the corresponding ratio of Gd1 to avidin indicated the number of probes bound to each avidin (Fig. S21†). Upon the addition of four equivalents of Gd1, no observable change in the absorption of HABA was noted. These results confirm complete and efficient displacement of HABA from avidin in the presence of four equivalents of Gd1, suggesting negligible influence of the macrocyclic chelate on binding of the distal biotin to avidin.
Having established the desired 4:1 binding stoichiometry, we investigated the ability to target Gd1 to cells overexpressing TfR. The corresponding Gd1:avidin:Tf (3:1:1) macromolecular construct was generated and cellular uptake subsequently examined in 9L cells known to overexpress TfR.11 Cells incubated with targeted agent (20 μM Gd(III), 6.6 μM Tf) and untargeted agent (20 μM Gd(III), 0 μM Tf) for 1, 2, 4, 8, or 24 hours show higher labelling for the targeted agent of approximately 0.3–0.4 fmol Gd(III) per cell at all time points (Fig. 3). Additionally, at short incubation times of 1 and 2 hours, the magnitude of labelling by the untargeted agent is less than 0.1 fmol Gd(III) per cell indicating that short incubation times are necessary to minimize the magnitude of non-specific binding. We therefore examined uptake in cells incubated with increasing concentrations of targeted and untargeted contrast agents for 2 hours (Fig. 3). At each concentration, there is significant preferential uptake of the targeted contrast agent. However, there is no significant increase in uptake of targeted contrast agent between incubations of 22 and 45 μM Gd(III) indicating that cells have been saturated with the agent. These findings demonstrate that we are well within the limit of detection necessary for bioactivatable Gd(III)-based agents (vide infra).33–35 However, attempts to visualize combinatorial biomarkers through colocalization of TfR-targeted Gd1 and EGFR-targeted β-gal and subsequent activation of the former in vitro were unsuccessful. We conjectured that this finding was a result sluggish MR activation.
 | ||
| Fig. 3 Time- (top) and concentration-dependent uptake (bottom) of TfR-targeted and untargeted Gd1 illustrate significant uptake when complexed with biotinylated transferrin. | ||
We investigated the kinetics associated with biotinylated β-gal activation required for subsequent targeted-reporter imaging. Biotinylated β-gal activity was established using ortho-nitrophenyl-β-galactoside (ONPG) through spectrophotometric measurements (Fig. S22†). Initial rates were determined and the reciprocal initial velocities (1/V0) plotted against the corresponding inverse concentrations (1/[ONPG]) to afford a Lineweaver–Burk plot (Fig. S23†). We examined ONPG activation by this enzyme while complexed with 0.25 equivalents of avidin (Fig. S24 and S25†). The corresponding host–guest interaction was found to exert little influence on the observed kcat, indicating this platform's suitability for subsequent targeted-reporter imaging applications.
Spectrophotometric measurements were repeated in the presence of Gd1 (Fig. S26 and S27†). However, complexation of this agent with avidin (4:1) afforded negligible activation during the time course of the experiment, likely given the steric bulk of these complementing fragments. Despite this finding, we were able to visualize MR contrast enhancement following β-gal activation of Gd1 complexed with avidin in solution (Fig. 4).
 | ||
| Fig. 4 T 1-weighted MR phantom images at 1.5 T. (A) Gd1 (0.15 mM, 100 mM MOPS, pH = 7.4, 37 °C) alone, (B) Gd1 complexed to avidin (20 U), and (C) Gd1 complexed to avidin in the presence of β-gal (2.6 mU) after 4 h. | ||
The detection limit of Gd(III) in vivo depends on a variety of factors. These include the r1 of the agent, the intrinsic T1 values of the tissue being imaged, and the field strength of the magnet. The detection limit must therefore be determined for each imaging scenario. However, the limit at 9.4 T is commonly approximated to be 10 μM for a Gd(III) chelate with an r1 of c. 7 mM−1 s−1.34 Whole proteome quantification of HeLa cells has shown that only proteins in the top 1% of expression level achieve cellular concentration of 10 μM.35 As a result, protein detection using Gd(III) requires either a significant payload of Gd(III) per binding event, or a target that amplifies the Gd(III) uptake beyond simple stoichiometry. If a cell can be approximated to be 1 pL, a pellet of densely packed cells would require a total uptake value of 0.01 fmol Gd(III)/cell. However, this value is unrealistic given the large degree of free space between cells. A more accurate approximation would be 0.1–1.0 fmol Gd(III) per cell, thus providing a dynamic range for visualizing Gd(III) in vivo. As stated above, although we are well within this range, the inability of targeted β-gal to activate targeted Gd1 is due to either poor colocalization of these complementing fragments or inefficient activation under the conditions examined.
We have developed two bioresponsive MR agents for combinatorial imaging. Gd1 and Gd2 incorporate a self-immolative linker able to modulate agent efficiency in response to β-gal. While complexation between Gd2 and avidin yielded an increase in r1 (124%), the resulting host–guest complex was incapable of undergoing enzyme activation. Complexation between Gd1 and avidin failed to result in an r1 increase owing to a concurrent decrease in q and increase in τR. However, this agent underwent a 130% increase in r1 following activation by β-gal while complexed with avidin. Moreover, Gd1 displayed exceptional targeted cellular uptake necessary for combinatorial imaging. Future work is focused on further improving the change in r1 upon enzyme activation, and accelerating the kinetics associated with this process.
Acknowledgements
This work was supported by the NIH (R01EB005866 and P01HL108795), a CBC Postdoctoral Award (J.O.M.), NSF GRFP and a Biophysics Training Grant (T32GM008382) from the NIGMS (S. G. K.), and NFCR (J. P. B.). Imaging was performed at the Center for Advanced Molecular Imaging (NU) generously supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. ICP-MS was performed at the Quantitative Bioelemental Imaging Center (NU) generously supported by the NASA Ames Research Center NNA06CB93G.Notes and references
- F. G. Giancotti and E. Ruoslahti, Science, 1999, 285, 1028 CrossRef CAS PubMed.
- R. Bei, A. Budillon, L. Masuelli, V. Cereda, D. Vitolo, E. Di Gennaro, V. Ripavecchia, C. Palumbo, F. Ionna, S. Losito, A. Modesti, M. H. Kraus and R. Muraro, J. Pathol., 2004, 204, 317 CrossRef CAS PubMed.
- D. B. Hoelzinger, L. Mariani, J. Weis, T. Woyke, T. J. Berens, W. McDonough, A. Sloan, S. W. Coons and M. E. Berens, Neoplasia, 2005, 7, 7 CrossRef CAS PubMed.
- M. Nakada, S. Nakada, T. Demuth, N. L. Tran, D. B. Hoelzinger and M. E. Berens, Cell. Mol. Life Sci., 2007, 64, 458 CrossRef CAS PubMed.
- C. Magkou, L. Nakopoulou, C. Zoubouli, K. Karali, I. Theohari, P. Bakarakos and I. Giannopoulou, Breast Cancer Res., 2008, 10, R49 CrossRef PubMed.
- F. R. Hirsch, M. Varella-Garcia, P. A. Bunn, M. V. Di Maria, R. Veve, R. M. Bremnes, A. E. Barón, C. Zeng and W. A. Franklin, J. Clin. Oncol., 2003, 21, 3798 CrossRef CAS PubMed.
- Ö. Alper, E. S. Bergmann-Leitner, T. A. Bennett, N. F. Hacker, K. Stromberg and W. G. Stetler-Stevenson, J. Natl. Cancer Inst., 2001, 93, 1375 CrossRef PubMed.
- G. Krähn, U. Leiter, P. Kaskel, M. Udart, J. Utikal, G. Bezold and R. U. Peter, Eur. J. Cancer, 2001, 37, 251 CrossRef.
- D. Högemann-Savellano, E. Bos, C. Blondet, F. Sato, T. Abe, L. Josephson, R. Weissleder, J. Gaudet, D. Sgroi, P. J. Peters and J. P. Basilion, Neoplasia, 2003, 5, 495 CrossRef.
- H. Habashy, D. Powe, C. Staka, E. Rakha, G. Ball, A. Green, M. Aleskandarany, E. C. Paish, R. Douglas Macmillan, R. Nicholson, I. Ellis and J. W. Gee, Breast Cancer Res. Treat., 2010, 119, 283 CrossRef CAS PubMed.
- A.-M. Broome, N. Bhavsar, G. Ramamurthy, G. Newton and J. P. Basilion, Mol. Pharmaceutics, 2010, 7, 60 CrossRef CAS PubMed.
- D. L. J. Thorek, A. Ogirala, B. J. Beattie and J. Grimm, Nat. Med., 2013, 19, 1345 CrossRef CAS PubMed.
- L. M. Manus, R. C. Strauch, A. H. Hung, A. L. Eckermann and T. J. Meade, Anal. Chem., 2012, 84, 6278 CrossRef CAS PubMed.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496 CrossRef CAS PubMed.
- R. A. Moats, S. E. Fraser and T. J. Meade, Angew. Chem., Int. Ed., 1997, 36, 726 CrossRef CAS.
- A. Y. Louie, M. M. Huber, E. T. Ahrens, U. Rothbacher, R. Moats, R. E. Jacobs, S. E. Fraser and T. J. Meade, Nat. Biotechnol., 2000, 18, 321 CrossRef CAS PubMed.
- W.-h. Li, S. E. Fraser and T. J. Meade, J. Am. Chem. Soc., 1999, 121, 1413 CrossRef CAS.
- J. L. Major, G. Parigi, C. Luchinat and T. J. Meade, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13881 CrossRef CAS PubMed.
- E. L. Que, E. Gianolio, S. L. Baker, A. P. Wong, S. Aime and C. J. Chang, J. Am. Chem. Soc., 2009, 131, 8527 CrossRef CAS PubMed.
- C. Tu, R. Nagao and A. Y. Louie, Angew. Chem., Int. Ed., 2009, 48, 6547 CrossRef CAS PubMed.
- S. Iwaki, K. Hanaoka, W. Piao, T. Komatsu, T. Ueno, T. Terai and T. Nagano, Bioorg. Med. Chem. Lett., 2012, 22, 2798 CrossRef CAS PubMed.
- J. L. Major and T. J. Meade, Acc. Chem. Res., 2009, 42, 893 CrossRef CAS PubMed.
- E. L. Que and C. J. Chang, Chem. Soc. Rev., 2010, 39, 51 RSC.
- J. A. Duimstra, F. J. Femia and T. J. Meade, J. Am. Chem. Soc., 2005, 127, 12847 CrossRef CAS PubMed.
- N. M. Green, in Advances in Protein Chemistry, ed. J. T. E. C.B. Anfinsen and M. R. Frederic, Academic Press, 1975, vol. 29, p. 85 Search PubMed.
- L. M. De León-Rodríguez, Z. Kovacs, A. C. Esqueda-Oliva and A. D. Miranda-Olvera, Tetrahedron Lett., 2006, 47, 6937 CrossRef.
- M. Giardiello, M. P. Lowe and M. Botta, Chem. Commun., 2007, 4044, 10.1039/B711989E.
- W. Szalecki, Bioconjugate Chem., 1996, 7, 271 CrossRef CAS PubMed.
- A. Renaud de la Faverie, F. Hamon, C. Di Primo, E. Largy, E. Dausse, L. Delaurière, C. Landras-Guetta, J.-J. Toulmé, M.-P. Teulade-Fichou and J.-L. Mergny, Biochimie, 2011, 93, 1357 CrossRef CAS PubMed.
- F. Arena, J. B. Singh, E. Gianolio, R. Stefanìa and S. Aime, Bioconjugate Chem., 2011, 22, 2625 CrossRef CAS PubMed.
- S. M. Vibhute, J. Engelmann, T. Verbic, M. E. Maier, N. K. Logothetis and G. Angelovski, Org. Biomol. Chem., 2013, 11, 1294 CAS.
- L. M. Urbanczyk-Pearson, F. J. Femia, J. Smith, G. Parigi, J. A. Duimstra, A. L. Eckermann, C. Luchinat and T. J. Meade, Inorg. Chem., 2008, 47, 56 CrossRef CAS PubMed.
- E. T. Ahrens, U. Rothbächer, R. E. Jacobs and S. E. Fraser, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8443 CrossRef CAS.
- K. Hanaoka, A. J. M. Lubag, A. Castillo-Muzquiz, T. Kodadek and A. D. Sherry, Magn. Reson. Imaging, 2008, 26, 608 CrossRef CAS PubMed.
- B. Schwanhausser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Nature, 2011, 473, 337 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02217d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |