
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of peptide-containing N5-unmodified neutral flavins that catalyze aerobic oxygenations†
Yukihiro
Arakawa
 a,
Ken
Yamanomoto
a,
Hazuki
Kita
a,
Keiji
Minagawa
ab,
Masami
Tanaka
c,
Naoki
Haraguchi
d,
Shinichi
Itsuno
d and
Yasushi
Imada
*a
a,
Ken
Yamanomoto
a,
Hazuki
Kita
a,
Keiji
Minagawa
ab,
Masami
Tanaka
c,
Naoki
Haraguchi
d,
Shinichi
Itsuno
d and
Yasushi
Imada
*a
aDepartment of Applied Chemistry, Tokushima University, Minamijosanjima, Tokushima 770-8506, Japan. E-mail: imada@tokushima-u.ac.jp
bInstitute of Liberal Arts and Sciences, Tokushima University, Minamijosanjima, Tokushima 770-8502, Japan
cFaculty of Pharmaceutical Sciences, Tokushima Bunri University, Yamashiro, Tokushima 770-8514, Japan
dDepartment of Environmental and Life Sciences, Toyohashi University of Technology, Toyohashi 441-8580, Japan
First published on 30th May 2017
Abstract
Simulation of the monooxygenation function of flavoenzyme (Fl-Enz) has been long-studied with N5-modified cationic flavins (FlEt+), but never with N5-unmodified neutral flavins (Fl) despite the fact that Fl is genuinely equal to the active center of Fl-Enz. This is because of the greater lability of 4a-hydroperoxy adduct of Fl, FlOOH, compared to those of FlEt+, FlEtOOH, and Fl-Enz, FlOOH-Enz. In this study, Fl incorporated into a short peptide, flavopeptide (Fl-Pep), was designed by a rational top-down approach using a computational method, which could stabilize the corresponding 4a-hydroperoxy adduct (FlOOH-Pep) through intramolecular hydrogen bonds. We report catalytic chemoselective sulfoxidation as well as Baeyer–Villiger oxidation by means of Fl-Pep under light-shielding and aerobic conditions, which are the first Fl-Enz-mimetic aerobic oxygenation reactions catalyzed by Fl under non-enzymatic conditions.
Introduction
Isoalloxazines, such as riboflavin and its analogues (Fig. 1a), show flexible redox activities as well as visible-light emission properties due to the specific conjugated heterocyclic structure, which are responsible for the catalytic functions of a variety of flavoenzymes such as flavin-containing monooxygenase (Fl-Enz, Fig. 1a), oxidase, and photolyase.1 Whereas a number of flavin-inspired catalytic reactions for organic synthesis have been developed with artificial isoalloxazines, N5-modified cationic flavins (FlEt+, Fig. 1b, upper),2 there has been much less progress in developing those with genuine isoalloxazines, N5-unmodified neutral flavins (Fl, Fig. 1b, lower), under non-enzymatic conditions despite their availability and the fact that nature actually utilizes them as catalysts. Recently, Fl has received increasing attention because of its economical as well as environmental friendliness and appeared as thermal-redox,3a–c photoredox,3d–i and photosensitizing catalysts.3j–m However, the use of Fl as oxygenation catalysts simulating the function of Fl-Enz has remained unexplored.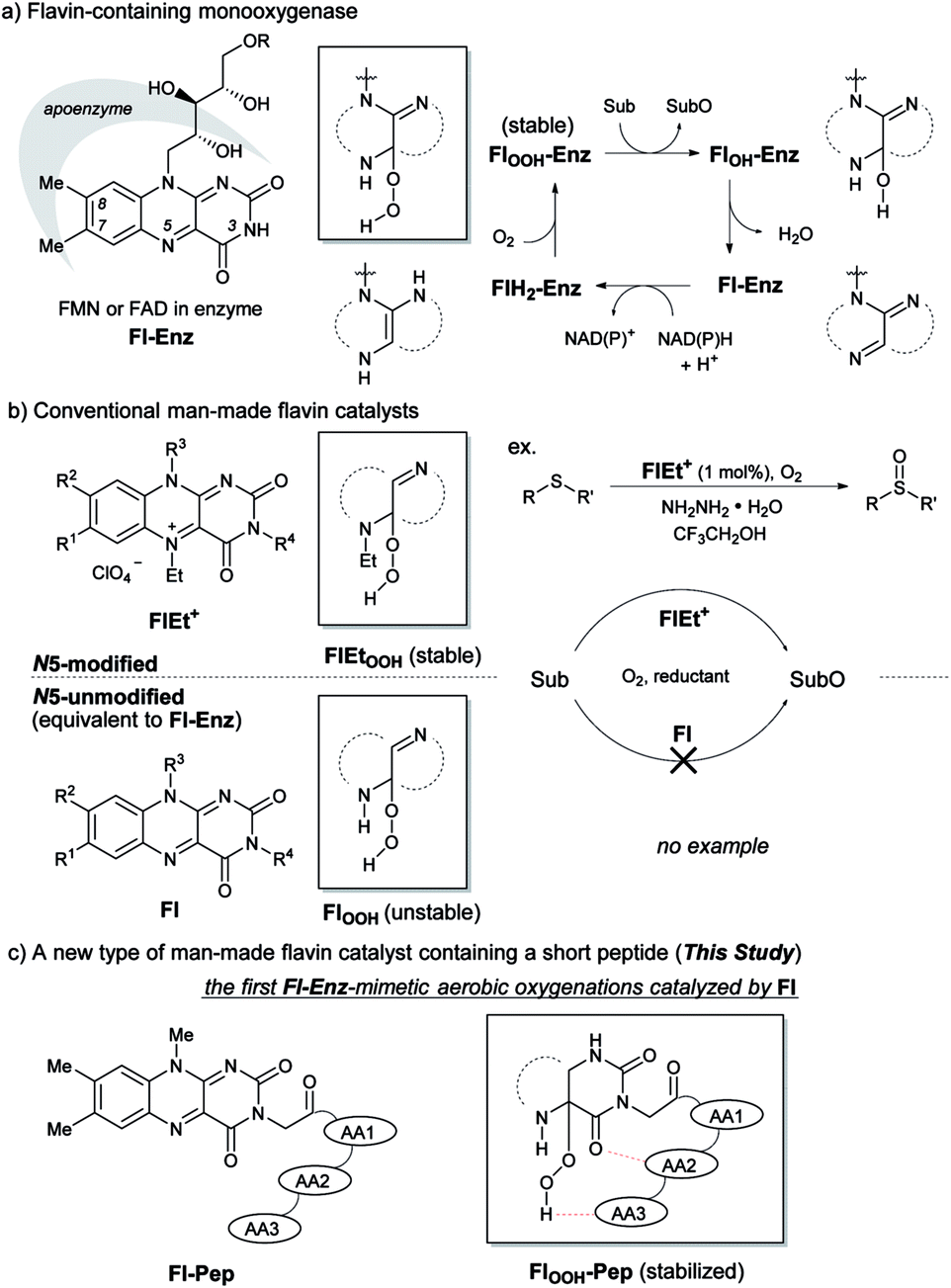 | ||
| Fig. 1 Flavin-catalyzed aerobic oxygenation reaction. | ||
The catalytic cycle of the oxygenation by Fl-Enz has been well understood (Fig. 1a) as a result of numerous early studies on flavin chemistry.1 A single oxygen atom is transferred from 4a-hydroperoxyflavin (FlOOH-Enz), a key active species in Fl-Enz catalysis, to a substrate (Sub) to give an oxidized product (SubO) and 4a-hydroxyflavin (FlOH-Enz), which eliminates H2O to form the oxidized flavin Fl-Enz. Then, Fl-Enz is reduced with NAD(P)H to afford the reduced flavin (FlH2-Enz), which finally reacts with molecular oxygen to regenerate FlOOH-Enz. Previously, we have successfully simulated this catalytic cycle using FlEt+ and hydrazine monohydrate instead of Fl-Enz and NAD(P)H, respectively (Fig. 1b, upper).4 For example, the aerobic oxygenation of sulfides was feasible in the presence of 1 mol% of 5-ethyl-3-methyllumiflavinium perchlorate (Fig. 1b, R1 = R2 = R3 = R4 = Me in FlEt+), 1 equivalent of hydrazine monohydrate, and 1 atm of O2 in 2,2,2-trifluoroethanol (TFE), in which TFE was crucial as a reaction solvent for predominant oxidation of sulfides in the coexistence of readily oxidizable hydrazine. By contrast, 3-methyllumiflavin (Fig. 1b, R1 = R2 = R3 = R4 = Me in Fl) was sluggish as a catalyst under the same reaction conditions, which was not surprising because of a kind of common knowledge that there is a huge difference in stability between the active species, 4a-hydroperoxyflavins FlOOH-Enz, FlEtOOH, and FlOOH (Fig. 1a and b). While FlOOH-Enz can be properly stabilized by hydrogen bonds between its FlOOH and peripheral proteins (Enz)5 and also FlEtOOH themselves are relatively stable,6 enzyme free FlOOH are typically so labile and readily decomposed to H2O2 and Fl. In 1988, Tamao and co-workers introduced Fl-catalyzed aerobic Tamao–Fleming oxidation, in which the eliminated H2O2 from FlOOH was utilized as an oxidant for the reaction.3a Very recently, König reported Fl-catalyzed oxidative chlorination of arenes under visible-light irradiation, in which the eliminated H2O2 from FlOOH was utilized for converting acetic acid to peracetic acid that subsequently oxidizes Cl− to OCl−, the active species for the chlorination.3i The only relevant work on FlOOH-related oxygenation was reported by Yoneda and co-workers who showed that an artificial Fl bearing a carboxyl group at C6 position could promote the oxidation of thioanisole, although the oxidant was H2O2 and its actual active species was not identified.7 As a result, the development of Fl-Enz mimetic aerobic oxygenation catalyzed by Fl has never been realized.
Herein, we present the first Fl-catalyzed aerobic oxygenation reactions under non-enzymatic conditions. To break through this long-standing challenge, we envisioned Fl containing a short peptide such as di- or tripeptides, flavopeptide (Fl-Pep), which might be stabilized in its 4a-hydroperoxy adduct (FlOOH-Pep) by intramolecular hydrogen bonds between FlOOH and Pep (Fig. 1c). Though peptides as catalysts have recently become powerful tools for organic synthesis with the advancement of combinatorial “bottom-up” screening methods using peptide libraries, the rational “top-down” design of peptidic catalysts from a large degree of molecular diversity is still highly challenging.8 In this study, we successfully designed Fl-Pep as efficient catalysts for aerobic sulfoxidation as well as aerobic Baeyer–Villiger oxidation by a top-down approach that simply consists of computational estimation9 followed by experimental fine-tuning of suitable structures.
Results and discussion
Computational design and synthesis of Fl-Pep
The design of Fl-Pep (Fig. 1c) was started by hypothesizing the following things: (i) Pep should be connected to the N3 position of Fl relatively close to the active site; (ii) readily available lumiflavin-3-acetic acid (3-FlC2)10 should be used as Fl and incorporated to the N terminus of Pep; (iii) a simple di- (AA1-AA2) or tripeptide (AA1-AA2-AA3) should be designed as Pep using inexpensive L-amino acids; (iv) L-proline residue should be placed at AA1 to induce constrained γ-turn structure and make the active site and AA2-AA3 spatially close; (v) AA2 and/or AA3 should be filled with acidic amino acid residues that can be expected to interact with the active site by intramolecular hydrogen bonds. In accordance with these design policies, we initially supposed 3-FlC2-Pro-AA2 and 3-FlC2-Pro-AA2-AA3 as the frameworks of Fl-Pep. To estimate appropriate structures for AA2/AA3 in Fl-Pep, lowest energy conformations of several FlOOH-Pep bearing different amino acid residues in vacuum were explored by DFT calculation at B3LYP/6-31G* level. Stable conformations of dipeptidic FlOOH-Pep, 3-FlC24a(R)OOH-Pro-Glu-NHMe, 3-FlC24a(R)OOH-Pro-Tyr-NHMe, and 3-FlC24a(R)OOH-Pro-Gly-NHMe had no desirable intramolecular hydrogen bonds in calculation. On the other hand, tripeptidic 3-FlC24a(R)OOH-Pro-Tyr-Glu-NHMe was suggested to be a promising sequence whose stable conformation includes ideal intramolecular hydrogen bonds between (1) CO neighboring to the nitrogen atom of Pro and NH of Tyr (γ-turn), (2) C(4)O of 3-FlC2 and OH in the side chain of Tyr, and (3) 4aOOH of 3-FlC2 and CO in the side chain of Glu (Fig. 2). Such a set of hydrogen bonds was not observed when Tyr-Glu in 3-FlC24a(R)OOH-Pro-Tyr-Glu-NHMe was replaced with other residues, Phe-Glu, Asp-Glu, and Tyr-Ser. In addition, replacement of either Pro with β-Ala or 3-FlC24a(R)OOH with 3-FlC24a(S)OOH also led to loosing effective hydrogen bonds. These results obtained from just the above 9 calculation samples (for more details see ESI†) led us to synthesize Fl-Pep consisting of the sequence of 3-FlC2-Pro-Tyr-Glu.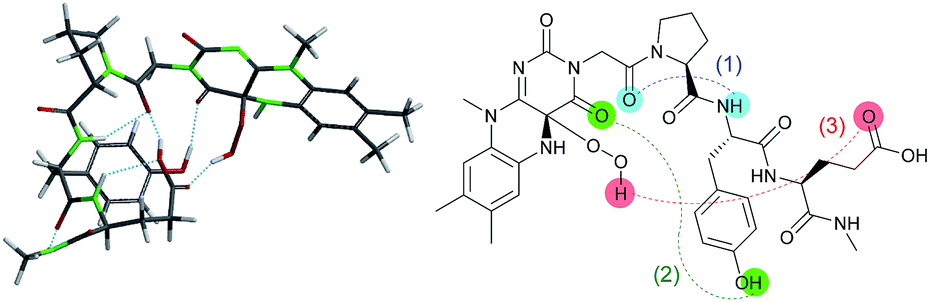 | ||
| Fig. 2 Lowest energy structure of 3-FlC24a(R)OOH-Pro-Tyr-Glu-NHMe estimated by DFT calculation (left) and graphical representation of remarkable hydrogen bonds (right). | ||
The synthesis of Fl-Pep was accomplished by standard solid phase peptide synthesis following Fmoc/tBu protocol using an amine-functionalized polystyrene resin (NH2-PS) (see ESI†).
Aerobic sulfoxidation catalyzed by Fl-Pep
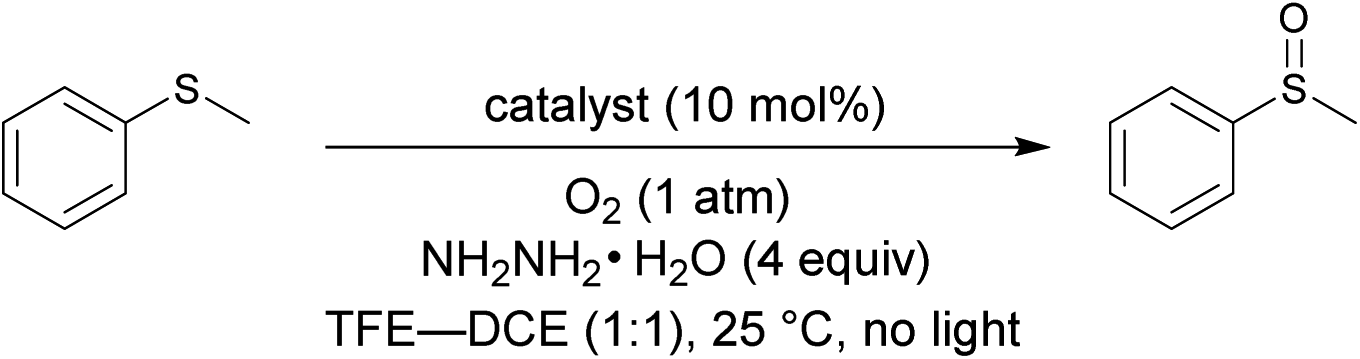
First of all, 3-FlC2-Pro-Tyr-Glu-βAla-NH-PS (Fl-Pep1-a, Fig. 3) bearing the peptide sequence designed by the above computational calculation was synthesized and tested as a polymer-supported peptide catalyst11 for aerobic oxidation of thioanisole under conditions that were previously developed by us for the reaction catalyzed by FlEt+.4 In the presence of 10 mol% of Fl-Pep1-a and 1 atm of O2 and 4 equivalents of hydrazine monohydrate in TFE, 9% of thioanisole was converted to methyl phenyl sulfoxide in 36 h (Table 1, entry 1), which was hopeful despite its low efficiency because the reaction did not proceed at all in the absence of the catalyst under otherwise identical conditions. As the efficiency of an insoluble polymer-supported catalyst can be strongly influenced by the nature of a reaction solvent,12 we subsequently used the mixed solvent of TFE and 1,2-dichloroethane (DCE) that can make polystyrene resin well swollen. As expected, the desired reaction was smoothly catalyzed by Fl-Pep1-a to give methyl phenyl sulfoxide in 60% yield in 36 h without any side reactions such as overoxidation to methyl phenyl sulfone (Table 1, entry 2). It should be noted that no reaction occurred in the absence of either TFE, O2, hydrazine (NH2NH2), or Fl-Pep, which indicates that all of them are essential. In addition, light was certainly shut out during the successful reaction, so that the involvement of singlet oxygen could be ruled out.3j,m Moreover, the excellent chemoselectivity, which is one of the feature of flavin catalyst,1,2 could leave the participation of peracid out and suggest FlOOH-Pep as a major oxidant. Furthermore, 3-methyllumiflavin as well as 3-FlC2-NH-PS was ineffective as a catalyst under the same reaction conditions (Table 1, entries 3 and 4), showing the Pro-Tyr-Glu sequence in Fl-Pep1-a is responsible for its catalytic activity.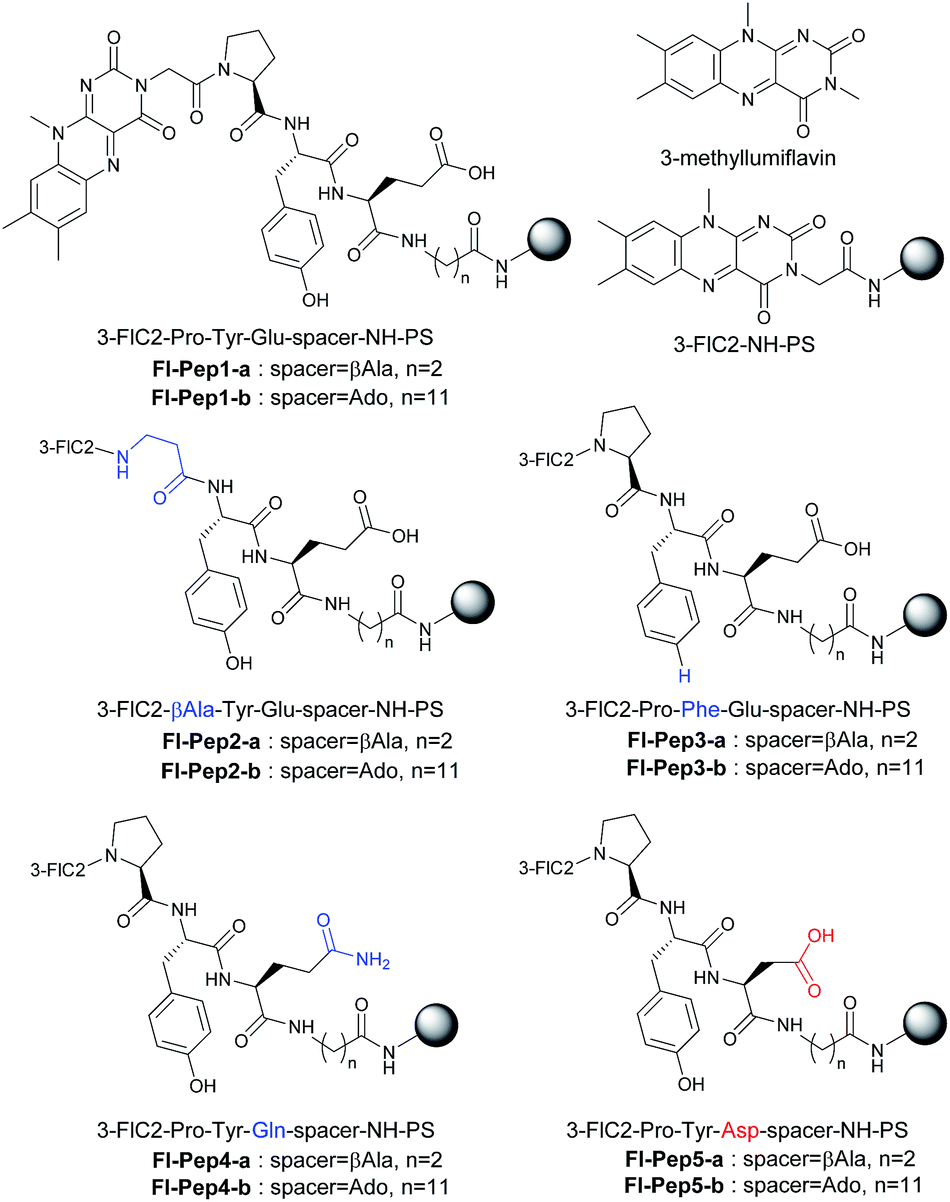 | ||
| Fig. 3 Structures of flavopeptides Fl-Pep1–Fl-pep5. | ||
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) |
a Reactions were performed using 0.1 mmol of thioanisole, 0.4 mmol of hydrazine monohydrate in 0.5 ml of a mixture of TFE and DCE (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the presence of 10 mol% of the catalyst under 1 atm of O2 at 25 °C.
b Determined by GC analysis.
c In TFE.
d Value after 36 h. :1) in the presence of 10 mol% of the catalyst under 1 atm of O2 at 25 °C.
b Determined by GC analysis.
c In TFE.
d Value after 36 h.
|
|||
| 1c | Fl-Pep1-a | 36 | 9 |
| 2 | Fl-Pep1-a | 24 | 36(60)d |
| 3 | 3-Methyllumiflavin | 36 | 2 |
| 4 | 3-FlC2-NH-PS | 36 | 1 |
| 5 | Fl-Pep2-a | 36 | 10 |
| 6 | Fl-Pep3-a | 36 | 25 |
| 7 | Fl-Pep3-a + 10 mol% phenol | 36 | 18 |
| 8 | Fl-Pep4-a | 36 | 15 |
| 9 | Fl-Pep4-a + 10 mol% AcOH | 36 | 16 |
| 10 | Fl-Pep5-a | 24 | 52(78)d |
| 11 | Fl-Pep1-b | 24 | 44 |
| 12 | Fl-Pep2-b | 24 | 18 |
| 13 | Fl-Pep3-b | 24 | 18 |
| 14 | Fl-Pep3-b + 10 mol% phenol | 24 | 16 |
| 15 | Fl-Pep4-b | 24 | 12 |
| 16 | Fl-Pep4-b + 10 mol% AcOH | 24 | 10 |
| 17 | Fl-Pep5-b | 24 | 62(99)d |
To explore structural and functional requirements for the catalytic activity of Fl-Pep1, we synthesized some analogues Fl-Pep2–Fl-Pep5 (Fig. 3) and compared their catalytic activity with Fl-Pep1 in the aerobic oxidation of thioanisole (Table 1). When Pro was replaced with βAla (3-FlC2-βAla-Tyr-Glu-βAla-NH-PS, Fl-Pep2-a), the catalytic activity dropped considerably (entry 5). Likewise, the replacement of Tyr with Phe (3-FlC2-Pro-Phe-Glu-βAla-NH-PS, Fl-Pep3-a), and that of Glu with Gln (3-FlC2-Pro-Tyr-Gln-βAla-NH-PS, Fl-Pep4-a) led to large decreases in reaction efficiency, respectively (entries 6 and 8), which were not improved even if a catalytic amount of phenol (entry 7) or acetic acid (entry 9) was used as an external additive. Interestingly, by contrast, enhancement of activity was observed (entry 10) when Glu was replaced with Asp (3-FlC2-Pro-Tyr-Asp-βAla-NH-PS, Fl-Pep5-a). These results indicate that the structure and functionality of all amino acid residues initially designed by the computational method was crucial for the efficient catalysis and, in particular, the carboxylic acid functionality of AA3 could play a significant role for fine-tuning of the activity. The same tendency on catalytic activities of Fl-Pep1–Fl-Pep5 was observed by using those immobilized on polystyrene resin having a longer alkyl spacer (Fl-Pep1-b–Fl-Pep5-b, Fig. 3) with rather better performance (entries 11–17), probably because both conformational flexibility of the immobilized Fl-Pep and its accessibility to the substrate are enhanced. The best efficiency was achieved with Fl-Pep5-b for the present reaction, which provided methyl phenyl sulfoxide in 99% yield in 36 h (entry 17).13 It should be noted that all reaction yields in Table 1 were determined by GC analysis without product isolation to precisely evaluate the catalytic activity of each Fl-Pep.14 In addition, no methyl phenyl sulfone was observed in any cases.
With these results in hand, we revisited the computational prediction of Fl-Pep to ensure its validity. We calculated 3-FlC24a(R)OOH-Pro-Tyr-Gln-NHMe, which was proven to be an ineffective sequence (Table 1, entries 8 and 15), and FlC24a(R)OOH-Pro-Tyr-Asp-NHMe, which was found to be the most effective sequence (Table 1, entries 10 and 17). In accordance with the experimental results, an effective set of hydrogen bonds (1), (2), and (3), similar to that highlighted in Fig. 2, were observed only in the lowest energy structure of FlC24a(R)OOH-Pro-Tyr-Asp-NHMe (Fig. 4, for others see ESI†). It seems obvious that the Asp-derivative (Fig. 4) has an even better coordination than the Glu-derivative (Fig. 2) between the carboxylic acid and the hydroperoxy moiety with an additional interaction (4).
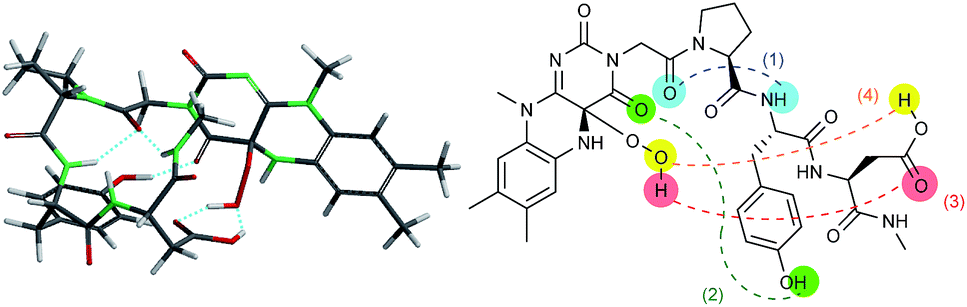 | ||
| Fig. 4 Lowest energy structure of 3-FlC24a(R)OOH-Pro-Tyr-Asp-NHMe estimated by DFT calculation (left) and graphical representation of remarkable hydrogen bonds (right). | ||
To gain an insight into active species for the oxygen transfer, we performed a Hammett study for the present aerobic sulfoxidation using Fl-Pep1-a. The relative reactivity values for p-substituted thioanisoles with respect to X = H (kX/kH) were determined, and the corresponding −log(kX/kH) versus the Hammett σ values were plotted to give ρ value of −1.54 (Fig. 5). The ρ value is similar to that of the stoichiometric oxidation of sulfides with FlEtOOH (ρ = −1.47)15 and also those of the aerobic (ρ = −1.60)4b as well as H2O2 sulfoxidation (ρ = −1.90)4b catalyzed by FlEt+. This result suggests that the present oxidation of sulfides takes place electrophilically with FlOOH-Pep as the active species.
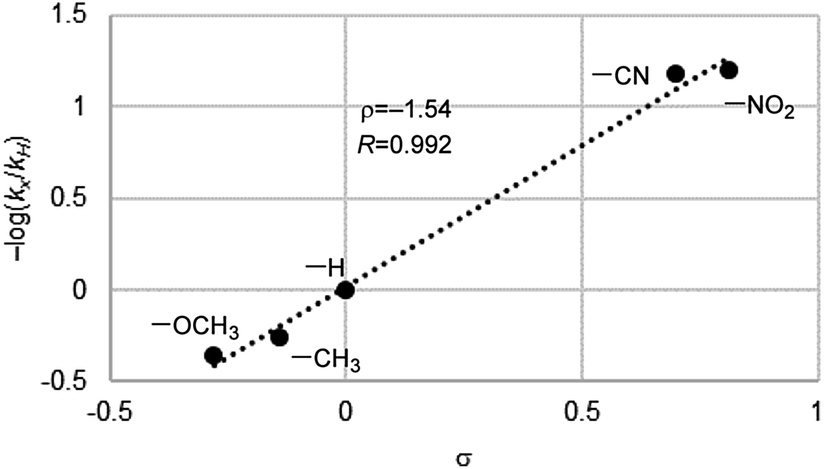 | ||
| Fig. 5 Hammett plot for aerobic oxidation of p-substituted methyl phenyl sulfides catalyzed by Fl-Pep1-a. | ||
Aerobic Baeyer–Villiger oxidation catalyzed by Fl-Pep
Encouraged by the above results we turned our attention to the Baeyer–Villiger oxidation for expanding the utility of Fl-Pep catalyst. Previously we developed this type of reaction catalyzed by FlEt+, which has so far been the sole example of organocatalytic Baeyer–Villiger oxidation using O2 as a terminal oxidant.16 The active species of nucleophilic FlEtOOH generated in situ allowed for selective Baeyer–Villiger oxidation of cyclobutanones into the corresponding γ-butyrolactones in the presence of alkene or sulfide functionality that could be readily oxidized with mCPBA, a typical oxidant for Baeyer–Villiger oxidation. Thus, Fl-Pep has also a great potential in the development of aerobic Baeyer–Villiger oxidation, and such chemoselectivity will be a strong evidence for the involvement of FlOOH-Pep as an active species.The Baeyer–Villiger oxidation of 3-phenylcyclobutanone into β-phenyl-γ-butyrolactone was used as a test reaction under conditions that were previously developed by us for the reaction catalyzed by FlEt+.16 In the presence of 5 mol% of Fl-Pep5-b, 1 atm of O2, 20 equivalents of H2O, and 3.5 equivalents of zinc dust in a mixed solvent of acetonitrile, toluene, and ethyl acetate (8:4:1), the desired oxidation proceeded smoothly to afford the target product in 72% yield in 7 h (Table 2, entry 1).17 Ethanol can be used instead of both CH3CN as a hydrophilic co-solvent and water as an essential proton source,16 however, toluene was crucial as a hydrophobic co-solvent that could render polystyrene resin properly swollen (see ESI†). As expected, 3-methyllumiflavin (entry 2) as well as 3-FlC2-NH-PS (entry 3) was totally inactive under the same reaction conditions. These results convinced us that, as in the case of the above sulfoxidation, an appropriate peptide sequence in Fl-Pep is essential for the catalysis involving the key stabilization of FlOOH-Pep as illustrated in Fig. 4.
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb (%) |
|
a Reactions were performed using 0.1 mmol of 3-phenylcyclobutanone, 0.35 mmol of zinc, 2.0 mmol of H2O in 1.0 ml of a mixture of acetonitrile, toluene, and ethyl acetate (8:4:1) in the presence of 5 mol% of the catalyst under 1 atm of O2 at 35 °C.
b Determined by NMR analysis using dodecane as an internal standard.
|
||
| 1 | Fl-Pep5-b | 72 |
| 2 | 3-Methyllumiflavin | <1 |
| 3 | 3-FlC2-NH-PS | 2 |

With the appropriate conditions in hand, we then carried out the Baeyer–Villiger oxidation of 3-phenylcyclobutanone catalyzed by Fl-Pep5-b in the presence of an equimolar amount of other reactive substrate. Cyclooctene as a competitor remained intact during the desired conversion of the ketone (eqn (1)), whereas the preferential formation of cyclooctene oxide has occurred under mCPBA-based conditions (eqn (2)). Such excellent chemoselectivity was also observed in a competitive oxygenation of the ketone and thioanisole (eqn (3) and (4)). These results strongly suggest that peracid does not participate in the Fl-Pep5 systems (eqn (1) and (3)) and, given that the ketone underwent oxidation predominantly, the corresponding FlOOH-Pep5 can be rather nucleophilic as opposed to the above chemoselective sulfoxidation.18
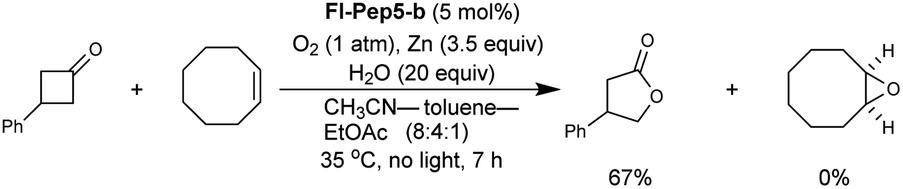 | (1) |
 | (2) |
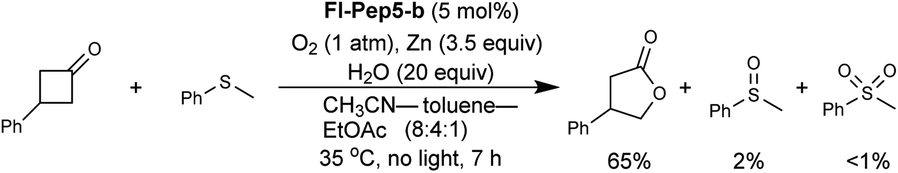 | (3) |
 | (4) |
Conformational analysis of Fl-Pep
We synthesized the soluble analogue of Fl-Pep5, 3-FlC2-Pro-Tyr-Asp-Ado-NH2, using Rink amide Resin to gain its conformational information by NMR spectroscopy (see ESI†). 3-FlC2-Pro-Tyr-Asp-Ado-NH2 was soluble in polar solvents such as dimethyl sulfoxide and methanol, but unfortunately, hardly soluble in less polar solvents such as acetonitrile, acetone, and chloroform. Thus, DMSO-d6 was inevitably used as the solvent, although it is quite unlike the actual reaction microenvironment that must be much less polar because of the hydrophobic nature of polystyrene resin. The NMR analysis showed that 3-FlC2-Pro-Tyr-Asp-Ado-NH2 forms two different conformers in DMSO-d6 at 25 °C in a ratio of ∼1.4:1, in which the major conformer has 3-FlC2-Pro amide bond in the trans conformation (58%), while that of the minor conformer is cis (42%). It should be noted that the observed trans–cis ratio for 3-FlC2-Pro-Tyr-Asp-Ado-NH2 in DMSO-d6 is similar to that for N-acetyl-L-proline N′-methylamide (Ac-Pro-NHMe, 65% trans)19 in the same solvent regardless of their large difference in structure and functionality. Given that Ac-Pro-NHMe predominantly favours the trans-C7 form (γ-turn) over other forms including the cis form in the gas phase and non-polar solvents,20 it is plausible that the flavopeptide moiety surrounded by the strongly hydrophobic environment in Fl-Pep5 also populates the γ-turn form as included in the predicted stable conformation of 3-FlC24a(R)OOH-Pro-Tyr-Asp-NHMe (Fig. 4). In fact, the catalytic activity of 3-FlC2-Pro-Tyr-Asp-Ado-NH2 was found to be much lower (7% yield in 24 h, see ESI†) than that of Fl-Pep5-b (Table 1, entry 17) in the sulfoxidation of thioanisole under the same conditions, showing the importance of the hydrophobic support resin that would make the flavopeptide conformationally profitable.21
Mechanistic aspects of Fl-Pep-catalyzed aerobic oxygenations
Given all the above experimental facts, it is plausible to consider that both the sulfoxidation and the Baeyer–Villiger oxidation catalyzed by Fl-Pep occur viaFl-Enz-like mechanism (Fig. 6). As for the sulfoxidation, since effective Fl-Pep1 and Fl-Pep5 possess a carboxyl group that can readily react with an equivalent of NH2NH2 to be the corresponding salt (Fl-Pep·NH2NH2) in situ, the catalytic cycle (Fig. 6a) can be initiated by reducing Fl-Pep·NH2NH2 with another molecule of NH2NH2 to afford FlH2-Pep·NH2NH2 and diazene (NH = NH). The resulting NH = NH can also be used to reduce Fl-Pep·NH2NH2 from the second cycle by releasing N2. Molecular oxygen can be then inserted into the C(4a) of FlH2-Pep·NH2NH2 to give FlOOH-Pep·NH2NH2. This hydroperoxy species may be effectively stabilized to perform subsequent monooxygenation of a substrate to give the corresponding 4a-hydroxy adduct (FlOH-Pep·NH2NH2), which finally undergoes dehydration to regenerate Fl-Pep·NH2NH2. The Hammett study (Fig. 5) shows that the oxygen transfer from FlOOH-Pep·NH2NH2 to a substrate is a rate-determining step of the proposed catalysis and takes place electrophilically. Although it is not trivial to verify the generation of the FlOOH-Pep species spectroscopically due to the insolubility of resin, for the present, it is reasonable to understand that FlOOH-Pep can be stabilized by means of intramolecular hydrogen bonds similar to those predicted by the DFT calculations (Fig. 2 and 4) including a probable coordination between C(4a)O of 3-FlC2 and +NH3NH2 to make the hydroperoxy moiety electrophilic (Fig. 6b). The fact that Asp instead of Glu in AA3 enhanced the catalytic activity (Table 1, entries 2 vs. 10 and entries 11 vs. 17) could be rationalized by assuming such stabilization model that allows for a spatially less-forced intervention of NH2NH2 in between the carboxyl group and the hydroperoxy group. We consider that the presence of +NH3NH2 is a key for stabilizing FlOOH-Pep, which is a similar situation to FlOOH-Enz that can be stabilized by complexation with NAD(P)+.5c Actually, an additional experiment on the effect of equivalents of hydrazine monohydrate for the present sulfoxidation revealed that the larger equivalents of NH2NH2, the faster reaction rate (see ESI†).22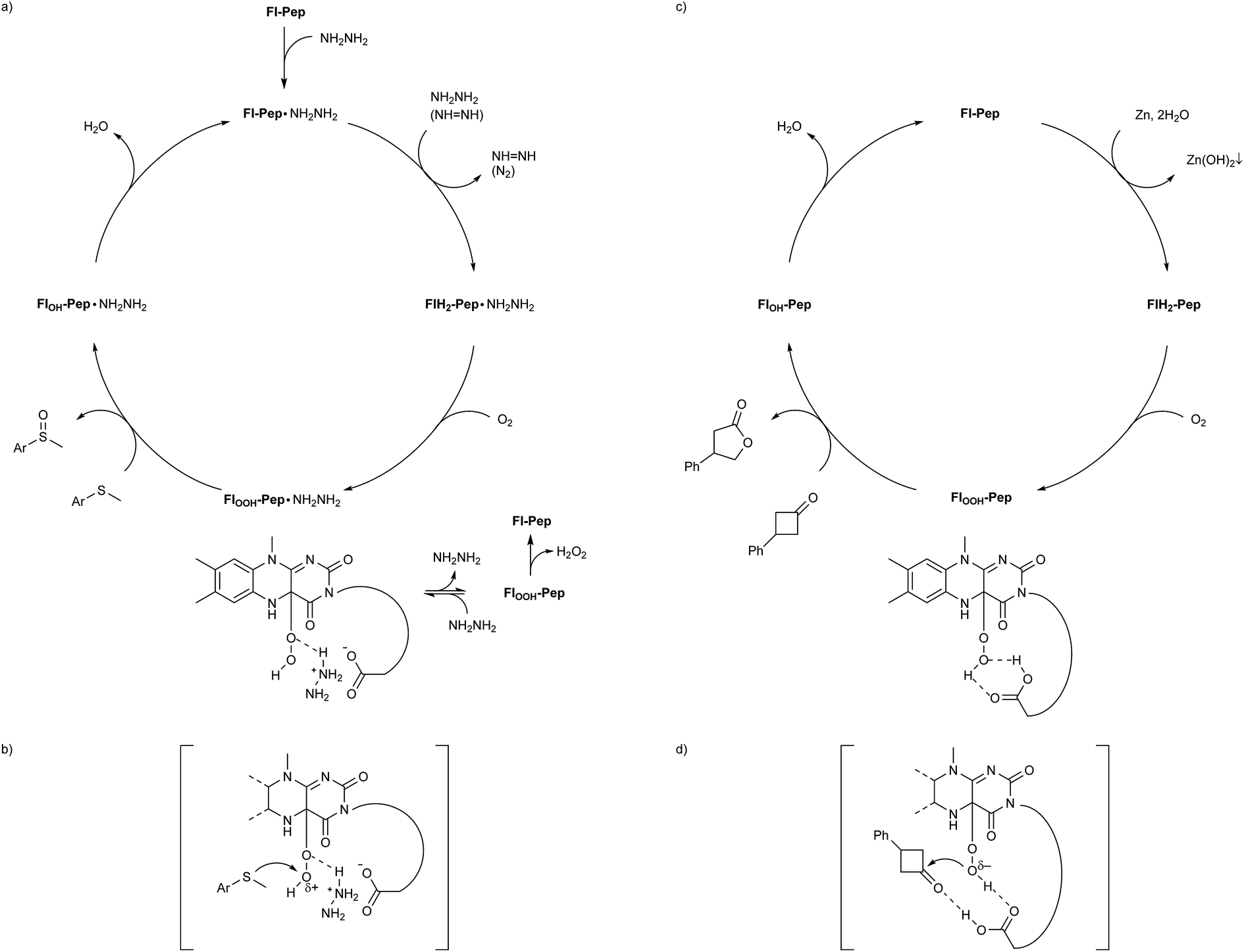 | ||
| Fig. 6 Proposed catalytic cycles and transition state models for Fl-Pep-catalyzed aerobic (a and b) sulfoxidation and (c and d) Baeyer–Villiger oxidation. | ||
On the other hand, FlOOH-Pep in the Baeyer–Villiger oxidation can be formed via the reduction of Fl-Pep with Zn and H2O into FlH2-Pep followed by the oxygen insertion to FlH2-Pep and stabilized by computationally predicted hydrogen bonds (Fig. 4) including a cyclic coordination between 4aOOH of 3-FlC2 and COOH in the side chain of Asp, which then selectively oxidizes the ketone into the lactone to give FlOH-Pep that finally release H2O to regenerate Fl-Pep (Fig. 6c). The nucleophilic activity of FlOOH-Pep can be explained by assuming a transition state model involving simultaneous activation of the hydroperoxy moiety and the keto-carbonyl moiety by the COOH group (Fig. 6d), although the involvement of Zn+(OH) instead of H+ cannot be excluded for the moment.
Conclusion
In conclusion, the first Fl-Enz-mimetic aerobic oxygenation reactions catalyzed by Fl under non-enzymatic conditions were realized. We predicted the structure of Fl-Pep that could stabilize the corresponding FlOOH-Pep by a computational method, and synthesized the most promising Fl-Pep1 and its analogies Fl-Pep2–Fl-Pep5 as resin-immobilized peptides. Exploring their catalytic activity for aerobic sulfoxidation using hydrazine monohydrate as terminal reductant revealed that the computational design of Fl-Pep catalyst was reasonable although the fine-tuned Fl-Pep5 showed superior activity than the original Fl-Pep1. On the other hand, the use of zinc as an alternative reductant under suitable conditions was found to allow for Fl-Pep5-catalyzed aerobic Baeyer–Villiger oxidation with excellent chemoselectivity. Multiple control experiment as well as mechanistic experiment suggested that both types of oxygenations could proceed viaFl-Enz-like mechanism and the active species could be FlOOH that had been efficiently used only in Fl-Enz so far. It is noteworthy that the electronic properties of the hydroperoxy moiety in FlOOH-Pep can be orthogonally controlled by reductants and reaction conditions, realizing electrophilic sulfoxidation as well as nucleophilic Baeyer–Villiger oxidation in a highly chemoselective manner.18 We believe that the results are so important for the research fields of both flavin chemistry and peptide chemistry, because they not only provide new possibilities for the development of flavin catalysts as well as the fundamental study on flavin-containing monooxygenase but also demonstrate great potential of computational chemistry for the rational design of peptide-based catalysts.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas ‘Advanced Molecular Transformations by Organocatalysts’ from MEXT.Notes and references
- (a) T. C. Bruice, Acc. Chem. Res., 1980, 13, 256–262 CrossRef CAS; (b) C. Walsh, Acc. Chem. Res., 1980, 13, 148–155 CrossRef CAS; (c) D. P. Ballou, in Flavins and Flavoproteins, ed. V. Massey and C. H. Williams, Elsevier, New York, 1982, p. 301 Search PubMed; (d) Chemistry and Biochemistry of Flavoenzymes, ed. F. Müller, CRC Press, Boston, 1991 Search PubMed; (e) R. B. Silverman, Acc. Chem. Res., 1995, 28, 335–342 CrossRef CAS; (f) N. M. Kamerbeek, D. B. Janssen, W. J. H. van Berkel and M. W. Fraaije, Adv. Synth. Catal., 2003, 345, 667–678 CrossRef CAS; (g) Flavins—Photochemistry and Photobiology, ed. E. Silva and A. M. Edwards, Royal Society of Chemistry, Cambridge, 2006 Search PubMed; (h) M. W. Fraaije and Janssen, in Modern Biooxidations—Enzymes, Reactions and Applications, ed. R. D. Schmid and V. B. Urlacher, Wiley-VCH, Weinheim, 2007, p. 77 Search PubMed; (i) M. Insińska-Rak and M. Sikorski, Chem.–Eur. J., 2014, 20, 15280–15291 CrossRef PubMed.
- (a) H. Iida, Y. Imada and S.-I. Murahashi, Org. Biomol. Chem., 2015, 13, 7599–7613 RSC; (b) R. Cibulka, Eur. J. Org. Chem., 2015, 915–932 CrossRef CAS; (c) G. de Gonzalo and M. W. Fraaije, ChemCatChem, 2013, 5, 403–415 CrossRef CAS; (d) Y. Imada and T. Naota, Chem. Rec., 2007, 7, 354–361 CrossRef CAS PubMed; (e) F. G. Gelalcha, Chem. Rev., 2007, 107, 3338–3361 CrossRef CAS PubMed; (f) J.-E. Bäckvall, in Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley-VCH, Weinheim, 2004, p. 193 Search PubMed.
- (a) K. Tamao, T. Hayashi and Y. Ito, J. Chem. Soc., Chem. Commun., 1988, 795–797 RSC; (b) Y. Imada, T. Kitagawa, T. Ohno, H. Iida and T. Naota, Org. Lett., 2010, 12, 32–35 CrossRef CAS PubMed; (c) Y. Imada, H. Iida, T. Kitagawa and T. Naota, Chem.–Eur. J., 2011, 17, 5908–5920 CrossRef CAS PubMed; (d) H. Schmaderer, P. Hilgers, R. Lechner and B. König, Adv. Synth. Catal., 2009, 351, 163–174 CrossRef CAS; (e) R. Lechner and B. König, Synthesis, 2010, 2010, 1712–1718 CrossRef; (f) R. Lechner, S. Kümmel and B. König, Photochem. Photobiol. Sci., 2010, 9, 1367–1377 RSC; (g) B. Mühldorf and R. Wolf, Chem. Commun., 2015, 51, 8425–8428 RSC; (h) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040–1045 CrossRef CAS PubMed; (i) T. Hering, B. Mühldorf, R. Wolf and B. König, Angew. Chem., Int. Ed., 2016, 55, 5342–5345 CrossRef CAS PubMed; (j) J. Dad’ová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620–623 CrossRef; (k) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2015, 137, 11254–11257 CrossRef CAS PubMed; (l) V. Mojr, E. Svobodová, K. Straková, T. Neveselý, J. Chudoba, H. Dvořáková and R. Cibulka, Chem. Commun., 2015, 51, 12036–12039 RSC; (m) T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654–1663 CrossRef.
- (a) Y. Imada, H. Iida, S. Ono and S.-I. Murahashi, J. Am. Chem. Soc., 2003, 125, 2868–2869 CrossRef CAS PubMed; (b) Y. Imada, H. Iida, S. Ono, Y. Masui and S.-I. Murahashi, Chem.–Asian J., 2006, 1, 136–147 CrossRef CAS PubMed.
- (a) L. L. Poulsen and D. M. Ziegler, J. Biol. Chem., 1979, 254, 6449–6455 CAS; (b) V. Massey and P. Hemmerich, Biochem. Soc. Trans., 1980, 8, 246–257 CrossRef CAS PubMed; (c) N. B. Beaty and D. P. Ballou, J. Biol. Chem., 1980, 255, 3817–3819 CAS.
- C. Kemal and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 995–999 CrossRef CAS.
- T. Akiyama, F. Simeno, M. Murakami and F. Yoneda, J. Am. Chem. Soc., 1992, 114, 6613–6620 CrossRef CAS.
- (a) E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759–5812 CrossRef PubMed; (b) H. Wennemers, Chem. Commun., 2011, 47, 12036–12041 RSC; (c) J. Duschmale, Y. Arakawa and H. Wennemers, in Science of Synthesis: Asymmetric Organocatalysis, ed. K. Maruoka, Thieme, Stuttgart, 2012, p. 741 Search PubMed.
- A splendid work on the structural analysis of catalytic peptides using DFT calculation together with X-ray crystallography and NMR spectroscopy was recently reported by Miller's group, see: A. J. Metrano, N. C. Abascal, B. Q. Mercado, E. K. Paulson, A. E. Hurtley and S. J. Miller, J. Am. Chem. Soc., 2017, 139, 492–516 CrossRef CAS PubMed.
- H. Ikeda, K. Yoshida, M. Ozeki and I. Saito, Tetrahedron Lett., 2001, 42, 2529–2531 CrossRef CAS.
- For selected studies on polymer-supported peptide catalysts, see: (a) K. Akagawa, S. Sakamoto and K. Kudo, Tetrahedron Lett., 2005, 46, 8185–8187 CrossRef CAS; (b) K. Akagawa and K. Kudo, Angew. Chem., Int. Ed., 2012, 51, 12786–12789 CrossRef CAS PubMed; (c) K. Akagawa, J. Sen and K. Kudo, Angew. Chem., Int. Ed., 2013, 52, 11585–11588 CrossRef CAS PubMed; (d) Y. Arakawa and H. Wennemers, ChemSusChem, 2013, 6, 242–245 CrossRef CAS PubMed; (e) K. Akagawa, N. Sakai and K. Kudo, Angew. Chem., Int. Ed., 2015, 54, 1822–1826 CrossRef CAS PubMed.
- For reviews, see: (a) Polymeric Chiral Catalyst Design and Chiral Polymer Synthesis, ed. S. Itsuno, Wiley, Hoboken, 2011 Search PubMed; (b) A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418–514 CrossRef CAS PubMed.
- The product was racemic. To achieve not only the stabilization of active species but also enantioselective reactions, the catalysts must be designed in a more elaborated perspective. However, it would be interesting to note that the oxidation of 2-(4-methoxyphenyl)-1,3-dithiane could also be promoted with Fl-Pep5-b under the optimal conditions found for the sulfoxidation of thioanisole to give 2-(4-methoxyphenyl)-1,3-dithiane 1-oxide in 88% yield (65 h), a diastereoselectivity of 24:1 (trans:cis), and an enantioselectivity of 3% ee (trans). This preliminary result shows a possibility of the development of asymmetric flavopeptidic catalysts.
- To demonstrate the feasibility of product isolation, we carried out the oxidation of thioanisole with Fl-Pep5-b under the optimized conditions in a larger reaction scale (1 mmol of thioanisole, see ESI†). The reaction was rather less efficient compared with the standard scale, possibly due to varied mixing efficiency of the gas-liquid-solid triphasic system, so that 4 equivalents of hydrazine monohydrate was added after 14 h to the reaction mixture. After 48 h in total, methyl phenyl sulfoxide was obtained in 94% GC yield and, after purification by silica gel column chromatography, in 85% isolated yield (119 mg, 0.85 mmol).
- S. Oae, K. Asada and T. Yoshimura, Tetrahedron Lett., 1983, 24, 1265–1268 CrossRef CAS.
- Y. Imada, H. Iida, S.-I. Murahashi and T. Naota, Angew. Chem., Int. Ed., 2005, 44, 1704–1706 CrossRef CAS PubMed.
- Similar reaction efficiency was observed on a larger scale (0.8 mmol of 3-phenylcyclobutanone) and the product was isolated in 66% yield (see ESI†).
- Similar orthogonal reactivities in catalytic oxygenations were previously reported by Miller's group with aspartyl-peptide oxidation catalysts, although their active species was the transiently formed peracid of the aspartyl group unlike that of our case, see: (a) J. S. Alford, N. C. Abascal, C. R. Shugrue, S. M. Colvin, D. K. Romney and S. J. Miller, ACS Cent. Sci., 2016, 2, 733–739 CrossRef CAS PubMed; (b) D. K. Romney, S. M. Colvin and S. J. Miller, J. Am. Chem. Soc., 2014, 136, 14019–14022 CrossRef CAS PubMed; (c) P. A. Lichtor and S. J. Miller, J. Am. Chem. Soc., 2014, 136, 5301–5308 CrossRef CAS PubMed.
- V. Madison and K. D. Kopple, J. Am. Chem. Soc., 1980, 102, 4855–4863 CrossRef CAS.
- S. Sul, D. Karaiskaj, Y. Jiang and N.-H. Ge, J. Phys. Chem. B, 2006, 110, 19891–19905 CrossRef CAS PubMed.
- Such an effect that gives rise to the unordinary catalytic function on polystyrene resin is still unusual. For a recent example, see: K. Goren, J. K. Kuks, Y. Shiloni, E. B. Kulbak, S. J. Miller and M. Portnoy, Chem.–Eur. J., 2015, 21, 1191–1197 CrossRef CAS PubMed.
- Hydroxylamine can also be used as an alternative reductant, although the reaction has required heating (60 °C) to be efficient (see ESI†). We suppose that hydroxylamine serves the same role as hydrazine (Fig. 6a and b) and the insufficient activity is ascribable to its lower oxidation potential than that of hydrazine. For the oxidation potentials of hydrazine and hydroxylamine, see: J. Li and X. Lin, Sens. Actuators, B, 2007, 126, 527–535 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc01933e |
| This journal is © The Royal Society of Chemistry 2017 |