
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent [{ONNN}Mg–Cl] complexes in highly active and living lactide polymerization†
Tomer
Rosen
a,
Israel
Goldberg
a,
Wanda
Navarra
b,
Vincenzo
Venditto
*b and
Moshe
Kol
 *a
*a
aThe School of Chemistry, Tel Aviv University, Ramat Aviv, Tel Aviv 69978, Israel. E-mail: moshekol@post.tau.ac.il
bDepartment of Chemistry and Biology A. Zambelli, INSTM Research Unit, University of Salerno, Via Giovanni Paolo II 132, I-84084 Fisciano, SA, Italy. E-mail: vvenditt@unisa.it
First published on 26th May 2017
Abstract
Chloro-magnesium complexes of divergent tetradentate-monoanionic {ONNN}-type ligands which formed as either mononuclear or dinuclear complexes are reported. These complexes catalyze the ring-opening polymerization of lactide with high activity and in a living manner, and enable the synthesis of well-defined stereo-diblock copolymers with Mn > 200 kDa, as well as stereo-n-block copolymers (n = tri, tetra) with high molecular weights by sequential monomer addition. Thermal and crystallographic characterizations revealed that even the very high molecular weight PLA copolymers crystallized in a stereocomplex phase, and that their degradation temperature was as high as 354 °C.
Introduction
Poly(lactic acid) (PLA), a biodegradable polyester having biomedical and other useful applications,1 has attracted considerable current interest, and is produced by the catalyzed ring opening polymerization (ROP) of lactide (LA), the cyclic dilactone of lactic acid.2 The stereoregularity of PLA has a strong influence on its chemical/physical properties.3 PLLA and PDLA, the enantiomeric isotactic forms of PLA which consist of repeat units of the homochiral lactides (L-LA or D-LA), are the favoured forms, due to their high crystallinity. PLLA and PDLA tend to co-crystallize in the stereocomplex phase (sc-PLA),4 which exhibits an enhanced melting transition and superior mechanical properties.5 However, this tendency reverts to the homochiral phase (hc-PLA) for high molecular weight PLA.6 To retain the stereocomplexation tendency, an isotactic stereo-block microstructure should be devised. Several strategies have been developed en route to this microstructure including the attachment of functionalized PLA segments,7 the multi-step sequential polymerization of lactide enantiomers,8 and the isoselective polymerization of racemic LA.9 A more straightforward method would be the one-pot sequential monomer addition to a truly living polymerization catalyst.10–13 However, such catalysts are extremely rare. Herein, we describe a family of chloro-magnesium complexes of readily accessible {ONNN} ligands that are highly active in the living/immortal ROP of lactide and are able to polymerize lactide stereoisomers sequentially in a one-pot reaction, to produce stereo-n-block PLA copolymers (n = di, tri, and tetra).Magnesium (and zinc) complexes show vast potentials as ROP catalysts due to their high activity, biocompatibility and low cost.14 Recently, we reported a chloro-magnesium complex supported by a sequential tetradentate monoanionic ligand (Fig. 1, left),12 which was proposed to operate by the uncommon activated-monomer mechanism,15 and the complex was found to exhibit exceptional activity in the living ROP of lactide, enabling the synthesis of precise stereo-n-block PLA copolymers. We hypothesized that such a mechanistic preference might prevail in other chloro-magnesium complexes, and targeted the readily accessible divergent monoanionic {ONNN} ligands consisting of a phenol and two pyridine side arms (Fig. 1, right). Zinc and magnesium complexes of these ligands were previously explored as catalysts in the ROP of lactide.16,17 While the chloro- and HMDS-zinc complexes showed acceptable activities, the enolato-magnesium complex was inactive. The corresponding chloro-magnesium complexes, described herein, were not investigated until now, to the best of our knowledge.
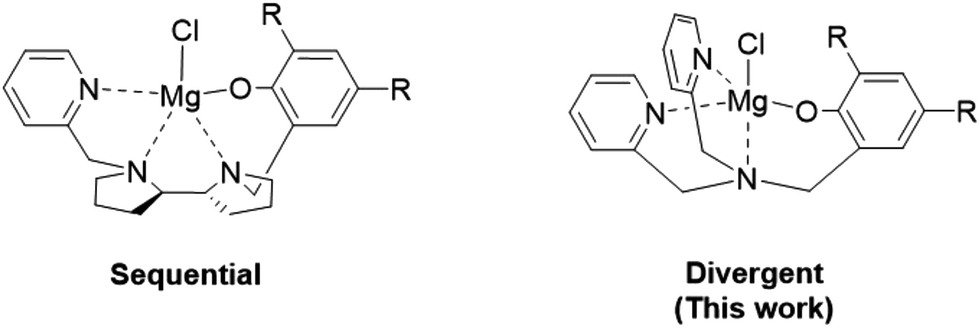 | ||
| Fig. 1 Sequential {ONNN}Mg–Cl complex vs. divergent {ONNN}Mg–Cl complex. | ||
Results and discussion
To address structure–activity relationships, five divergent monoanionic ligands were prepared (Lig1–5H, Scheme 1) having alkyl substituents of different steric bulk (H, Me, tBu, adamantyl and cumyl) in the ortho-positions of the phenolate arm. Out of the five ligands, Lig4H and Lig5H have not been described before.16,17 The ligands were readily synthesised by single-step procedures in high yields, by a modified reductive-amination reaction (Lig1,3–5H) or by the Mannich reaction (Lig2H) employing the commercially-available bis(2-pyridylmethyl)amine and a commercially- or readily-available substituted phenol/salicylaldehyde. The ligand precursors were obtained as colourless or yellow powders, and their identities were confirmed by NMR spectroscopy and high-resolution mass spectrometry (see the ESI†). | ||
| Scheme 1 Synthesis of the divergent {ONNN}Mg–Cl complexes. The steric character of the phenol substituents dictates the complex nuclearity. | ||
Following our previous synthesis of the sequential chloro-magnesium {ONNN}Mg–Cl complexes,12 the bulky ligand precursors Lig3–5H were reacted with one equiv. of benzyl magnesium chloride in toluene at room temperature and yielded the corresponding chloro-magnesium complexes as yellow powders in high to quantitative yields (Scheme 1). 1H NMR spectroscopy (in CDCl3) revealed a single set of peaks signifying mononuclear complexes of the type LignMg–Cl obtained as single stereoisomers of Cs-symmetry (Fig. 2, top, and Fig. S11–S16†). The magnesium complexes of the non-bulky divergent ligand precursors Lig1,2H were prepared likewise, and yielded the corresponding complexes in high yields as yellow powders as well. However, the 1H NMR spectra of these two complexes displayed different characteristics including two sets of peaks for the two pyridine rings and three AB systems for the three CH2 bridges. This reduced symmetry is attributed to the formation of dinuclear magnesium complexes of the type [(μ-Lign)Mg–Cl]2 for these sterically non-encumbered ligands (Fig. 2, bottom, and Fig. S7–S10 and S17–S20†) having either Ci- or Cs-averaged symmetry.
 | ||
| Fig. 2 1H NMR spectra (CDCl3, 3–9.5 ppm) of representative mononuclear (top, Lig3Mg–Cl) vs. dinuclear (bottom, [(μ-Lig1)Mg–Cl]2) chloro-magnesium complexes. | ||
Single crystals of the complexes [(μ-Lig1)Mg–Cl]2 and [(μ-Lig2)Mg–Cl]2 were grown from dichloromethane solutions at −35 °C, and their molecular structures were determined by X-ray diffraction studies. The two complexes were isostructural, featuring dinuclear chloro-magnesium complexes of octahedral geometry in which the phenolate oxygens of the two ligand units bridge between the two magnesium atoms, thus supporting the NMR findings (Fig. 3 and S21†). A crystallographic Ci-symmetry of the two complexes dictates a planar Mg–O–Mg–O ring, and the Mg–O(Ph) bond lengths are only slightly different, being 2.032 Å and 2.061 Å for [(μ-Lig1)Mg–Cl]2 and 2.026 Å and 2.062 Å for [(μ-Lig2)Mg–Cl]2. Despite our attempts, single crystals of the presumably mononuclear complexes Lig3–5Mg–Cl could not be obtained. The molecular structure of the previously reported magnesium enolato complex of Lig3 featured a pentacoordinate mononuclear magnesium centre,16 supporting our proposed mononuclear structure of Lig3–5Mg–Cl. Further evidence for these different coordination modes was provided by high-resolution mass spectrometry (HRMS) analysis, which supported the formation of the proposed mononuclear and dinuclear structures for Lig3–5Mg–Cl and Lig1,2Mg–Cl, respectively. Such a coordination pattern in which the steric bulk of the phenolate substituent dictates the complex nuclearity was previously observed with similar divergent {ONOO} zinc complexes.18
 | ||
| Fig. 3 Molecular representation of the crystallographic structure of [MgCl(μ-Lig1)]2. | ||
We investigated the catalytic activity of the magnesium complexes in the ROP of L-LA. Polymerization runs were performed in dichloromethane solution at RT by adding lactide to the LignMg-Cl complex as the catalyst and benzyl alcohol as the initiator. Under these conditions, the polymerization of 300 equiv. of lactide by the mononuclear complexes Lig3–5Mg–Cl led to almost full consumption of the monomer within 2–5 min (see Tables 1 and S1†). Although slightly less active than the related chloro-magnesium catalyst of the sequential bipyrrolidine based {ONNN} ligand, these complexes still represent some of those with the highest activities ever reported for lactide polymerization.19 The PLLA samples obtained were characterized by gel permeation chromatography (GPC) analysis and were found to have remarkably low PDI values of ≤1.08, with molecular weights (Mn) in agreement with the monomer/initiator molar ratios. The performance of these catalysts was also explored under ‘immortal conditions’ (namely, using an initiator/catalyst ratio > 1, which may lead to the production of more than one single polymer chain per catalyst unit),20 which led to PLLA samples with very narrow PDIs and Mn values consistent with the monomer/benzyl alcohol initiator ratios. Monomer loadings of up to 2000 equiv. were attempted giving almost full consumption after 5 min, and yielding monodispersed PLLA of high Mn. These divergent {ONNN}Mg–Cl complexes were found to be highly active ROP catalysts for L-LA even in the absence of benzyl alcohol (see Table S1†). NMR spectra of mixtures of the LignMg-Cl complexes and benzyl alcohol in CDCl3 revealed that no reaction took place. Consistently, an intact Lig3Mg–Cl species rather than a Lig3Mg-OBn species was observed in the HRMS measurements of a mixture of Lig3Mg–Cl and benzyl alcohol. All of these findings indicate that a magnesium-alkoxo species does not form, and support our proposal that these ROP catalysts operate by the activated monomer mechanism, rather than the common coordination insertion mechanism.
| En | Initiator | [I]/[BnOH]/[LA] | Timeb | Conv.c | M n | PDI |
|---|---|---|---|---|---|---|
| a Polymerizations performed in CH2Cl2 (5 mL) at RT employing 5 μmol of catalyst. b Polymerization time given in minutes. c Determined by 1H NMR spectroscopy (500 MHz). d Determined by GPC analysis calibrated with polystyrene standards and multiplied by a correction factor of 0.58. Values are given in g mol−1. | ||||||
| 1 | Lig3Mg–Cl | 1/1/300 | 5 | 0.98 | 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 900 |
1.04 |
| 2 | Lig4Mg–Cl | 1/1/300 | 2 | 0.98 | 46500 |
1.08 |
| 3 | Lig4Mg–Cl | 1/4/600 | 3 | 0.98 | 20200 |
1.04 |
| 4 | Lig4Mg–Cl | 1/1/2000 | 5 | 0.96 | 271500 |
1.06 |
| 5 | Lig5Mg–Cl | 1/1/300 | 5 | 0.95 | 35900 |
1.08 |
| 6 | [MgCl(μ-Lig1)]2 | 1/1/300 | 15 | 0.90 | 30400 |
1.08 |
| 7 | [MgCl(μ-Lig2)]2 | 1/1/300 | 15 | 0.88 | 39200 |
1.07 |
The dinuclear complexes, [(μ-Lig1)Mg–Cl]2 and [(μ-Lig2)Mg–Cl]2, were also active in the immortal ROP of L-LA, however longer reaction times were required to obtain high conversions (see Tables 1 and S1†). Notably, the addition of benzyl alcohol to the dinuclear complex [(μ-Lig1)Mg–Cl] revealed that no reaction had occurred, in common with what was observed for the mononuclear complexes. Assuming that the activated-monomer mechanism governs the polymerization process with these complexes as well, the ability of a lactide monomer to penetrate through the congested coordination sphere of the dinuclear complexes and be activated by a magnesium centre having a reduced Lewis acidity is intriguing. Possibly, a low concentration of a mononuclear magnesium complex is responsible for the ROP activity, however, such a mononuclear species could not be detected in the NMR spectra, so, if it does exist, its concentration is very low.
The preparation of perfect stereo-block copolymers requires that following the full consumption of the first monomer, the addition of a second monomer portion leads to a continued growth of all polymer chains. In addition, side reactions and in particular trans-esterifications, which may broaden the PDIs of the polymers and hamper their blocks’ integrity, should be minimal. To find out whether the preparation of stereo-block copolymers would be viable with the current magnesium catalysts, we first attempted the sequential polymerization of two portions of 200 equiv. of L-LA under the above conditions, with a period of several minutes between monomer additions. We chose Lig3Mg–Cl and Lig4Mg–Cl, the catalysts that exhibited the highest activities. 1H NMR analysis of the polymerization mixtures revealed that very high monomer conversions (>0.98) were attained after both the first and the second lactide additions. Attempts to reach full first monomer conversion by prolonging the reaction time did not lead to significant improvement, but resulted in the broadening of the PDIs (according to GPC analysis). Namely, block-copolymerizations with these catalysts require the optimization of the polymerization time between monomer additions, to reach the highest conversion while minimizing possible side-reactions.
With this guideline, we attempted the synthesis of stereo-diblock copolymers by adding L-LA to a stirring mixture of either Lig3Mg–Cl or Lig4Mg–Cl and benzyl alcohol in dichloromethane, followed by addition of D-LA several minutes later (Tables 2 and S2†). Although not reaching a full first monomer conversion, stereo-diblock copolymers having block lengths of up to 500 repeat units each were easily prepared in short periods of time of ca. 10 min. These stereo-diblock copolymers featured very narrow PDIs and their molecular weights coincided with the monomer/initiator ratios according to GPC analysis. This was particularly valid for Lig4Mg–Cl, featuring the adamantyl-phenolate group which enabled the synthesis of stereo-diblock copolymers with a PDI of ≤1.07, and very high block integrities according to their very high degrees of isotacticity (Pm ≥ 0.98). Evidently, the short polymerization times kept the side reactions to a minimum. To test the limits of this methodology we attempted the synthesis of even longer blocks: we prepared an L(800)-b-D(800) stereo-diblock copolymer in 20 min, and found that it featured a very high block integrity according to GPC and NMR characterization. To the best of our knowledge, this is the longest well-defined PLA stereo-diblock copolymer ever reported.5,6
| Entry | Initiator | Type | Composition | Timeb | M n(calc) | M n | PDI | T m | ΔHme |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerizations performed in CH2Cl2 (5 mL) at RT employing 5 μmol of the catalyst and 1 equiv. of BnOH. Almost full conversion (>0.98) was confirmed by 1H NMR (500 Hz). b Total polymerization time given in minutes. 5–10 min were maintained between each monomer addition, depending on the monomer amount and length of the polymer chain. c Calculated from monomer conversion assuming full benzyl alcohol participation. Values are given in g mol−1. d M n was determined by GPC analysis with CHCl3 as the eluent. e Melting temperature and enthalpy of first DSC run. Values are given in °C and J g−1, respectively. | |||||||||
| 1 | Lig3Mg–Cl | Di block | L(100)-b-D(100) | 10 | 28800 |
26700 |
1.11 | 213 | 75 |
| 2 | Lig3Mg–Cl | Di block | L(200)-b-D(200) | 10 | 57600 |
62670 |
1.20 | 213 | 64 |
| 3 | Lig4Mg–Cl | Di block | L(100)-b-D(100) | 10 | 28800 |
22900 |
1.06 | 212 | 59 |
| 4 | Lig4Mg–Cl | Di block | L(300)-b-D(300) | 10 | 86400 |
85800 |
1.04 | 214 | 76 |
| 5 | Lig4Mg–Cl | Di block | L(500)-b-D(500) | 11 | 144000 |
149110 |
1.07 | 214 | 79 |
| 6 | Lig4Mg–Cl | Di block | L(800)-b-D(800) | 20 | 228100 |
202300 |
1.09 | 214 | 67 |
| 7 | Lig4Mg–Cl | Tri block | L(100)-b-D(100)-b-L(100) | 15 | 43200 |
45860 |
1.08 | 202 | 36 |
| 8 | Lig4Mg–Cl | Tri block | L(300)-b-D(300)-b-L(300) | 18 | 129600 |
120590 |
1.12 | 201 | 40 |
| 9 | Lig4Mg–Cl | Tetra block | L(100)-b-D(100)-b-L(100)-b-D(100) | 22 | 57600 |
64690 |
1.10 | 202 | 49 |
| 10 | Lig4Mg–Cl | Tetra block | L(200)-b-D(200)-b-L(200)-b-D(200) | 24 | 115200 |
104230 |
1.13 | 205 | 56 |
The results of the thermal analysis of the stereo-diblock copolymers, carried out on dichloromethane (DCM) cast films, are reported in Tables 2 and S3† and in Fig. 4. Regardless of the block lengths, both melting temperatures and enthalpies (Tm, ΔHm) have high values which range, in the first DSC heating run, from 211 to 215 °C, and from 59 to 87 J g−1, respectively. These Tm and ΔHm values support the high copolymer stereoregularities and the assumed high block integrity of the copolymers. This assumption is also valid for the exceptionally long stereo-diblock copolymer L(800)-b-D(800), whose Tm and ΔHm are comparable with those of the shorter copolymers (Tm and ΔHm of the stereo-diblock copolymers are compared in Fig. S30†). All of the diblock copolymers crystallized either from the polymerization solution, DCM solution or melt during the DSC cooling run, and are in stereocomplex crystal form (wide-angle X-ray diffraction (WAXD) patterns of the copolymers are shown in Fig. 5 and differently crystallized L(800)-b-D(800) samples are reported in Fig. S31 and S32,† respectively). To the best of our knowledge, the L(800)-b-D(800) stereo-diblock copolymer is the first example of a PLLA–PDLA system having a MW > 200 kDa (either PLLA–PDLA blends or L-LA/D-LA stereo-diblock copolymers) which fully crystallizes in the stereocomplex form only, rather than as a mixture with the homochiral form.5,6 Aiming to validate the assumption that the isotactic stereo-block microstructure is a prerequisite for the crystallization in the stereocomplex phase for polymers of such high molecular weights, we prepared high molecular-weight PLLA and PDLA homochiral samples, mixed them in a 1:1 ratio in DCM solution, casted films from them, and then analyzed them with WAXD. Samples of PLLA and PDLA of ca. 800 repeat units corresponding to each block of the L(800)-b-D(800) stereo-diblock copolymer, as well as samples of ca. 1600 repeat units, were synthesized and tested. Fig. 5 compares the WAXD patterns of the films, annealed at 100 °C for 10 min, of the L(800)-b-D(800) stereo-diblock copolymer and the two homochiral polymer mixtures. The diblock copolymer pattern only shows the reflections of the stereocomplex crystal form (at 2θ ≈ 12, 21, 24°) while the spectra of the two mixtures only show the reflections of the homochiral crystal form (at 2θ ≈ 16.8, 19, 22.4°), supporting the above assumption. Notably, the L(800)-b-D(800) stereo-diblock copolymer has a high degradation temperature of 354 °C (TDTG, valued by the weight loss derivative maximum), the highest among the copolymers prepared (thermogravimetric curves of selected diblock copolymers are reported in Fig. S33†), due to the unzipping depolymerization mechanism operating during degradation.5a The relationship between TDTG and Mn determined by GPC, for the stereo-diblock copolymers listed in Table S2,† is reported in Fig. S34† and in Table S4,† which includes the TDTG of the mixture of PLLA and PDLA homopolymers of ca. 1600 repeat units, for comparison. The TDTG of the stereo-diblock copolymers increases linearly with Mn for molecular weights ranging from 20 to 150 kDa, while for higher Mns it reaches the plateau value of 350 °C.
 | ||
| Fig. 4 Thermograms of the second DSC heating run of stereo-diblock copolymers having (a) 100, (b) 300 and (c) 800 mer block lengths. | ||
 | ||
| Fig. 5 WAXD patterns of the films, annealed at 100 °C for 10 min. Blends 1 and 2, the mixtures of the homochiral polymers having ca. 800 and 1600 repeat units, respectively are compared with L(800)-b-D(800) stereo-diblock copolymer. | ||
The synthesis of higher stereo-n-block copolymers, namely stereo tri- and tetra-block copolymers, was attempted by sequential monomer additions according to the above conditions and employing Lig4Mg–Cl. It might be anticipated that the accumulation of stereoerrors that result from incomplete monomer conversion and possible trans-esterification involvement would increase as a function of the block number n, and the polymerization time. Surprisingly, this catalyst enabled the synthesis of stereo-n-block (n = 3, 4) copolymers of different block lengths whose integrities were very high judging by their high degrees of isotacticity (Pm ≥ 0.96).
Thermal analysis results are summarized in Tables S5 and S6,† and thermograms (second DSC heating runs) and WAXD spectra of the di-, tri- and tetra-block copolymers having 200 mer block lengths, are compared in Fig. S35 and S36,† respectively. These revealed high Tm and ΔHm values, in perfect agreement with the high polymer stereoregularities which supported our hypothesis of high block integrity even for these tri- and tetra-block copolymers.21 Notably, as reported in ref. 12, copolymers with enantiomeric unpaired blocks (stereo-triblock copolymers in the current study) show lower ΔHms than those of enantiomeric paired blocks (stereo-diblock and tetra-block copolymers).
Conclusions
We introduced a new family of chloro-magnesium complexes based on the readily available divergent tetradentate-monoanionic {ONNN}-type ligands. Either mononuclear or dinuclear complexes had formed as a direct consequence of the steric bulk of the phenolate substituents. All complexes, and in particular the mononuclear complexes, were found to be highly active in the ring opening polymerization catalysis of homochiral lactides consuming up to 2000 equiv. of monomer within five min and giving rise to monodispersed poly(lactic acid)s of expected molecular weights. Moreover, these catalysts enabled the preparation of stereo-diblock copolymers of very high lengths and stereo-n-block copolymers consisting of up to four blocks in short periods of time by sequential monomer addition. All copolymers crystallized exclusively in the stereocomplex crystal form and showed high Tm and ΔHm. In particular, the L(800)-b-D(800) stereo-diblock copolymer was found to crystallize in the stereocomplex phase resulting in a very high melting transition, and to degrade at a very high temperature, a combination of features that cannot be attained by mixtures of the homochiral polymers of high molecular weights or by stereo-block copolymers of lower molecular weights. These catalysts are proposed to operate by the uncommon activated monomer mechanism. It would thus appear that successful catalysts that lead to tailor-made polymeric materials, which are difficult to attain by alternative catalysts, and are derived from readily available ligands and the common metal magnesium are at hand. We are currently investigating the structure–activity properties of complexes of these divergent ligands, and are attempting to develop polymeric materials of uncommon microstructures.Acknowledgements
This work was funded by grant No. 650/14 of the Israel Science Foundation and by grant No. 2012187 of the United States – Israel Binational Science Foundation, and by project SEC-242387 of the FP7 European Commission.Notes and references
- (a) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS; (b) R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed; (c) A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed; (d) M. Brzeziński and T. Biela, Polym. Int., 2015, 64, 1667–1675 CrossRef; (e) E. Castro-Aguirre, F. Iñiguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS PubMed.
- (a) J. Pretula, S. Slomkowski and S. Penczek, Adv. Drug Delivery Rev., 2016, 107, 3–16 CrossRef CAS PubMed; (b) B. J. O’keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC; (c) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (d) R. H. Platel, L. M. Hodgson and C. K. Wiliiams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS; (e) M. H. Chisholm, Pure Appl. Chem., 2010, 82, 1647–1662 CrossRef CAS; (f) J. M. Becker, R. J. Pounder and A. P. Dove, Macromol. Rapid Commun., 2010, 31, 1923–1937 CrossRef CAS PubMed; (g) M. J. L. Tschan, E. Brule, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC; (h) S. Slomkowski, S. Penczek and A. Duda, Polym. Adv. Technol., 2014, 25, 436–447 CrossRef CAS.
- (a) M. J. Sanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC; (b) S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- (a) H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed; (b) K. Fukushima and Y. Kimura, Polym. Int., 2006, 55, 626–642 CrossRef CAS; (c) H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS PubMed; (d) Z. Li, B. H. Tan, T. Lin and C. He, Prog. Polym. Sci., 2016, 62, 22–72 CrossRef CAS.
- M. Hirata, K. Kobayashi and Y. Kimura, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 794–801 CrossRef CAS.
- (a) K. Masutani, C. W. Lee and Y. Kimura, Polym. J., 2013, 45, 427–435 CrossRef CAS; (b) L. Han, Q. Xie, J. Bao, G. Shan, Y. Bao and P. Pan, Polym. Chem., 2017, 8, 1006–1016 RSC.
- (a) M. Hirata, K. Kobayashi and Y. Kimura, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 794–801 CrossRef CAS; (b) H. Tsuji and T. Tajima, Macromol. Mater. Eng., 2014, 299, 1089–1105 CAS.
- (a) N. Spassky, M. Wisniewski, C. Pluta and A. Le-Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS; (b) N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem.Soc., 2002, 124, 5938–5939 CAS.
- (a) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS PubMed; (b) N. Othman, C. Xu, P. Mehrkhodavandi and S. G. Hatzikiriakos, Polymer, 2012, 53, 2443–2452 CrossRef CAS; (c) D. C. Aluthge, C. Xu, N. Othman, N. Noroozi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2013, 46, 3965–3974 CrossRef CAS.
- C. Fliedel, V. Rosa, F. M. Alves, A. M. Martins, T. Avilés and S. Dagorne, Dalton Trans., 2015, 44, 12376–12387 RSC.
- T. Rosen, I. Goldberg, V. Venditto and M. Kol, J. Am. Chem. Soc., 2016, 138, 12041–12044 CrossRef CAS PubMed.
- For a zinc analogue of the above complex, see: T. Rosen, Y. Popowski, I. Goldberg and M. Kol, Chem.–Eur. J., 2016, 22, 11533–11536 CrossRef CAS PubMed.
- (a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed; (b) M. H. Chisholm, N. W. Eilerts, J. C. Huffmann, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845–11854 CrossRef CAS; (c) M. L. Shueh, T. S. Wang, B. H. Huang, C. Y. Kuo and C. C. Lin, Macromolecules, 2004, 37, 5155–5162 CrossRef CAS; (d) H. Y. Tang, H. Y. Chen, J. H. Huang and C. C. Lin, Macromolecules, 2007, 40, 8855–8860 CrossRef CAS; (e) V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827 RSC; (f) V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2011, 40, 523–534 RSC; (g) Y. Wang, W. Zhao, X. Liu, D. Cui and E. Y.-X. Chen, Macromolecules, 2012, 45, 6957–6965 CrossRef CAS; (h) L. F. Sanchez-Barba, A. Garces, J. Fernandez-Baeza, A. Otero, C. Alonso-Moreno, A. Lara-Sanchez and A. M. Rodríguez, Organometallics, 2011, 30, 2775–2789 CrossRef CAS; (i) S. Song, H. Ma and Y. Yang, Dalton Trans., 2013, 42, 14200–14211 RSC; (j) H. Xie, Z. Mou, B. Liu, P. Li, W. Rong, S. Li and D. Cui, Organometallics, 2014, 33, 722–730 CrossRef CAS; (k) H. Wang, Y. Yang and H. Ma, Macromolecules, 2014, 47, 7750–7764 CrossRef CAS.
- (a) M. Normand, V. Dorcet, E. Kirillov and J. F. Carpentier, Organometallics, 2013, 32, 1694–1709 CrossRef CAS; (b) Y. Sun, Y. Cui, J. Xiong, Z. Dai, N. Tang and J. Wu, Dalton Trans., 2015, 44, 16383–16391 RSC.
- Z. J. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, C. M. Thomas, T. Roisnel, O. Casagrande and J.-F. Carpentier, New J. Chem., 2008, 32, 2279–2291 RSC.
- M. J.-L. Tschan, J. Guo, S. K. Raman, E. Brulé, T. Roisnel, M.-N. Rager, R. Legay, G. Durieux, B. Rigaud and C. M. Thomas, Dalton Trans., 2014, 4550–4564 RSC.
- S. Groysman, E. Sergeeva, I. Goldberg and M. Kol, Eur. J. Inorg. Chem., 2006, 2739–2745 CrossRef CAS.
- (a) L. Wang and H. Ma, Macromolecules, 2010, 43, 6535 CrossRef CAS; (b) Y. Gao, Z. Dai, J. Zhang, X. Ma, N. Tang and J. Wu, Inorg. Chem., 2014, 53, 716–726 CrossRef CAS PubMed; (c) A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- T. Aida and S. Inoue, Acc. Chem. Res., 1996, 29, 39–48 CrossRef CAS.
- We attribute the lower Tm and ΔHm of the 4 × 300 mer stereo-tetrablock copolymer to the accumulation of mis-insertions of unreacted monomer from previous steps in this very high MW polymer (Table S6†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data regarding the synthesis and characterization of the ligands, complexes and PLA polymers. CCDC 1537631–1537632. For ESI and crystallographic data in CIF format or other electronic format see DOI: 10.1039/c7sc01514c |
| This journal is © The Royal Society of Chemistry 2017 |