
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ground-state dioxygen undergoes metal-free [3 + 2]-annulations with allenes and nitrosoarenes under ambient conditions†
Jinxian
Liu‡
ab,
Manisha
Skaria‡
a,
Pankaj
Sharma
a,
Yun-Wei
Chiang
 a and
Rai-Shung
Liu
*a
a and
Rai-Shung
Liu
*a
aDepartment of Chemistry, National Tsing-Hua University, Hsinchu, Taiwan, Republic of China. E-mail: rsliu@mx.nthu.edu.tw
bCollege of Chemistry & Materials Science, Longyan University, Fujian, China
First published on 24th May 2017
Abstract
The cycloadditions of molecular dioxygen with neutral π-bond motifs rely heavily on singlet-state 1O2, whereas ground state 3O2 is chemically inactive. Here we report novel [3 + 2]-annulations among ground-state 3O2 (1 bar), allenes, and nitrosoarenes at low temperatures, efficiently yielding dioxygen-containing oxacycles. With less hindered 1-arylallene derivatives, these dioxygen species undergo skeletal rearrangement to 3-hydroxy-1-ketonyl-2-imine oxides. These cycloadditions represent valuable one-pot O,N,O-trifunctionalizations of allenes. Our EPR experiments confirm the presence of 1,4-diradical intermediates from an allene/nitrosoarene mixture, which manifest the hidden diradical properties of nitrosoarenes.
Introduction
Cycloadditions of two or three π-bond molecules are powerful tools to access carbo- or heterocycles. Ground-state 3O2 has low-lying LUMO orbitals, but its triplet state greatly reduces its chemical reactivity toward neutral molecules1 unless a metal catalyst is present. The cycloadditions of 3O2 dioxygen rely nearly exclusively on prior photo-activation to form singlet-state 1O2 (ref. 1) that reacts with dienes,2 olefins3 or even arenes4 in [n + 2]-cycloadditions (n = 2 and 4, Scheme 1, eqn (1)). This photolytic process requires a sensitizer in a cold bath (−40 °C) over a protracted period (>12 h) because highly energetic 1O2 might produce byproducts from the oxygen-ene reactions5 and oxidative C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C cleavages.6 In the case of allenes, singlet dioxygen afforded a complicated mixture of undesired compounds.7a,b
C cleavages.6 In the case of allenes, singlet dioxygen afforded a complicated mixture of undesired compounds.7a,b
 | ||
| Scheme 1 Cycloadditions of unsaturated hydrocarbons with 1O2 and 3O2. | ||
As ground-state 3O2 is a free π-molecule and is available everywhere; its metal-free [n + 2]-cycloadditions with commonly used unsaturated hydrocarbons would provide a clean and cheap synthesis of valuable 1,n-diols, although there is no literature precedence. As far as we are aware, only 1,4-diradical precursors such as o-benzocyclobutanes,8 1,2,6,7-octatetraenes,9 2,3-dimethylenebicyclo[2.2.0]hexane10 and other 1,4-diazo species11 reacted with ground-state 3O2 in thermal [4 + 2]-cycloadditions; these precursors are too uncommon to show general utility. We recently achieved metal-catalyzed annulations of N-hydroxy allenylamines with nitrosoarenes via a single radical process.7d In search of a breakthrough in dioxygen chemistry, we developed facile [3 + 2]-cycloadditions among nitrosoarenes, allenes and ground-state 3O2 to efficiently afford N-(1,2-dioxolan-4-ylidene)aniline oxides (eqn (2)). Particularly notable are the ambient conditions: −15 to 0 °C, 3O2 (1 bar), no light, no catalyst and no additive. Importantly, these facile spin-forbidden dioxygen annulations reveal a new role of nitrosoarenes as effective diradical precursors that is synthetically significant in nitroso chemistry.12 In the context of nitroso/alkene and nitroso/alkyne reactions,13 theoretical calculations by Houk12e,f suggested the intermediacy of the diradical species, but these transient species could not be trapped with dioxygen or other small molecules.
2-Amino-1,3-diols are present in numerous natural products with diverse biological activity (Fig. 1).14 Catalytic O,N,O-trifunctionalization of allenes is a new appealing tool to assess these motifs, as noted by the work of Schomaker, who reported Rh-catalyzed intramolecular cyclizations of homoallenylsulfamate esters via a two-step sequence.15a In contrast, our one-pot intermolecular O,N,O-functionalizations employ common and cheap nitrosoarenes, allenes and oxygen.
 | ||
| Fig. 1 O,N,O-Trifunctionalizations of allenes and selected natural products. | ||
Results and discussion
Table 1 presents the optimized yields of a O,N,O-trifunctionalized molecule 3a from a mixture of allene 1a, nitrosobenzene 2a (n equiv.) and O2 (1 bar). When 1.5 equiv. of nitrosobenzene 2a was used in cold THF (−15 °C), the yield was 43% (entry 1). The yield of 3a increased to 63% with nitrosobenzene in three fold proportions (entry 2). In other solvents, the yields of 3a were 50% in toluene, 54% in CH3CN, and 58% in DCM (entries 3–5). The yield of 3a decreased substantially to 10% in THF at 25 °C (entry 6). The reaction under N2 failed to yield the desired product 3a in a traceable amount (entry 7).16 Compound 3a assumes an E-configuration with its hydroxyl cis to the nitrone oxygen to form a hydrogen bond. This structure was inferred from X-ray diffraction measurements of its relative 3b17 (Table 2 entry 1).
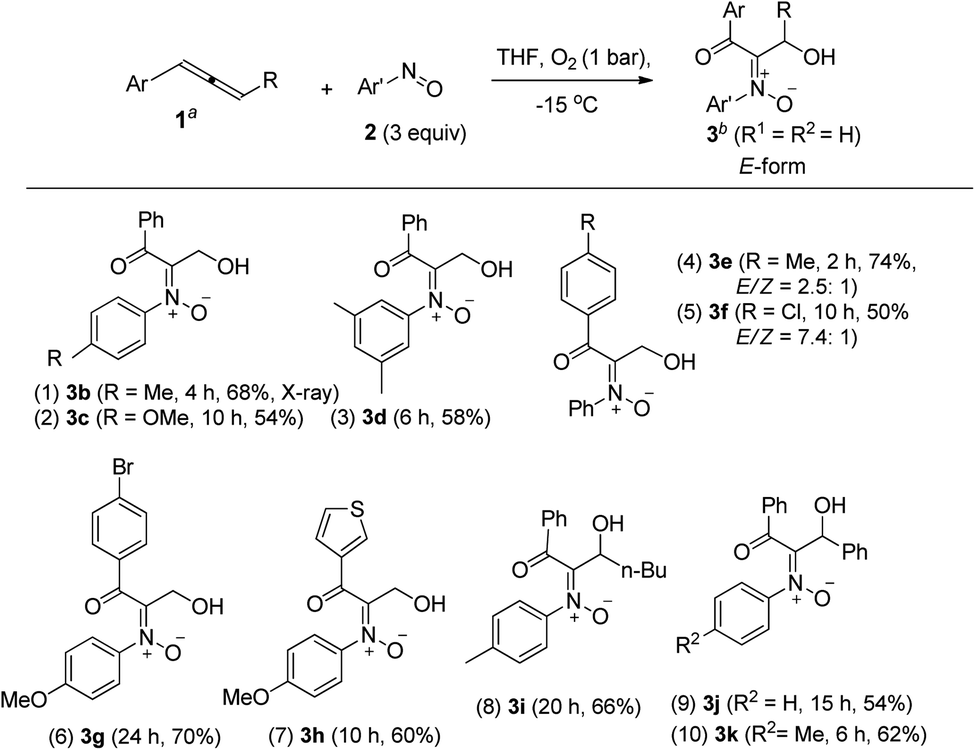
To assess the reaction scope, we applied these optimized conditions to additional mono- and 1,3-disubstituted allenes 1b–1g; Table 2 summarizes the results. For phenylallene 1a, its corresponding reactions with 4-methyl-, 4-methoxy- and 3,5-dimethylphenylnitroso species afforded 3-hydroxy-1-ketonyl-2-imine oxides 3b–3d in 54–68% yields (entries 1–3). Varied arylallenes 1b–1e (Ar = 4-MeC6H4, 4-ClC6H4, 4-BrC6H4 and 3-thienyl) yielded desired compounds 3e–3h in satisfactory yields (50–74%, entries 4–6). 3-Substituted phenylallenes 1f and 1g (R = n-Bu and Ph) were also effective substrates for these cycloadditions (entries 8–10).
Notably, the reaction of sterically hindered 3-cyclohexyl-1-phenylallene 1i with 4-methoxyphenylnitroso 2c and O2 (1 bar) afforded dioxygen-containing oxacycle 4a together with desired product 3l; the yields were 45% and 28%, respectively. Species 4a assumes an anti-configuration (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) according to its 1H NOE spectra; this new compound was efficiently converted to compound 3l in hot THF (eqn (3)), via a Kornblum–DeLaMare rearrangement.22
:1) according to its 1H NOE spectra; this new compound was efficiently converted to compound 3l in hot THF (eqn (3)), via a Kornblum–DeLaMare rearrangement.22
 | (3) |
The kinetic stability of dioxygen-containing oxacycle 4a is enhanced with a suitable steric environment. We further tested the reactions on various 1-aryl-1-methylallenes 1j–1m with 4-methoxyphenylnitroso 2c and O2 (1 bar) in THF (0 °C), generating dioxygen-containing compounds 4b–4e (Ar = 4-RC6H4, R = H, Me, MeO, Br) in satisfactory yields (Table 3, entries 1–4). The molecular structure of compound 4b was confirmed by its X-ray diffraction pattern.17 Various 1-aryl-3,3-dimethylallenes 1n–1q (Ar = 4-RC6H4, R = H, Me, MeO, Br), electron-rich nitrosoarenes and O2 were also amenable to such cycloadditions, yielding desired compounds 4f–4m in satisfactory yields (60–72%, entries 5–12) except 4k in only 38% yield. This dioxygen cycloaddition was applicable to cyclohexylidene-derived phenylallene 1r, affording compound 4n in 66% yield (entry 13). Compounds 4 serve as the first examples of the cycloadditions of ground-state 3O2 with unsaturated hydrocarbons at low temperatures.

An electron-deficient nitrosoarene is an inapplicable substrate, as shown by eqn (4). Under O2, the reaction of trisubstituted allene 1p with 4-chlorophenylnitroso species 2f in cold THF (0 °C) afforded nitroso-containing cycloadduct 5a in 53% yield; the dioxygen-containing product, ca. 5%, was unstable for isolation (eqn (4)). In contrast, the same allene 1p could deliver dioxygen-containing species 4j and 4k using electron-rich nitrosoarenes under the same conditions (entries 9–10, Table 3).
 | (4) |
 | (5) |
Under nitrogen, trisubstituted allene 1p reacted with 4-methylphenylnitroso 2b in cold THF to form nitroso-containing cycloadduct 5b in 60% yield (eqn (5)). The stereochemistry and its E-configuration of this new compound was confirmed by its X-ray diffraction pattern.17 Such a new reaction represents a new and useful O,N,N-functionalization of allenes. A preliminary survey of the reaction scope is summarized in Table 4. We tested the reactions on 1,3-di- and 1,1,3-trisubstituted allenes 1g and 1t that reacted with nitroso-arenes (R = H, Cl, CO2Et) to afford nitroso-containing cycloadducts 5c–5g in reasonable yields (58–83%). Furthermore, the anti-configuration of compound 5c was determined by X-ray diffraction.17

Dioxygen-containing heterocycles 4 are readily reduced with Pd/C, H2 (1 atm) in MeOH (23 °C)18 to cleave their O–O bonds, satisfactorily yielding desired 1,3-dihydroxy-2-imine oxides 6. These reductions highlight the utility of molecular oxygen to afford 1,3-dihydroxy-2-amino derivatives. Several instances of affording tertiary 1,3-alcohol derivatives are illustrated in eqn (6) and (7); their chemical yields exceed 65%. Under these reductions, the valuable nitrone functionalities of these acyclic 1,3-diols remain intact as indicated by their HRMS and 13C-NMR spectra.
 | (6) |
 | (7) |
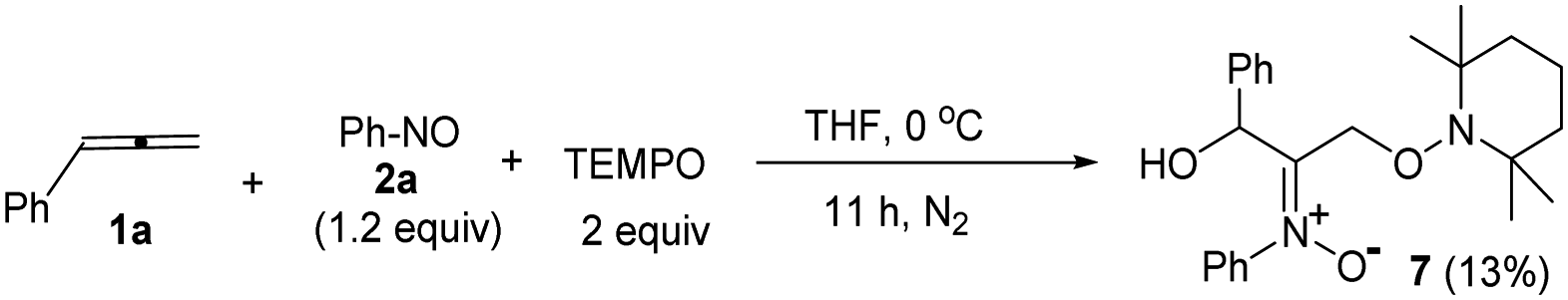
The facile cycloadditions among allenes, nitrones and ground-state O2 are very astonishing because an intersystem crossing (ISC) must be involved for one key intermediate. To investigate the mechanism, we examined the reaction of 1-phenyl-3-cyclopropylallene 1s with 4-methylphenylnitroso species 2b under O2, yielding compound 3m in 71% yield; this transformation did not induce cyclopropane cleavage because of the stability of the phenylallylic radical A (eqn (8)).19 We thus exclude the intermediacy of the dicarbon radical A′, although analogous carbon radicals were postulated for the o-quinodimethine species.8 We isolated compound 7 in 13% yield from the reaction of 1-phenylallene 1a with PhNO (1.2 equiv.) and TEMPO (2 equiv.) under N2, indicating the formation of diradical intermediates (eqn (9)). We employed EPR to characterize the diradical species from a mixture of 3,3-dimethyl-1-phenylallene 1n and nitrosobenzene 2a in THF at 0 °C (0.5 h). Fig. 2 (top) shows the EPR signal of the diradical species; the intensity of this signal remains unchanged for 5 h under N2. The simulation analysis was performed using the EasySpin program.20 The satisfactory fit was achieved with a two-component simulation (bottom). The abundant component (70%) corresponds to nitrogen-centered diradicals (g = 2.00616, aN = 10.7 G and 3.0 G).21 The minor component corresponds to a monoradical nitroxide with aN = 10.7 G. Notably, when recorded at T < 130 K, the spectrum exhibits a well-known nitroxide rigid-limit lineshape in accordance with the above simulation result; the coupling of unpaired electrons with the nitrogen center is evident.
 | (8) |
 | (9) |
 | ||
| Fig. 2 Observed and simulated EPR spectra. | ||
Scheme 2 depicts a plausible mechanism to rationalize the remarkable facility of such dioxygen annulations. We postulate that allene 1 reacts initially with nitrosobenzene to form 1,4-diradical species A, which is likely to be a major component, as detected in the EPR spectra; its nitroxy and allylic radicals are expected to couple with nitrogen in two magnitudes, i.e. aN = 10.7 G and 3.0 G respectively.21 The capture of molecular dioxygen 3O2 by 1,4-diradical species A forms peroxy diradical B in a triplet state, as the two radical centers of species B are remote from each other, rendering an intersystem crossing (ISC) feasible. After a change of spin state, singlet-state diradical B′ is expected to form primary 1,2-oxaziridine diradical C through a 3-exo-trig cyclization that is more feasible than an alternative 5-endo-trig cyclization.23 A final radical–radical coupling of resulting species C forms precursor D, and ultimately yields desired 1,2-dioxolanes 4. This proposed path rationalizes the formation of compound 7 from the TEMPO experiment (eqn (9)) well. The trapping of the 1,4-biradical generates single radical species F that undergoes a rapid 3-exo-trig cyclization to form benzylic radical G. A second trapping of this species with the TEMPO radical is expected to yield species I that is prone to hydrolysis on a silica column to yield observed product 7.
 | ||
| Scheme 2 A plausible mechanism. | ||
Conclusions
Prior to this work, singlet state oxygen 1O2 failed to react with allenes to give useful oxygenated products.7 This study reports the first examples of metal-free [3 + 2]-cycloadditions among allenes, nitrosoarenes and ground-state 3O2 (1 bar) at low temperatures, efficiently yielding dioxygen-containing oxacycles.24 With less hindered 1-arylallene derivatives, the resulting oxacycles undergo skeletal rearrangement to 3-hydroxy-1-ketonyl-2-imine oxides. These transformations highlight a cheap, efficient and clean synthesis of 1,3-dihydroxy-2-amino derivatives. Our experimental data indicate that an initial attack of a nitrosoarene at an allene generates a diradical species that is detectable with EPR. We envisage that the concept of nitrosoarenes as diradical precursors will inspire new synthetic concepts.Acknowledgements
The authors thank the National Science Council and the Ministry of Education, Taiwan, for supporting this work.Notes and references
- For reviews see: (a) P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181 RSC; (b) J. Sivaguru, M. R. Solomon, T. Poon, S. Jockusch, S. G. Bosio, W. Adam and N. Turro, Acc. Chem. Res., 2008, 41, 387 CrossRef CAS PubMed; (c) A. Greer, Acc. Chem. Res., 2006, 39, 797 CrossRef CAS PubMed; (d) A. G. Leach and K. N. Houk, Chem. Commun., 2002, 1243 RSC.
- (a) J. A. Celaje, D. Zhang, A. M. Guerrero and M. Selke, Org. Lett., 2011, 13, 4846 CrossRef CAS PubMed; (b) E. Salamci, H. Seçen, Y. Sütbeyaz and M. Balci, J. Org. Chem., 1997, 62, 2453 CrossRef CAS PubMed; (c) K. M. Davis and B. K. Carpenter, J. Org. Chem., 1996, 61, 4617 CrossRef CAS PubMed.
- (a) K. Ohkubo, T. Nanjo and S. Fukuzumi, Org. Lett., 2005, 7, 4265 CrossRef CAS PubMed; (b) W. Adam, C. R. Saha-Moeller and S. B. Schambony, J. Am. Chem. Soc., 1999, 121, 1834 CrossRef CAS; (c) K. A. Zaklika, B. Kaskar and A. P. Schaap, J. Am. Chem. Soc., 1980, 102, 386 CrossRef CAS.
- (a) M. Klaper and T. Linker, Chem.–Eur. J., 2015, 21, 8569 CrossRef CAS PubMed; (b) W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071 CrossRef CAS PubMed; (c) H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999 CrossRef CAS PubMed.
- (a) A. Eske, B. Goldfuss, A. G. Griesbeck, A. Kiff, M. Kleczka, M. Leven, J.-M. Neudorfl and M. Vollmer, J. Org. Chem., 2014, 79, 1818 CrossRef CAS PubMed; (b) W. Adam and M. J. Richter, J. Org. Chem., 1994, 59, 3335 CrossRef CAS; (c) L. M. Stephenson, Acc. Chem. Res., 1980, 13, 419 CrossRef CAS.
- W. Adam and H. Rebollo, Tetrahedron Lett., 1981, 22, 3049 CrossRef CAS.
- (a) K. Gollnick and A. Schnatterer, Tetrahedron Lett., 1985, 26, 5029 CrossRef CAS; (b) I. Erden and T. R. Martinez, Tetrahedron Lett., 1991, 32, 1859 CrossRef CAS; (c) R. K. Howe, J. Org. Chem., 1968, 33, 2848 CrossRef CAS; (d) P. Sharma and R. S. Liu, Org. Lett., 2016, 18, 412 CrossRef CAS PubMed.
- (a) J. Drujon, R. Rahmani, V. Heran, R. Blanc, Y. Carissan, B. Tuccio, L. Commeiras and J. Parrain, Phys. Chem. Chem. Phys., 2014, 16, 7513 RSC; (b) W. R. Roth, T. Ebbrecht and A. Beitat, Chem. Ber., 1988, 121, 1357 CrossRef CAS.
- (a) W. R. Roth, R. Longer, M. Bartmann, B. Stevermann, G. Maier, H. P. Reisenauer, R. Sustmann and W. Müller, Angew. Chem., Int. Ed., 1987, 26, 256 CrossRef; (b) W. R. Roth, B. P. Scholz, R. Breuckmann, K. Jelich and H. W. Lennartz, Chem. Ber., 1982, 115, 1934 CrossRef CAS.
- W. R. Roth and B. P. Scholz, Chem. Ber., 1982, 115, 1197 CrossRef CAS.
- (a) W. Adam, S. Grabowski and H. Platsch, J. Am. Chem. Soc., 1989, 111, 751 CrossRef CAS; (b) W. Adam, K. Hannemann and R. M. Wilson, J. Am. Chem. Soc., 1986, 108, 929 CrossRef CAS; (c) W. Adam, K. Hannemann and R. M. Wilson, J. Am. Chem. Soc., 1984, 106, 1646 CrossRef; (d) W. R. Roth, M. Biermann, G. Erker, K. Jelich, W. Gerhartz and H. Görner, Chem. Ber., 1980, 113, 586 CrossRef CAS.
- (a) H. Yamamoto and N. Momiyama, Chem. Commun., 2005, 3514 RSC; (b) W. Adam and O. Krebs, Chem. Rev., 2003, 103, 4131 CrossRef CAS PubMed; (c) P. Zuman and B. Shah, Chem. Rev., 1994, 94, 1621 CrossRef CAS; (d) K. Mikami and M. Shimizu, Chem. Rev., 1992, 92, 1021 CrossRef CAS; (e) A. G. Leach and K. N. Houk, Org. Biomol. Chem., 2003, 1, 1389 RSC; (f) A. G. Leach and K. N. Houk, J. Am. Chem. Soc., 2002, 124, 14820 CrossRef CAS PubMed; (g) D. J. Fisher, G. L. Burnett, R. Velasco and J. R. de Alaniz, J. Am. Chem. Soc., 2015, 137, 11614 CrossRef CAS PubMed.
- (a) G. Ieronimo, A. Mondelli, F. Tibiletti, A. Maspero, G. Palmisano, S. Galli, S. Tollari, N. Masciocchi, K. M. Nicholas, S. Tagliapietra, G. Cravotto and A. Penoni, Tetrahedron, 2013, 69, 10906 CrossRef CAS; (b) A. Penoni, G. Palmisano, Y. L. Zhao, K. N. Houk, J. Volkman and K. M. Nichols, J. Am. Chem. Soc., 2009, 131, 653 CrossRef CAS PubMed.
- (a) M. Kurano, K. Tsukamoto, M. Hara, R. Ohkawa, H. Ikeda and Y. Yatomi, J. Biol. Chem., 2015, 290, 2477 CrossRef CAS PubMed; (b) E. Ogier-Denis, A. Blais, J. J. Houri, T. Voisin, G. Trugnan and P. Codogno, J. Biol. Chem., 1994, 269, 4285 CAS; (c) O. N. Kostopoulou, E. C. Kouvela, G. E. Magoulas, T. Garnelis, I. Panagoulias, M. Rodi, G. Papadopoulos, A. Mouzaki, G. P. Dinos, D. Papaioanmou and D. L. Kalpaxis, Nucleic Acids Res., 2014, 42, 8621 CrossRef CAS PubMed; (d) E. N. Glaros, W. S. Kim, B. J. Wu, C. Suarna, C. M. Quinn, K.-A. Rye, R. Stocker, W. Jessup and B. Garner, Biochem. Pharmacol., 2007, 73, 1340 CrossRef CAS PubMed.
- Triple functionalizations of allenes are focused extensively on their double epoxidations,15c–e and other reactions are very few.15a,b See: (a) C. S. Adams, R. D. Grigg and J. M. Schomaker, Chem. Sci., 2014, 5, 3046 RSC; (b) W. Zhao and J. Montgomery, J. Am. Chem. Soc., 2016, 138, 9763 CrossRef CAS PubMed; (c) C. S. Adams, C. D. Weatherly, E. G. Burke and J. M. Schomaker, Chem. Soc. Rev., 2014, 43, 3136 RSC; (d) S. D. Lotesta, S. Kiren, R. R. Sauers and L. J. Williams, Angew. Chem., Int. Ed., 2007, 46, 7108 CrossRef CAS PubMed; (e) P. Ghosh, S. D. Lotesta and L. J. Williams, J. Am. Chem. Soc., 2007, 129, 2438–2439 CrossRef CAS PubMed.
- R. K. Howe, J. Org. Chem., 1968, 33, 2848 CrossRef CAS.
- Crystallographic data of 3b, 4b, 5b and 5c were deposited at the Cambridge Crystallographic Data Centre (3b CCDC 1507478, 4b CCDC 1507477, 5b CCDC 1510902, 5c CCDC 1540299).
- T. V. Robinson, D. S. Pedersen, D. K. Taylor and R. T. Tiekink, J. Org. Chem., 2009, 74, 5093 CrossRef CAS PubMed.
- The rearrangement of cyclopropylmethyl radicals to homoallylic radicals is seriously affected by the radical substituents; this process is reversible. Previous studies by Bowry indicate that stable cyclopropylbenzylic radicals are reluctant to form the corresponding homoallylic radicals. In our system, key intermediate A is a very stable phenylallylic radical that has many resonance forms. The equilibrium of this rearrangement is expected to be favourable for initial radicals A that can be trapped by O2 to yield the observed product 3m. For the nature of this radical rearrangement, see the leading reference, A. J. Beckwith and V. W. Bowry, J. Am. Chem. Soc., 1994, 116, 2710–2716 CrossRef CAS
. - S. Stoll and R. D. Britt, Phys. Chem. Chem. Phys., 2009, 11, 6614 RSC.
- (a) L. Jonkman, H. Muller and J. Kommandeur, J. Am. Chem. Soc., 1971, 93, 5833 CrossRef CAS; (b) V. Branchadell, J. Font, A. G. Moglioni, C. O. de Echagulen, A. Oliva, M. Rosa, R. M. Ortuno, J. Veciana and J. Vidal-Gancedo, J. Am. Chem. Soc., 1997, 119, 9992 CrossRef CAS; (c) P. Astolfi, P. Carloni, E. Damiani, L. Greci, M. Marini, C. Rizzoli and P. Stipa, Eur. J. Org. Chem., 2008, 3279 CrossRef CAS.
- (a) N. Kornblum and H. E. DeLaMare, J. Am. Chem. Soc., 1951, 73, 880 CrossRef CAS; (b) S. T. Staben, X. Linghu and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 12658 CrossRef CAS PubMed.
- (a) J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC; (b) J. E. Baldwin, J. Cutting, W. Dupont, L. Kruse, L. Silberman and R. C. Thomas, J. Chem. Soc., Chem. Commun., 1976, 736 RSC; (c) C. Chatgilialoglu, C. Ferreri, M. Guerra, V. Timokhin, G. Froudakis and T. Gimisis, J. Am. Chem. Soc., 2002, 124, 10765 CrossRef CAS PubMed.
- For metal-catalyzed oxidations of 5-hydroxy-1-enes with O2 to yield tetrahydrofuran products, see (a) S. Inoki and T. Mukayama, Chem. Lett., 1990, 67–70 CrossRef CAS; (b) C. Palmer, N. M. Morra, A. C. Stevens, B. Bajtos, B. P. Machin and B. L. Pagenkopf, Org. Lett., 2009, 11, 5614–5617 CrossRef CAS PubMed; (c) R. M. Trend, Y. K. Ramtohul, E. M. Ferreira and B. M. Stoltz, Angew. Chem., Int. Ed., 2003, 42, 2892 CrossRef CAS PubMed; (d) X. Xie and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3767 CrossRef CAS PubMed; (e) S. L. Zultanski, J. Zhao and S. S. Stahl, J. Am. Chem. Soc., 2016, 138, 6416 CrossRef CAS PubMed.

Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1507477, 1507478, 1510902 and 1540299. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01770g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |