
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface engineering of graphitic carbon nitride polymers with cocatalysts for photocatalytic overall water splitting
Guigang
Zhang
,
Zhi-An
Lan
and
Xinchen
Wang
 *
*
State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350002, China. E-mail: xcwang@fzu.edu.cn
First published on 6th June 2017
Abstract
Graphitic carbon nitride based polymers, being metal-free, accessible, environmentally benign and sustainable, have been widely investigated for artificial photosynthesis in recent years for the photocatalytic splitting of water to produce hydrogen fuel. However, the photocatalytic stoichiometric splitting of pure water into H2 and O2 with a molecular ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 is far from easy, and is usually hindered by the huge activation energy barrier and sluggish surface redox reaction kinetics. Herein, we provide a concise overview of cocatalyst modified graphitic carbon nitride based photocatalysts, with our main focus on the modulation of the water splitting redox reaction kinetics. We believe that a timely and concise review on this promising but challenging research topic will certainly be beneficial for general readers and researchers in order to better understand the property–activity relationship towards overall water splitting, which could also trigger the development of new organic architectures for photocatalytic overall water splitting through the rational control of surface chemistry.
:1 is far from easy, and is usually hindered by the huge activation energy barrier and sluggish surface redox reaction kinetics. Herein, we provide a concise overview of cocatalyst modified graphitic carbon nitride based photocatalysts, with our main focus on the modulation of the water splitting redox reaction kinetics. We believe that a timely and concise review on this promising but challenging research topic will certainly be beneficial for general readers and researchers in order to better understand the property–activity relationship towards overall water splitting, which could also trigger the development of new organic architectures for photocatalytic overall water splitting through the rational control of surface chemistry.
1. Introduction
Photocatalytic overall water splitting using nanoparticulate semiconductors is regarded as a potentially scalable and economically feasible method to convert the cost-free and earth-abundant solar energy into clean and renewable hydrogen fuel.1–6 Substantial research efforts have mainly focused on improving the photocatalytic activities of the semiconductors, and mostly focus on modification of the composition, structure, texture and morphology of the materials.7–15 However, until now, most of the materials that have been reported for water splitting reactions have been based on inorganic semiconductors,16,17 which are usually composed of rare elements and are not suitable for large-scale sustainable development. It is therefore reasonable to explore the use of earth-abundant materials, namely materials that are accessible, cost-affordable, and environmentally benign, and their textural and optical properties which can also be easily tailored to achieve photocatalytic water splitting with high performance.In very recent years, organic conjugated polymers have attracted particular research interest since melon based carbon nitride polymers and crystalline graphitic carbon nitrides (both traditionally named as g-C3N4 for simplicity), which have been reported to be promising visible light photocatalysts, are accessible to many different researchers. They can be prepared in large amounts and can be used in studies on reactor and process design.18–27 Compared with traditional inorganic semiconductors, conjugated polymers possess more advantages, including being metal-free, nontoxic and low-cost, and the fact that their composition, structure and properties can also be readily tuned by adjusting the building blocks of versatile organic protocols. Furthermore, the organic texture of carbon nitride enables it to exhibit some unique properties, such as an abundance of nitrogen lone-pair electrons and grain boundary defects, thus making it very suitable for the construction of metal/carbon nitride heterojunctions for fast charge transfer at the interface. The first publication on carbon nitride photocatalysis has been cited >3000 times since 2009, as reported by Google Scholar. This has motivated extensive research into artificial photosynthesis using polymers, and a series of conjugated polymers have been investigated as excellent organic semiconductors for photocatalytic H2 production from water.18–27 In the presence of cocatalysts (e.g. Pt or Pd) and electron donors (e.g. triethanolamine or methanol), some of the conjugated polymers could achieve sufficient activities for visible light H2 production. Until now, the apparent quantum yield (AQY) of conjugated polymers for H2 evolution under visible light irradiation (λ > 420 nm) that could be obtained was as high as 38.8%.28 However, only a few of those investigated conjugated polymers have exhibited activity towards water oxidation, due to being restrained by their weak stability and insufficient oxidation ability of the valence holes. Therefore, it is observed that only a limited number of conjugated polymers can be utilized for overall water splitting.
By virtue of their well configured band structures (CB: −1.3 V; VB: 1.4 V vs. NHE, pH = 6.8) and robust stability, both of which are normally regarded as the two prerequisites for water splitting, melon-based g-C3N4 polymers have demonstrated promising capability for photocatalytic overall water splitting. In order to improve their photocatalytic activities, strategies such as doping,29–34 copolymerization,35–39 nanostructure engineering,40–45 hybridization46–50 and sol–gel51 modification have been developed to modify the composition, structure and optical properties of g-C3N4 polymers. It should be noted that pure g-C3N4 only absorbs visible light below ca. 470 nm; expansion of the optical absorption threshold through the use of strategies such as doping or copolymerization is therefore regarded as a feasible way to improve the photocatalytic efficiency from the viewpoint of enhanced energy input. However, in most cases, pure g-C3N4 without cocatalysts cannot exhibit sufficient photoactivity due to the huge activation energy and sluggish surface reaction kinetics. Thus, suitable H2 and O2 evolution cocatalysts, i.e. Pt,52–55 Rh,56 MoS2,57–60 and Co3O4,61–63 are indispensable to separately promote the H2 and O2 evolution reaction rate. On the one hand, cocatalysts could serve as charge carrier trap centres in order to quickly extract electrons or holes from the bulks of semiconductors to the interfaces of the cocatalysts. Because a Schottky barrier can be formed at the metal/g-C3N4 interface, noble metals with larger work functions, that is, a lower Fermi level, should trap electrons more readily. Thus Pt, among the many noble metals, with the largest work function, is the best cocatalyst for trapping electrons. Consequently, most of the light excited charge carriers would immediately participate in the subsequent surface redox reaction without recombination. On the other hand, a volcano relationship between the exchange current for H2 evolution and the M–H strength (M is a transition metal) was observed. Among the investigated transition metals, Pt was at the peak of the volcano, and showed the lowest activation energy for hydrogen evolution. It is thus desirable to imagine that the cocatalyst plays key roles in controlling the surface redox reaction kinetics, which are normally confirmed to represent the rate determining step of the overall water splitting process. Actually, the physicochemical properties of the cocatalysts, including their electron trapping ability, exchange current for H2 evolution, structure and morphology, are closely related to their photocatalytic activity.64–67 It is therefore of pivotal importance to investigate and conclude the property–activity relationships of H2 and O2 evolution cocatalysts.
Herein, we review the recent developments of g-C3N4 based polymers for the water splitting reaction, which mainly focus on cocatalyst modifications in order to tune the surface reaction kinetics and thus optimize the photocatalytic activities. We believe that a timely and systematic review into this very interesting and hot research topic will certainly be beneficial for general researchers to gather more experimental and theoretical details for a better understanding of the water splitting reaction mechanism. This will also open up new avenues to guide the development of new artificial photosynthesis systems by systematically configuring and engineering other promising organic architectures from the viewpoint of surface reaction kinetics modulation.
2. Fundamentals of water splitting
Photocatalytic overall water splitting for the stoichiometric generation of H2 and O2 in a molecular ratio of 2:1 is a thermodynamically uphill reaction that requires additional energy input, usually solar energy, to drive this multiple-electron-proton transfer and hugely energy consuming process. In principle, semiconductors that are theoretically capable of catalyzing the overall water splitting reaction are restricted:68,69 (i) the band gap energy should at least be larger than 1.23 eV (to meet the thermodynamic barrier, ideally the band gap should be 2.0 eV after considering the kinetic barriers);70 (ii) the conduction (valence) band potentials should be sufficiently negative (positive) for the water reduction (oxidation) half reactions; (iii) semiconductors should be robust enough towards light and solution corrosion.71 Except for these strictly defined requirements, polymers should also be well designed to capture sufficient visible photons for the excitation and generation of charge carriers (electrons and holes), which are involved in the subsequent water splitting reaction.6 In addition, conjugated polymers should exhibit excellent structural and textural properties, which are required for the fast separation and transfer of light-induced charge carriers to the interfaces of the photocatalysts without recombination. Therefore, it is certainly a great challenge to achieve overall water splitting through the rational construction of conjugated polymers.
As shown in Fig. 1, photocatalytic overall water splitting by a nanoparticulate photocatalyst generally involves three major steps: (i) semiconductors absorb incident light at an energy larger than the band gap for the excitation and generation of charge carriers; (ii) charge carrier separation, migration and transfer to the interfaces of semiconductors without recombination; (iii) electrons and holes separately react with the adsorbed protons and water for H2 and O2 evolution at the interface. It should be noted that the first two steps are closely related to the thermodynamic properties, such as optical properties, specific surface area, texture and microstructure of the semiconductors.68 Over the past few decades, substantial efforts have been devoted to developing promising semiconductors with enhanced optical absorption efficiency and optimized charge carrier behaviour.52,55 For instance, a variety of well-designed chemical strategies have been demonstrated to enhance the optical absorption, enlarge the surface area, and optimize the electronic and texture properties, thus improving the water splitting activities.63 However, the reaction rate of the third step is confirmed to be much slower than that of the first two steps, which normally involve the use of cocatalysts to decrease the reaction activation energy and accelerate the H–O bond breaking and O–O bond formation. Evidently, the last step is the major step for kinetic control of the overall water splitting efficiency. It is thus of pivotal importance to develop suitable cocatalysts to improve the water redox reaction efficiency and reinforce the stability of the semiconductors.
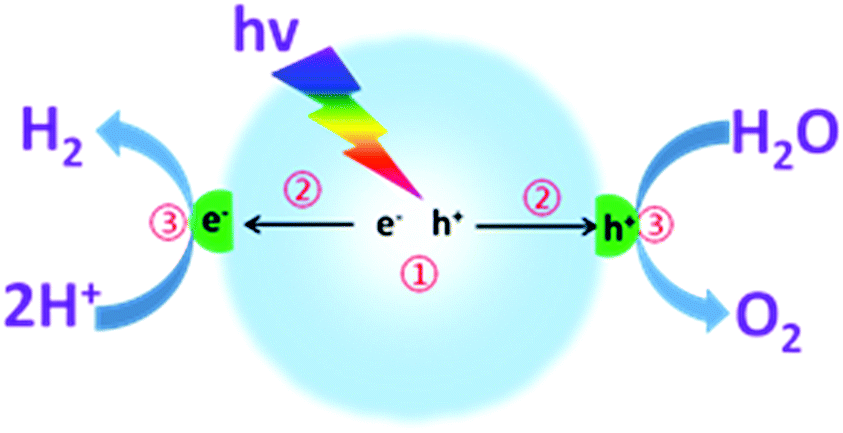 | ||
| Fig. 1 Schematic illustration of photocatalytic overall water splitting over a semiconductor photocatalyst modified with both H2- and O2-evolution cocatalysts. | ||
In principle, as is shown in Fig. 2, the Gibbs free energy change for the decomposition of 1 mol H2O to 1 mol H2 and 1/2 mol O2 under standard conditions is 237 kJ (corresponding to 1.23 eV). This reaction, despite only generating two singular molecules, is a thermodynamically uphill reaction that is non-spontaneous and usually calls for huge additional energy input (e.g. solar energy) to drive it.3–5 Furthermore, besides the additional energy input required, a huge energy barrier, i.e. the activation energy, usually hinders the water splitting reaction. The deposition of cocatalysts has been reported to promote photocatalytic activity.64,65 This is because, under light irradiation, the cocatalysts not only function as kinetic promoters to catalyse the evolution rate of the gases, but also serve as charge trap centres to extract electrons and holes from the photo-excited semiconductors. Evidently, the overall water splitting activity depends on the catalytic performance of the cocatalysts. In addition, in comparison with the water reduction half reaction, the water oxidation half reaction, which involves the transfer of four-electrons accompanied by O–H bond breaking and O–O bond formation, is usually restrained by the huge activation energy (∼700 mV) and sluggish O–O bond formation kinetics.69 Thus, the water oxidation process is usually more restrained in comparison with the water reduction half reaction and is thus regarded as the key step to achieving an efficient overall water splitting reaction.
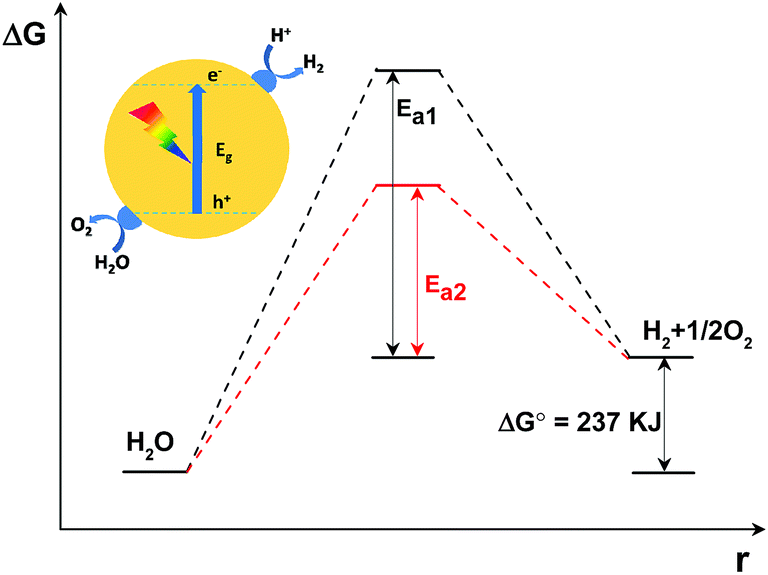 | ||
| Fig. 2 Schematic description of the energy diagram of a semiconductor modified with both H2 and O2 evolution cocatalysts under light irradiation for non-sacrificial photocatalytic water splitting. Ea1: activation energy without cocatalysts; Ea2: activation energy with cocatalysts; ΔG: Gibbs free energy change; ΔG0: Gibbs free energy change under standard conditions; r: redox reaction process. | ||
In order to achieve overall water splitting, it is advisable to individually investigate the water reduction and water oxidation half reactions at first. After careful examination of the property–activity relationships of the cocatalysts in sacrificial H2 or O2 evolution reactions, it is thus reasonable to carry out an overall water splitting investigation. It should be noted that the factors dominating the quantum efficiency should not be identical for non-sacrificial water splitting accompanied by a large increase in the Gibbs energy and sacrificial hydrogen or oxygen evolution reactions. For instance, the loading technology for cocatalysts may make different contributions to the overall water splitting and sacrificial hydrogen or oxygen evolution reactions. Furthermore, the reverse reaction is always considered to be a critical issue in an overall water splitting system, while it is not taken into consideration in sacrificial H2 or O2 evolution systems. Nevertheless, we are able to learn of some key factors and useful experience from the sacrificial half reactions. Such knowledge is very helpful for investigations into overall water splitting. Following this line of inquiry, in this perspective we firstly investigate the water reduction and water oxidation reactions, mainly focusing on the control of the surface kinetics of g-C3N4 polymers. After that, we demonstrate how to achieve overall water splitting by careful surface modification of g-C3N4 polymers with well-designed H2 and O2 evolution promoters.
3. Water reduction half reaction
To investigate the water reduction half reaction, electron donors (such as TEOA, methanol, ethanol, etc.) are usually added into the system to accelerate the hole oxidation process (Fig. 3).52,55,72 In most cases, the semiconductors themselves can only act as light transducers to absorb incident photons for the excitation and generation of charge carriers. Most of them require an additional catalyst, namely a cocatalyst, to accelerate the surface reaction kinetics and promote photocatalytic activity.64,65 In principle, cocatalysts could extract electrons from the interfaces of semiconductors, thus prolonging the charge carrier lifetime and restraining charge carrier recombination, both of which are largely beneficial for promoting the photocatalytic activity. Furthermore, noble metals, owing to their large work functions for trapping electrons and lowest activation energy for H2 evolution, are promising candidates for the catalysis of photocatalytic water splitting.64 Aside from their good catalytic properties, the cocatalysts could also suppress photo-corrosion and increase the stability of the semiconductor photocatalysts. | ||
| Fig. 3 Schematic illustration of photocatalytic water reduction for H2 evolution in the presence of an electron donor driven by a semiconductor modified with H2 evolution cocatalysts. TEOA: triethanolamine. | ||
Many factors could affect the capability of H2 evolution cocatalysts to promote the photocatalytic water reduction reaction, such as the composition, loading contents, particle size, morphology and structure of the cocatalysts.50–54 Traditional cocatalysts that are suitable for promoting H2 evolution activity are mainly based on noble metals such as Pt,50–54 Pd,73 Rh,56 Au74 and Ag.75,76 Noble metal cocatalysts usually exhibit very low overpotentials, even being close to zero for the H2 evolution reaction, and thus demonstrate excellent performance for increasing the H2 evolution activity. However, their high cost, toxicity and scarcity restrict their applications when scaling up. Hence, it is urgent to develop alternative cocatalysts that use earth-abundant elements for sustainable solar energy conversion. Recently, some transition metals (NiO,77 MoS2,57–60 WS2,78,79 Ni(OH)280–82 and CoP83) have also demonstrated an excellent ability to promote photocatalytic H2 evolution activity. Compared with noble metals, transition metals possess more advantages, such as their abundance, low-cost and low toxicity, thus making them suitable candidates for sustainable applications. Furthermore, other types of cocatalysts, such as graphene and carbon quantum dots,48,84 can also serve as versatile cocatalysts to promote H2 evolution activity. In this section, we will discuss the effects of different cocatalysts on the promotion of H2 evolution activity, with the aim to better understand their property–activity relationships.
3.1 Noble metal cocatalysts
As was discussed above, many different noble metals have been investigated as effective cocatalysts to promote photocatalytic H2 evolution activity. However, Pt proves to be the most effective cocatalyst for H2 evolution due to its largest work function for trapping electrons and its lowest activation energy for H2 evolution. For instance, when a Ru nanoparticle was deposited on g-C3N4 polymers as a H2 evolution cocatalyst, the visible light H2 evolution rate that was achieved was 2.1 μmol h−1.56 On the contrary, when the same amount of Pt cocatalyst was loaded onto the surface of pure g-C3N4, the visible light H2 evolution rate was increased to as high as 7.3 μmol h−1, which was 3.5 times higher than that of the Ru cocatalyst, thus indicating the great advantage of Pt in promoting H2 evolution activity.56 Furthermore, the loading contents of the H2 evolution cocatalysts could also largely affect the photocatalytic activity (Fig. 4). At lower loading contents, the activity increases when adding higher amounts of cocatalyst, which is mainly due to there being more active sites available for redox reactions. However, when excessive amounts of cocatalyst were deposited on the surfaces of the semiconductors, fewer surface active sites were exposed to absorb the reactive species. Furthermore, excessive cocatalyst amounts on the surfaces of semiconductors will shield the incident light and prevent light absorption. In addition, excessive cocatalyst amounts would stimulate particle aggregation which is certainly bad for the activity of the cocatalyst. Finally, high loading amounts of cocatalyst may also function as the charge carrier recombination centre and thus shorten the lifetime of the electron–hole pairs.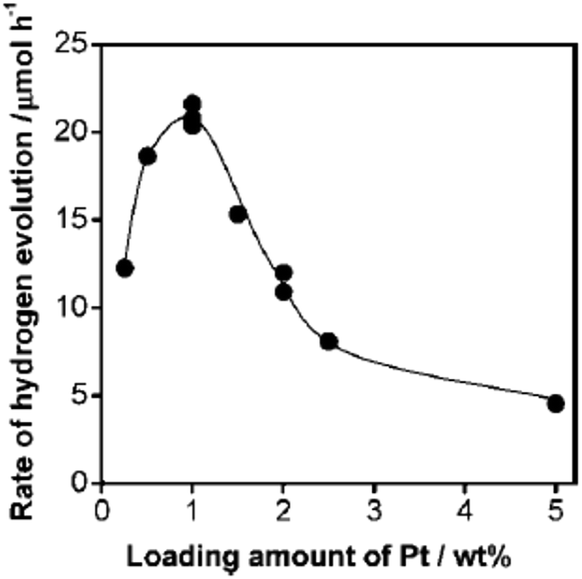 | ||
| Fig. 4 Dependence of the H2 evolution rate, when using Pt-loaded g-C3N4 under visible light, on the loading amount of Pt. Reprinted with permission from ref. 56. Copyright 2009, American Chemical Society. | ||
In general, noble metals are usually prepared by the reasonable reduction of metal precursors (e.g., H2PtCl6) from a liquid solution.29 Depending on the reduction methodology used, the as-prepared cocatalysts always exhibit quite different abilities for the promotion of the photocatalytic H2 evolution reaction. Up until now, the most popular method for obtaining noble metal cocatalysts has been through in situ photo-deposition from aqueous noble metal precursors, due to the facile fabrication process and highly efficient activities. For instance, when a certain amount of H2PtCl6 aqueous solution was added into the photocatalytic reaction system, homogeneous Pt species with particle sizes of about 3–5 nm were deposited on the surface of carbon nitride.56 In this case, the semiconductor absorbs light to generate excited electrons and holes. The adsorbed Pt6+ is reduced to metallic Pt by light excited electrons and is subsequently deposited in situ on the surface of carbon nitride. These metallic Pt particles could act as efficient cocatalysts to extract electrons from the bulk and reduce the H2 evolution over-potential. Compared to carbon nitride without modification by a Pt cocatalyst, the as-prepared Pt–g-C3N4 samples exhibited greatly enhanced activity, increased by a factor of 7, towards H2 evolution, thus reflecting the fact that Pt is an excellent cocatalyst for promoting H2 evolution activity.56
Another popular method for noble metal cocatalyst deposition consists of impregnation followed by subsequent reduction under hot H2 flow. The particle size of metallic Pt prepared by this H2 reduction is also ultrafine, with the particles homogeneously deposited on the surface of g-C3N4 polymers, which is a similar outcome to that prepared by in situ photo-reduction (Fig. 5a and b), thus enabling it to be an active cocatalyst for the promotion of H2 evolution. Furthermore, the heating treatment during H2 reduction creates tight contact between the Pt nanoparticles and the carbon nitride polymers, which obviously favours the interface charge carrier transfer and improves the H2 evolution activity. As is shown in Fig. 5c, the Pt nanoparticles prepared by H2 reduction exhibited much higher H2 evolution activity than that of the Pt nanoparticles obtained by in situ photoreduction. Furthermore, the results also demonstrate good stability over the long reaction time, thus demonstrating it is promising for the promotion of photocatalytic H2 production activity. However, the photoreduction strategy was always used in previous reports for examination of the activities of the g-C3N4 polymers due to the facile but effective operation process. Until now, by modifying the properties and surface reaction kinetics, the highest H2 evolution apparent quantum yield (AQY) of the g-C3N4 based polymers that can be achieved is as high as 50.7% (ref. 28) at 405 nm and 38.8% (ref. 28) at 420 nm, while Pt nanoparticles are deposited in situ as H2 evolution cocatalysts in the presence of TEOA as the electron donor.
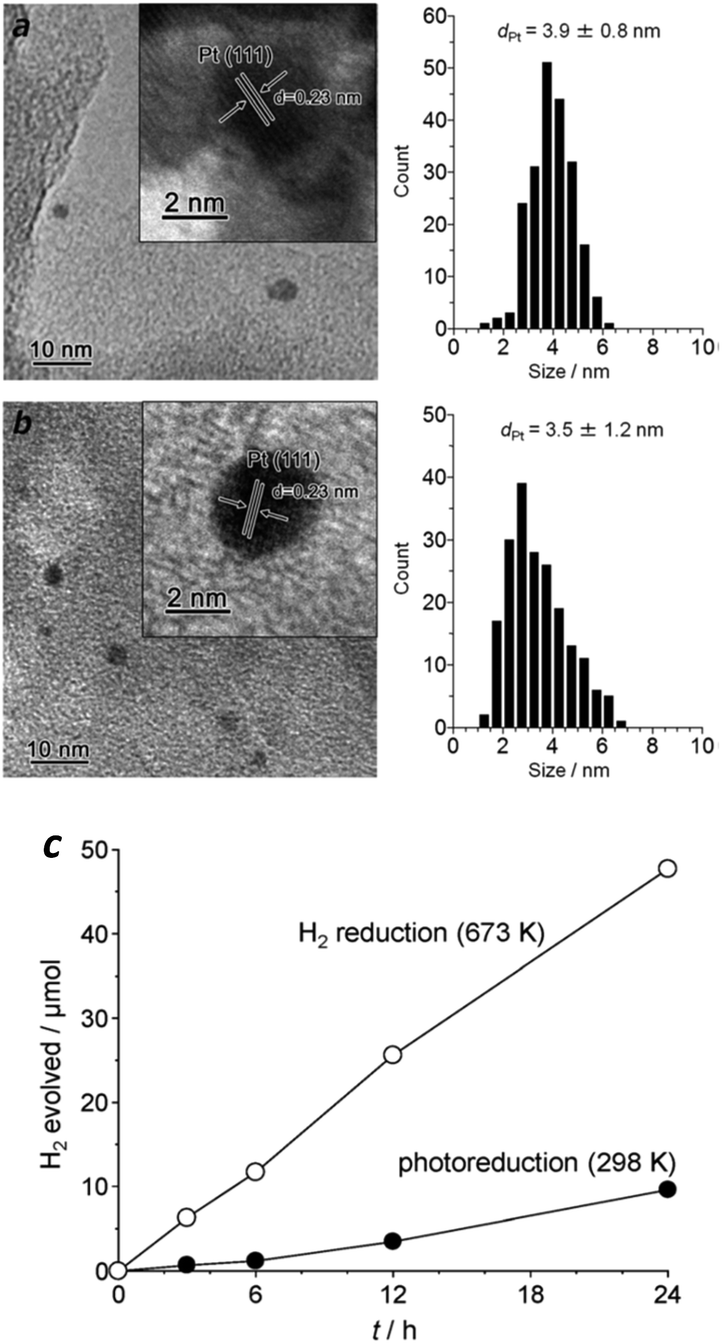 | ||
| Fig. 5 Size distribution of Pt particles prepared by (a) photoreduction (298 K) and (b) H2 reduction (673 K); (c) H2 evolution activities in the presence of an electron donor. Reprinted with permission from ref. 86. Copyright 2014, Royal Society of Chemistry. | ||
Noble metals do however usually have some limitations, such as high cost, toxicity and scarcity, which largely restrict their applications when scaling up. Therefore, further investigations are desired to develop new materials that use sustainable, low-cost and environmentally benign components to promote the H2 evolution activity of g-C3N4 polymers.
3.2 Transition metal based cocatalysts
Transition metals (including Fe3+, Co2+ and Ni2+) always demonstrate promising properties for heterogeneous catalysis due to their versatile chemical states and good stability towards solution corrosion. These transition metals can also act as kinetic promoters to improve the catalytic activity. Recently, some transition metal compounds, such as NiO,77 MoS2,57–60 Ni(OH)2,80–82 CoP,83 NiSx (ref. 87 and 88) and WS2,78,79 have been reported to exhibit excellent activities for the photocatalytic H2 evolution reaction. Depending on the composition, morphology and properties of the metal composites, they can also exhibit comparable activities to those of noble metals, thus demonstrating that they are promising candidates for a wide range of applications. In addition, compared with traditional noble metals, these transition metal complexes possess many advantages, such as low-cost, low-toxicity, abundant resources and a variety of properties. In this section, we will discuss the positive roles of different transition metals in the promotion of the photocatalytic H2 evolution reaction, with the major purpose of developing efficient and sustainable H2 evolution cocatalysts. | ||
| Fig. 6 (a) The rate of H2 production over mpg-C3N4 loaded with different amounts of MoS2 or Pt, (b) TEM image of MoS2/g-C3N4 and (c) illustration of the deposition of layered MoS2 on the surface of g-C3N4 for photocatalytic H2 evolution. Reprinted with permission from ref. 57. Copyright 2013, Wiley-CVH. | ||
3.2.2 Transition metal oxides or hydroxide based cocatalysts
Compared to metal sulfides, metal oxides exhibit much better stability towards solution corrosion. For instance, when a Ni@NiO core@shell nanostructure was constructed on the surface of g-C3N4 polymers, it demonstrated robust activity over a long reaction time.77 The NiO shell could act as a protective layer to prevent the size accumulation of the Ni core. Furthermore, only protons could penetrate the NiO shell and adsorb on the surface of the Ni core for H2 evolution. Other large sized species, such as O2, could not penetrate the shell and thus the backward reaction is avoided. Evidently, this could largely decrease the H2 evolution activation energy and promote the H2 evolution activity.Other than metal oxides, layered hydroxides, such as Ni(OH)2,80–82 are also excellent H2 evolution cocatalysts. For instance, Ni(OH)2 can function as a H2 evolution cocatalyst to combine with g-C3N4 for photocatalytic H2 production in the presence of an electron donor.80 As is shown in Fig. 7a, when Ni(OH)2 was deposited on g-C3N4 the binary composite demonstrated clearly enhanced H2 evolution activities in comparison with pure g-C3N4 without any cocatalyst. An adaptive interface developed between g-C3N4 and Ni(OH)2; this could promote interface charge carrier transfer and decrease recombination. Evidently, this contributes to promoting H2 evolution activity. Furthermore, the H2 evolution activities of the binary catalysts are closely related to the Ni(OH)2 loading amounts. Optimum activity was obtained when the loading percentage of Ni(OH)2 was 0.5 wt%. Meanwhile, the catalyst demonstrated comparable activities to that of the noble-metal Pt modified one. Furthermore, these Ni(OH)2 modified polymers also exhibit robust stability towards light and solution corrosion, which is necessary for practical applications. It is thus advisable to develop a noble-metal free system using all earth abundant elements for sustainable H2 production from water. Further improvements in the photocatalytic activity could potentially be obtained by reasonable adjustments of the composition, structure, morphology and properties of the transition-metal cocatalysts.
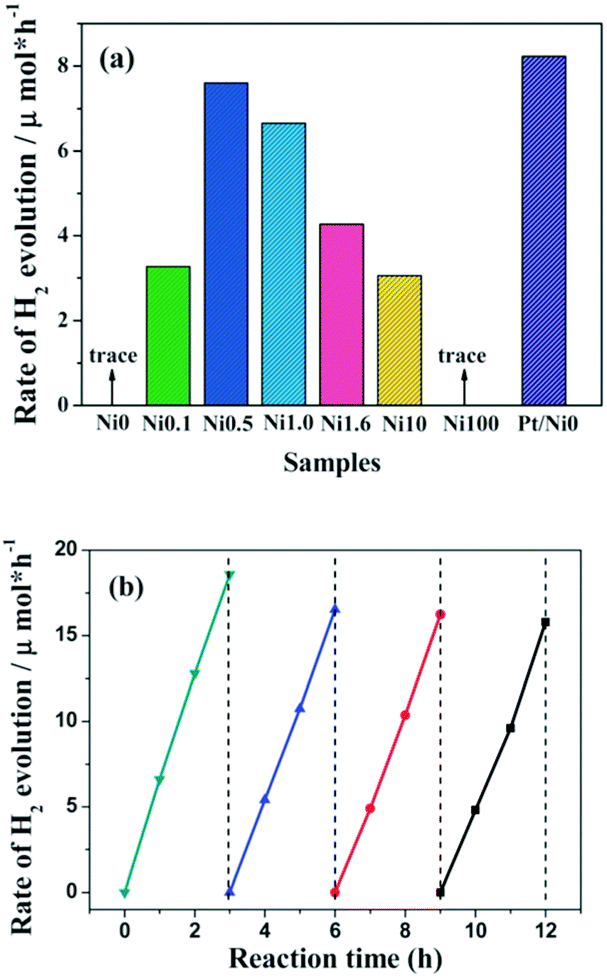 | ||
| Fig. 7 (a) Comparison of the photocatalytic activity of samples with different amounts of Ni(OH)2 loaded on g-C3N4 polymers, (b) cyclic H2-evolution curve for the Ni0.5 sample. Reprinted with permission from ref. 80. Copyright 2013, Royal Society of Chemistry. | ||
3.2.3 Artificial molecular based cocatalysts
Recently, some metal (Co, Ni and Fe) molecular systems have been developed which mimic the active sites of natural hydrogenases in plants, with high catalytic efficiencies for the reversible reduction of protons to molecular hydrogen.92,93 Being H2 active catalysts, hydrogenases possess unique properties, such as low activation energies for H2 evolution and a wide range of O2 sensitivities, while using organometallic catalytic sites composed of earth-abundant elements (e.g. Fe, Ni, S, C, N and O). However, one of the general problems of the currently used molecular systems is their instability upon long-term irradiation due to the existence of solution sensitive chemical bonds, i.e. the S–S bond. To overcome this issue, semiconductors with suitable band structures and robust stabilities are desired, both of which are believed to improve their efficiency and recyclability over long reaction times.As a prototypical example, Sun et al. have developed a series of Co-, Ni- and Fe-based molecular systems to combine with g-C3N4 for photocatalytic H2 evolution.94 The molecular structure of the cocatalysts and their H2 evolution activities are shown in Fig. 8a and b. It can be seen in the figure that the Ni-based system demonstrates the best activity in comparison with the activities of the Co- and Fe-based catalysts for visible light H2 evolution, while pure g-C3N4 without any cocatalyst only generates trace amounts of H2. After adding 4 wt% of acriflavine as a photosensitizer to enhance the visible light absorption, the H2 evolution rate further increased and the amount of the evolved H2 gas reached 72 μmol over 8 h of irradiation, which corresponds to a TON of 106 based on Ni. This molecular catalytic system has been proven to be active towards photocatalytic H2 evolution over more than 60 h in aqueous solution, which is much longer than that of the previously reported molecular catalytic system with organic dyes or metal-containing complexes. Another water-soluble and functional synthetic hydrogenase, [NiII(PPh2{NPhCH2P(O)(OH)2}2)2]Br2 (labelled as NiP for simplification), has been developed for use as a H2 evolution cocatalyst to combine with g-C3N4 for H2 production.95 The entirely synthetic g-C3N4–NiP system displays an unprecedentedly high TOF (109 h−1) and TON (166) for a hybrid system made of a molecular cocatalyst with g-C3N4 in aqueous solution. This synthetic g-C3N4–NiP system was active for 3 h of light irradiation. It is very interesting to observe that this system contains all sustainable elements (C, N, P and Ni) and no noble metals and organic solutions are used as solvents or electron donors. Obviously, this represents a green strategy for sustainable H2 production from the abundant resource of water.
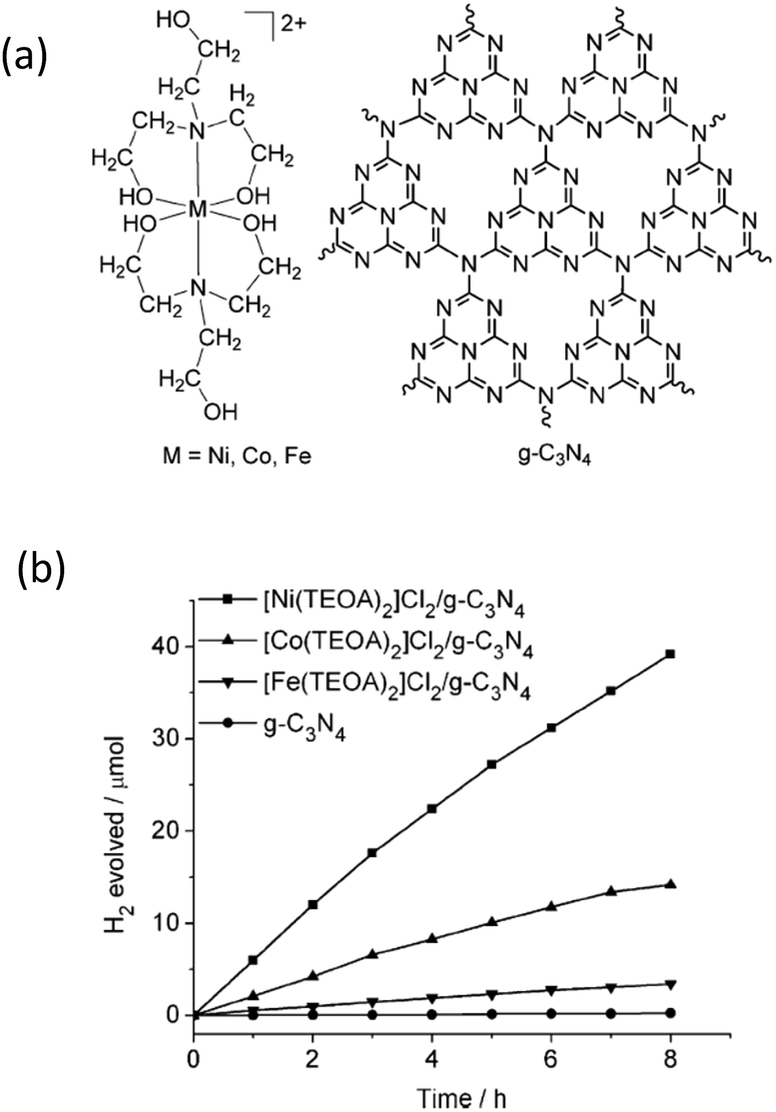 | ||
| Fig. 8 (a) Structures of Ni-, Co-, and Fe-based cocatalysts and g-C3N4, (b) photocatalytic H2-evolution for different cocatalyst modified g-C3N4 samples. Reprinted with permission from ref. 94. Copyright 2012, Wiley-CVH. | ||
However, up to now, most of the current examined molecular systems have only demonstrated limited lifetimes for reactions with long times, and are therefore not suitable for practical applications. Furthermore, the H2 production activities are still at a relatively low level. This main issue can be attributed to the poor interface charge carrier transfer between the water soluble molecular catalytic system and the insoluble solid state light transducers. Further investigations are required to facilitate the interface charge carrier transfer and prolong the lifetime of the molecular catalytic system in order to improve the photocatalytic H2 evolution activity.
3.3 Carbon based composite cocatalysts
Carbon-based materials, such as 0D carbon dots,84 1D carbon nanotubes (CNTs),85 and 2D nanosheets,48 always exhibit excellent electronic properties, thus making them promising candidates for electronic applications. Recently, these carbon-based materials have also found wide applications in the field of photocatalysis.48,85,96 This is because carbon-based materials can exhibit comparable conductivity to that of conductors, thus enabling them to act as an electron transport “highway” in order to accelerate charge carrier transfer and prolong the charge carrier lifetime. Evidently, these play determining roles in promoting the photocatalytic H2 evolution activity. Furthermore, carbon-based materials themselves can serve as H2 evolution cocatalysts or couple with other H2 evolution cocatalysts to further improve the photocatalytic H2 production activity. For instance, 2D graphene nanosheets incorporated with g-C3N4 polymers can greatly decrease the charge carrier recombination rate.48 Thus, more excited electrons are provided at the surfaces of the H2 evolution cocatalysts, which can be used for the proton reduction reaction. The optimum activity for photocatalytic H2 evolution was achieved when the deposition amount of graphene was determined as 1 wt%, and exceeded that of pure g-C3N4 by more than 3.07 times (Fig. 9a). The incorporated graphene nanosheet could quickly transfer excited electrons from the light transducer to the surface of the cocatalyst. In this case, the charge carrier recombination process is greatly decreased, which is certainly beneficial for the subsequent water redox reactions.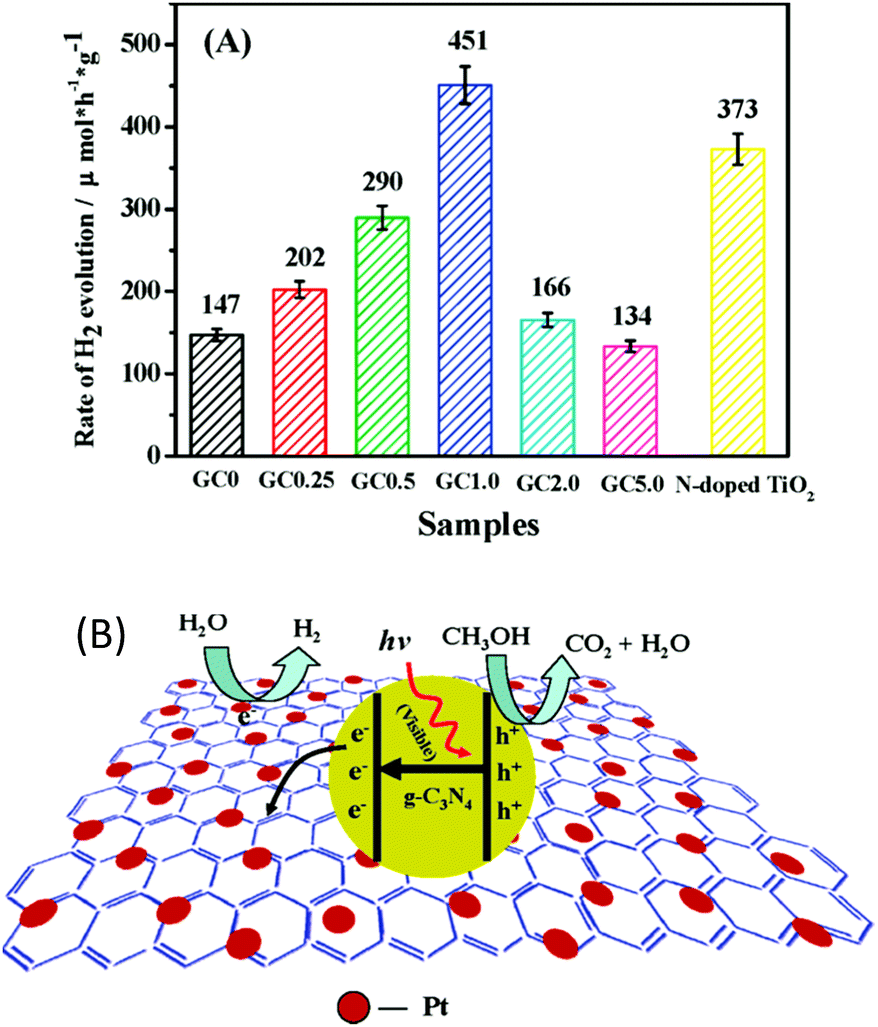 | ||
| Fig. 9 (A) Comparison of the photocatalytic activities of samples with different loading amounts of graphene on g-C3N4 polymers, (B) proposed mechanism for the enhanced electron transfer process of the graphene/g-C3N4 polymers. Reprinted with permission from ref. 48. Copyright 2011, American Chemical Society. | ||
Very recently, Qu et al. developed a metal-free 3D graphitic carbon nitride/nitrogen-rich carbon nanofiber composite photocatalyst by in situ freeze drying fabrication.85 The open 3D system enlarges the specific surface area and provides more active cites for the surface H2 evolution reaction. On the one hand, the nitrogen-rich carbon nanofibers could function as charge carrier transfer mediators to accelerate the charge carrier mobility. On the other hand, they could also act as a H2 evolution cocatalyst to reduce the H2 evolution overpotential and facilitate the reduction kinetics of surface protons. When the composite photocatalyst was tested for photocatalytic H2 evolution in the presence of TEOA as the electron donor, the visible light H2 evolution activity of the 3D nanocomposites reached as high as 168.85 μ mol h−1, which was 18.3 times higher than that of the pure g-C3N4 polymer. It should be noted that no other noble metal materials are deposited as H2 evolution cocatalysts, thus revealing the positive role of the carbon nanofibers in the promotion of the H2 evolution activity. This increased activity mainly arises from the synergistic effect of the two composites, which results in evidently improved charge carrier transfer, an enlarged surface area, a decreased energy barrier and enhanced visible light absorption. It should be noted that the H2 production rate of the g-C3N4@C binary photocatalysts is 3.6 times higher than that of Pt/g-C3N4 (46.65 μ mol h−1), thus indicating the potential of noble-metal free carbon nanofibers for promoting H2 evolution. A remarkable AQY of 14.3% at 420 nm for the g-C3N4@C sample was achieved, which is much higher than that of pure g-C3N4 polymer photocatalysts, even in the presence of Pt-based cocatalysts.
Based on the above discussions, we can conclude that loading a small amount of appropriate cocatalyst on the surfaces of light excited photocatalysts could largely decrease the surface activation energy and improve the surface redox reaction rate. Therefore, it could largely promote the photocatalytic water reduction activity. However, the composition, morphology, properties and loading technique could largely affect the photocatalytic activities. In order to better compare the roles of different H2 evolution cocatalysts, we then analysed the photocatalytic performances of the cocatalysts. As is shown in Table 1, it is clear to see that different cocatalysts always exhibit very different photocatalytic performances. Noble metals, transition metals and even carbon-based materials could all be developed for use as H2 evolution cocatalysts. In most cases, Pt was utilized to evaluate the photocatalytic H2 evolution activities due to the better activity and facile fabrication process. Until now, the optimum photocatalytic H2 evolution activity (38.8%) was obtained when Pt was deposited on the melon-based g-C3N4 under visible light irradiation.28 Thus, for a theoretical study, it is reasonable to deposit Pt nanoparticles as the H2 evolution cocatalysts for the overall water splitting study.
| Entry | Sample | Cocatalyst | Electron donor | λ [nm] | AQY [%] | Ref. |
|---|---|---|---|---|---|---|
| 1 | g-C3N4 | Ni(OH)2 | TEOA | 420 | 1.1 | 80 |
| 2 | g-C3N4 | β-Ni(OH)2 | TEOA | 402 | 1.48 | 81 |
| 3 | g-C3N4 | NiS | TEOA | 440 | 1.9 | 88 |
| 4 | mpg-C3N4 | MoS2 | Lactic acid | 420 | 2.1 | 57 |
| 5 | EB-g-C3N4 | MoSx | TEOA | 545 | 8.3 | 60 |
| 6 | g-C3N4 | Graphene + Pt | Methanol | 400 | 2.6 | 48 |
| 7 | g-C3N4 | C nanofibers | TEOA | 420 | 14.3 | 85 |
| 8 | CNU-ATCN | Pt | TEOA | 420 | 8.8 | 37 |
| 9 | NS-g-C3N4 | Pt | TEOA | 420 | 9.6 | 45 |
| 10 | CN-NS | Pt | TEOA | 420 | 26.1 | 21 |
| 11 | g-CN-1 | Pt | TEOA | 405 | 50.7 | 28 |
| 420 | 38.8 | 28 | ||||
| 11 | g-CN-1 | Pt | TEOA | 420 | 38.8 |
4. Water oxidation half reaction
Water oxidation is a multiple complex reaction, and involves 4-electron transfer, O–H bond breaking and O–O molecular bond formation. Therefore, this process is always constrained by its large energy consumption and sluggish reaction kinetics, which generate the current moderate activity.69 Furthermore, as water oxidation takes place at a high oxidation potential (theoretically this is 1.23 V), this always induces oxidative corrosion of the light transducer photocatalyst. Therefore, it is necessary to increase the selectivity of hole oxidation. In principle, cocatalyst modification could decrease the energy barrier and restrict charge recombination. This also calls for improvement of the O2 evolution selectivity, which contributes to maintaining the intrinsic catalytic properties of the photocatalysts.Normally, suitable cocatalysts that can dramatically decrease the activation energy and thus promote the water oxidation activity are highly desired (Fig. 10). Noble metals, such as RuO2 and IrO2, are known to be good catalysts for the water oxidation reaction.65 However, they always present high costs and toxicity, both of which greatly limit their sustainable applications for scaling up. Inspired by the plant cubene-like Mo4CaOx cluster as the water oxidation active site in photosystem II, substantial efforts have been devoted to developing artificial water oxidation catalysts with sustainable components. Very recently, some first-row transition metal based materials e.g. CoPi,97,98 CoOx,62,99,100 and Co(OH)2 (ref. 101 and 102) have been reported to demonstrate comparable properties to those of Ru- and Ir-based noble metals for the water oxidation reaction, thus making them candidates for sustainable water oxidation development. For instance, when cobalt oxide nanoparticles were deposited on the surface of LaTiO2N, they exhibited much higher water oxidation activities than those of samples modified with IrO2.103 The AQY of the cobalt-modified LaTiO2N for O2 evolution at 440 nm reached as high as 27.1 ± 2.6%. This is mainly because the cobalt oxides can largely prolong the charge carrier lifetime. Consequently, more charge carriers are available for use in the subsequent photocatalytic water redox reactions. This certainly contributes to the improvement in the photocatalytic water oxidation activities. Importantly, compared with the Ru- and Ir-based noble metals, the cobalt based species possess many advantages, such as low-cost, low toxicity, abundant resources, versatile chemical states (Co2+ and Co3+) and excellent catalytic activities. Therefore, it is advised that cobalt based materials should be utilized as water oxidation cocatalysts in order to improve the photocatalytic water oxidation activities of g-C3N4 polymers.
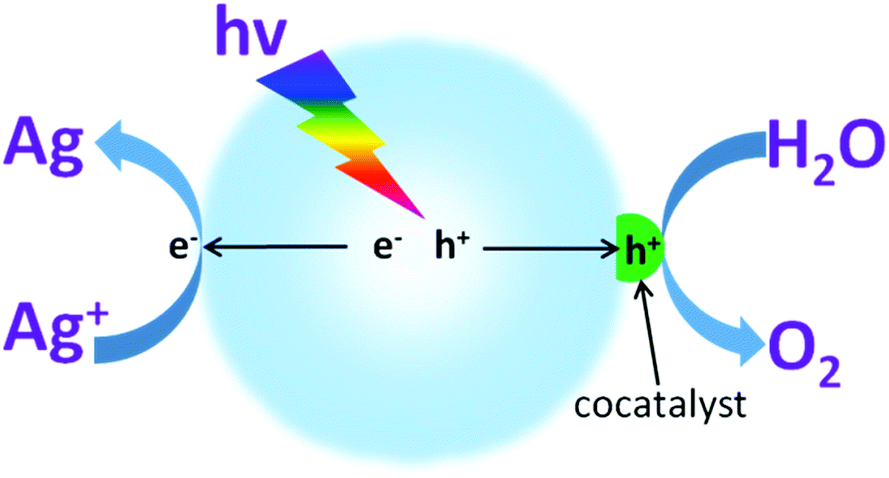 | ||
| Fig. 10 Schematic illustration of photocatalytic water oxidation for O2 evolution in the presence of an electron acceptor driven by a semiconductor modified with O2 evolution cocatalysts. | ||
In principle, pure g-C3N4 without any cocatalyst modification exhibit very low photocatalytic water oxidation activity even under strong UV light irradiation.18,56 This is mainly because the valence band potential of g-C3N4 is not positive enough to provide a sufficient driving force for the non-spontaneous water oxidation reaction. Furthermore, pure g-C3N4 is always defined by the fast charge carrier recombination rate, which decreases the water oxidation activity.104 In addition, the water oxidation process is hindered by the rather slow surface reaction kinetics, which are mainly attributed to the complex multielectron oxidation process and the huge activation energy barrier for O–O bond formation. In order to improve the photocatalytic water oxidation activities of the g-C3N4 polymers, it is advisable to deposit suitable cocatalysts on the surface of g-C3N4 to decrease the overpotential and accelerate the reaction kinetics.
As g-C3N4 has many lone-pair electrons, it can be used as an organic ligand to incorporate transition metals.105 Therefore, we firstly selected different transition metal ions (e.g. Fe3+, Ni2+ and Co2+) to incorporate with g-C3N4, with the aiming of fabricating an efficient water oxidation system containing sustainable elements.106 It is exciting to observe that cobalt modification could indeed improve the water oxidation activity of g-C3N4, thus indicating the positive role of cobalt in improving the water oxidation kinetics. However, the water oxidation activity of the system is closely related to the composition, structure, and properties of the cocatalysts, which are usually affected by the preparation strategy. Therefore, the cocatalyst modification technique should be optimized to further improve the water oxidation activities of the g-C3N4 polymers.
We then investigated the effect arising from cocatalyst modification. As is shown in Fig. 11a, two different modification techniques, based on bulk doping and surface deposition, have been developed to study the property–activity relationship.107 It should be noted that surface deposition with cobalt demonstrated great advantages in terms of improving the water oxidation activity in comparison with the bulk doping modification. As is shown in Fig. 11b, pure g-C3N4 exhibited low activity towards O2 evolution (5 μmol h−1) under UV light irradiation. When cobalt was doped into the framework of g-C3N4, the O2 evolution rate increased up to a value of 46 μmol h−1, which is 9 times higher than that of pure g-C3N4. The O2 evolution activity was further enhanced to 75.6 μmol h−1 when the same amounts of cobalt were deposited on the surface of g-C3N4. This is not difficult to understand, because more active sites would be exposed on the surfaces of the polymers, which is believed to maximize the activity. On the contrary, most of the active sites would be embedded into the bulk of the polymer framework, which decreases the interface water oxidation activity. The same enhanced water oxidation activities were also obtained when the samples were examined with visible light (Fig. 11c), thus elucidating well the huge advantage of surface deposition modification.
 | ||
| Fig. 11 (a) Schematic illustration of surface modification and bulk doping modification of CoOx with g-C3N4. Photocatalytic water oxidation activities of the g-C3N4, B–Co–g-C3N4 and S–Co–g-C3N4 samples under (b) UV (λ > 300 nm) and (c) visible light (λ > 420 nm) irradiation. Reprinted with permission from ref. 107. Copyright 2016, American Chemical Society. | ||
Except for the cobalt oxides, other cobalt based materials deposited on the surface of g-C3N4 could also decrease the energy barrier and accelerate the water oxidation reaction rate. For instance, when layered Co(OH)2 is deposited on the surface of g-C3N4 nanosheet,102 it will creates an intense adhesion between light transducer and water oxidation cocatalyst due to the similar 2D layered geometry, which to some extent will favour the interfacial charge carrier transfer and promote the water oxidation activities. Indeed, Co(OH)2 modified g-C3N4 showed obviously increased O2 evolution activities in comparison with the pristine g-C3N4. After 4 hours of persistent reaction under visible light irradiation, the total amounts of the evolved O2 gases could be reached 14 μmol for Co(OH)2 modified g-C3N4, while only 1.3 μmol of O2 gases were examined for pure g-C3N4. Other metal hydroxides, i.e. Fe(OH)3, Ni(0H)2, and Cu(OH)2 have also been deposited on the interface of g-C3N4 for water oxidation study, however, they only showed slightly or no increase in the water oxidation activities, demonstrating the advantage of cobalt based materials in promoting the water oxidation activities.
In addition, TMDs-based cobalt species, such as CoS2 and CoSe2, have also been developed to act as excellent water oxidation cocatalysts for improving the O2 evolution rate.108,109 By virtue of the different kinds of the cobalt species, they always exhibited different behaviours in promoting the water oxidation activities. Till now, the highest AQY of 1.1% at 420 nm for visible light driven water oxidation have been obtained on Co3O4 nanoparticles modified sulfur-mediated g-C3N4 polymers.62 Note that pristine g-C3N4 was barely active for water oxidation even under intensive UV irradiation. It is thus of pivotal interest to observe that cobalt based materials could indeed advance the interface charge carrier mobility and optimize the photocatalytic water oxidation performance.
Based on the above discussions, we can conclude that the cobalt based materials are excellent cocatalysts to promote the photocatalytic water oxidation activities of the g-C3N4 polymers in terms of largely decreasing the huge energy barrier and facilitating the sluggish reaction kinetics. Therefore, with the major purpose of achieving the 4-electron overall water splitting process, cobalt based materials are certainly regarded as the promising O2 evolution cocatalysts to improve the water oxidation half reaction.
5. Overall water splitting
Based on the above experimental observations, we can conclude that Pt and cobalt based materials are the most favourable H2 and O2 evolution cocatalysts, respectively, to separately promote the water reduction and water oxidation half reactions of g-C3N4 polymers. It is expected that overall water splitting should be achieved when the optimal H2 and O2 evolution cocatalysts are simultaneously deposited on the surfaces of g-C3N4 polymers to separately promote the water reduction and oxidation half reactions, respectively. Following this hypothesis, we intend to control the surface reaction kinetics of the g-C3N4 polymers through rational modification of the H2 and O2 evolution cocatalysts.Traditionally, the catalytic activities of cocatalysts are highly dependant on their physicochemical properties, such as particle size, morphology and structure.65 Therefore, the loading technique may be of pivotal significance for achieving photocatalytic overall water splitting. Three traditional reduction techniques, based on NaBH4 reduction, H2 reduction and in situ photo-reduction, were developed to deposit Pt-based species on the surface of g-C3N4 for a water splitting study.110 Excitingly, it was found that, different from the water reduction activity in the presence of sacrificial agents, only when Pt species are homogeneously loaded on the g-C3N4 polymers by in situ photo-reduction can overall water splitting with persistent H2 and O2 evolution be realized. Otherwise, only very small amounts of H2 evolution and nearly no O2 evolution were observed. High resolution XPS analysis revealed that both Pt and PtOx were generated by in situ photo-deposition, whereas only Pt was generated for the H2 and NaBH4 reduction, prepared using an immersion strategy. It was previously found that oxidized platinum could to some extent promote the water oxidation process. Therefore, both H2 and O2 evolution cocatalysts could be generated by in situ photodeposition, which may play a key role in triggering overall water splitting, otherwise only H2 evolution cocatalysts could be created. Under these circumstances, without the assistance of O2 evolution cocatalysts, the hole oxidation process is rather slow and the water oxidation process is extremely impeded, which would therefore increase the charge carrier recombination rate and generally decrease the overall water splitting activity. Moreover, in the absence of water oxidation cocatalysts, the water splitting reverse reaction, namely spontaneous water formation, could also immediately occur, which may also further decrease the overall water splitting reaction.
Therefore, the simultaneous formation of dual Pt and PtOx as H2 and O2 evolution cocatalysts by in situ photodeposition is believed to be a requirement for overall water splitting, as they can be selectively deposited on the active sites of g-C3N4 polymers as kinetic promoters in order to promote the water splitting reaction. HR-TEM images (Fig. 12a) further reveal that ultrafine Pt nanoparticles with average sizes of about 2–3 nm were homogeneously generated by in situ photo-deposition, and were believed to exhibit excellent activities for catalytic reactions. Conversely, large sized Pt particles were obtained when Pt was prepared by rough reduction from H2 or NaBH4 solution. Evidently, the large particle sizes of the catalysts usually decrease the number of available active sites and have proven to be insufficient for improving the water splitting activity.
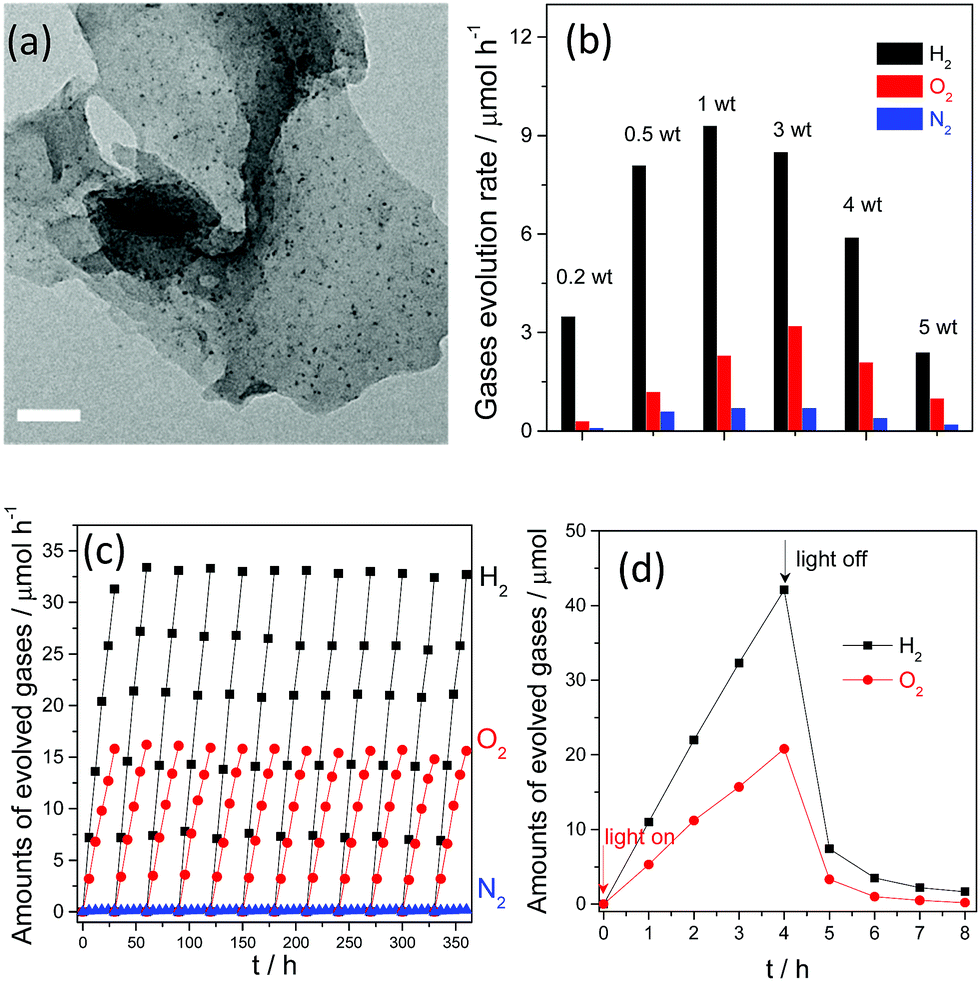 | ||
| Fig. 12 (a) TEM image of Pt deposited g-C3N4 polymers prepared by in situ photo-reduction; (b) overall water splitting activities of g-C3N4 polymers deposited with different amounts of Pt; (c) long reaction time overall water splitting by a Pt–Co–g-C3N4 polymer; (d) overall water splitting activities with the light on and off. Reprinted with permission from ref. 110. Copyright 2016, Royal Society of Chemistry. | ||
Furthermore, the molecular ratio of the evolved H2 and O2 gases, which in fact represents the water reduction and oxidation half reaction rates, could be finely tuned by simply varying the initial loading amounts of the platinum precursors. As is shown in Fig. 12b, the molecular ratio of H2 and O2 evolution clearly changed when the Pt and PtOx loading amounts were varied from 0.2 wt% to 5 wt%. When the loading content was increased from 0.2 wt% to 1 wt%, both the H2 and O2 evolution rates increased. Further increasing the loading amount decreased the H2 evolution rate, while the optimal O2 evolution rate was observed when the cocatalyst amount was confirmed as 3 wt%. It can also be seen that the molecular ratio of H2 and O2 evolution is slightly lower than 2:1, and is in close proximity to the stoichiometric ratio of overall water splitting. However, accompanying H2 and O2 evolution, evident N2 evolution was observed, which is probably due to self-oxidation of the g-C3N4 polymers by the valence band holes, and this would certainly decrease the O2 evolution selectivity and activity. This self-oxidation of the photocatalyst indeed promotes catalyst corrosion and is harmful to the overall efficiency from the viewpoint of the atom economy, which is mainly due to the absence of efficient water oxidation cocatalysts. Despite the fact that PtOx can promote the water oxidation reaction, it only demonstrates low efficiency and low selectivity for O2 evolution. Thus, it should be expected that the photocatalytic overall water splitting performance, especially the O2 evolution selectivity, could be further optimized when cobalt oxides are deposited as water oxidation cocatalysts. Actually, nearly no N2 evolution was obtained when a small amount (1 wt%) of CoOx was co-loaded as a water oxidation cocatalyst (Fig. 12c). A similar optimization was also observed when CoP was used as the water oxidation cocatalyst for overall water splitting.111 Meanwhile, the simultaneous loading of Pt and Co as excellent H2 and O2 evolution cocatalysts would greatly benefit the stability of the g-C3N4 polymers. After 500 hours of persistent reaction, nearly no evident decrease in the activity was observed for the Pt–Co–g-C3N4 photocatalyst, thus indicating its robust stability toward solution corrosion. Finally, overall water splitting with a stoichiometric ratio of 2:1 for H2 and O2 evolution could be achieved after in situ modification with Pt, PtOx and CoOx as the H2 and O2 evolution promoters. The currently achieved AQY of the system under visible light irradiation is only 0.3%. The relatively low efficiency is probably hindered by the fast and spontaneous reverse reaction, namely water formation (Fig. 12d). This is because although noble metals such as Pt and Rh are excellent promoters for H2 evolution, they can also function as good catalysts to promote the water formation reaction, which is thermodynamically spontaneous and is much easier to achieve than the non-spontaneous water splitting reaction. The evolved H2 and O2 gases immediately react on the surface of the cocatalyst to drive the water formation reaction. Therefore, further investigations based on surface nanostructure engineering of both cocatalysts and the photocatalysts to prevent the reverse reaction are believed to be an efficient route to improving the overall water splitting activity.
A prototypical example of this is a hollow sphere carbon nitride (HSCN) with a Janus interface to individually deposit Pt and Co3O4 nanoparticles on the inside and outside interfaces, thus spatially separating the H2 and O2 evolution active sites and so avoiding the reverse reaction.40 As is shown in Fig. 13, when the Pt and Co3O4 nanoparticles were separately deposited on the inside and outside interfaces of HSCN, the photocatalytic H2 and O2 evolution activity was much higher than that with both of the cocatalysts loaded on the outside interface, thus reflecting the fact that the reverse reaction was indeed restricted.
 | ||
| Fig. 13 Time course of the photocatalytic evolution of H2 and O2 using (a) Co3O4/HCNS/Pt and (b) (Co3O4 + Pt)/HCNS under UV irradiation (λ > 300 nm). Reprinted with permission from ref. 40. Copyright 2016, Wiley-CVH. | ||
6. Conclusions and outlook
Overall water splitting for the stoichiometric generation of H2 and O2 has been achieved by rational cocatalyst modification of g-C3N4 polymers to modulate the surface redox reaction kinetics. It was found that Pt and CoOx are excellent H2 and O2 evolution cocatalysts that decrease the activation energy barrier and accelerate the reaction kinetics of the g-C3N4 polymers. The current apparent quantum yield (AQY) for overall water splitting is calculated to be 0.3% at 405 nm. It should be noted that the reverse reaction, namely water formation, is also found to accompany the water splitting reaction. Furthermore, the competing reverse reaction is a thermodynamically downhill reaction; it can occur spontaneously and largely decreases the overall water splitting activity. Hence, future research should be directed towards restricting the reverse reaction, with the aim of further improving the overall water splitting activity. In addition, by virtue of the wide variety of conjugated polymers and their versatile structural variations from solution based organic protocols, future development of sustainable water splitting systems composed of new organic conjugated polymers are therefore envisaged. Furthermore, a careful and in depth understanding of the property–activity relationships between cocatalysts and carbon nitride would also provide promising new insight which will aid in the design of efficient systems for other catalytic reactions, such as CO2 reduction. Such a research topic is indeed very interesting, and some fascinating results have recently been achieved by scientists.112–116Acknowledgements
This work is financially supported by the National Basic Research Program of China (973 Program) (2013CB632405) and the National Natural Science Foundation of China (21425309 and 21761132002).Notes and references
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- N. Lewis and D. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- G. Zhang, Z. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and L. Can, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- H. Yang, C. Sun, S. Qiao, J. Zou, G. Liu, S. Smith, H. Cheng and G. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- F. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- M. Schröder, K. Kailasam, J. Borgmeyer, M. Neumann, A. Thomas, R. Schomäcker and M. Schwarze, Energy Technol., 2015, 3, 1014–1017 CrossRef.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- G. Liu, T. Wang, H. Zhang, X. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561–13565 CrossRef CAS PubMed.
- P. Niu, L. Yin, Y. Yang, G. Liu and H. Cheng, Adv. Mater., 2014, 26, 8046–8052 CrossRef CAS PubMed.
- D. Martin, P. Reardon, S. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- Q. Han, B. Wang, Y. Zhao, C. G. Hu and L. Qu, Angew. Chem., Int. Ed., 2015, 54, 11433–11437 CrossRef CAS PubMed.
- C. Yang, B. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- M. G. Schwab, M. Hamburger, X. L. Feng, J. Shu, H. W. Spiess, X. C. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- K. Kailasam, J. Schmidt, H. Bildirir, G. Zhang, S. Blechert, X. Wang and A. Thomas, Macromol. Rapid Commun., 2013, 34, 1008–1013 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed.
- Z. Lin and X. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS PubMed.
- J. Ran, T. Ma, G. Gao, X. Du and S. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 CAS.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 55, 1830–1834 CrossRef CAS PubMed.
- J. Hong, Xi. Xia, Y. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006–15012 RSC.
- Z. Lan, G. Zhang and X. Wang, Appl. Catal., B, 2016, 192, 116–125 CrossRef CAS.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- J. Zhang, G. Zhang, X. Chen, S. Lin, L. Möhlmann, G. Dołęga, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 3183–3187 CrossRef CAS PubMed.
- M. Zhang and X. Wang, Energy Environ. Sci., 2014, 7, 1902–1906 CAS.
- G. Zhang and X. Wang, J. Catal., 2013, 307, 246–253 CrossRef CAS.
- J. Qin, S. Wang, H. Ren, Y. Hou and X. Wang, Appl. Catal., B, 2015, 179, 1–8 CrossRef CAS.
- D. Zheng, X. Cao and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 11512–11516 CrossRef CAS PubMed.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11817 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, X. Ye, F. Guo and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 11926–11930 CrossRef CAS PubMed.
- Y. Jun, J. Park, S. Lee, A. Thomas, W. Hong and G. Stucky, Angew. Chem., Int. Ed., 2013, 52, 11083–11087 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121–4126 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, R. Sun and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145–10149 CrossRef CAS PubMed.
- L. Ge, F. Zuo, J. Liu, Q. Ma, C. Wang, D. Sun, L. Bartelsand and P. Feng, J. Phys. Chem. C, 2012, 116, 13708–13714 CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- C. Pan, J. Xu, Y. Wang, D. Li and Y. Zhu, Adv. Funct. Mater., 2012, 22, 1518–1524 CrossRef CAS.
- D. Zheng, G. Zhang and X. Wang, Appl. Catal., B, 2015, 179, 479–488 CrossRef CAS.
- J. Zhang, M. Zhang, L. Lin and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 6297–6301 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- D. Martin, K. Qiu, S. Shevlin, A. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- Y. Guo, J. Li, Y. Yuan, L. Li, M. Zhang, C. Zhou and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 14693–14697 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CAS.
- Y. Hou, A. Laursen, J. Zhang, G. Zhang, Y. Zhu, X. Wang, S. Dahl and I. Chorkendorff, Angew. Chem., Int. Ed., 2013, 52, 3621–3625 CrossRef CAS PubMed.
- D. Zheng, G. Zhang, Y. Hou and X. Wang, Appl. Catal., A, 2016, 521, 2–8 CrossRef CAS.
- Y. Hou, Z. Wen, S. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS PubMed.
- J. Xu, Y. Li and S. Peng, Int. J. Hydrogen Energy, 2015, 40, 353–362 CrossRef CAS.
- C. Han, L. Ge, C. Chen, Y. Li, X. Xiao, Y. Zhang and L. Guo, Appl. Catal., B, 2014, 147, 546–553 CrossRef CAS.
- J. Zhang, M. Grzelczak, Y. Hou, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Sci., 2012, 3, 443–446 RSC.
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95–108 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- G. Zhang, Z. Lan and X. Wang, ChemCatChem, 2015, 7, 1422–1423 CrossRef CAS.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- J. Zhang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 7230–7232 CrossRef CAS PubMed.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2955–2959 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 CAS.
- Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- N. Cheng, J. Tian, Q. Liu, C. Ge, A. Qusti, A. Asiri, A. Al-Youbi and X. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6815–6819 CAS.
- Y. Xu and W. Zhang, ChemCatChem, 2013, 5, 2343–2351 CrossRef CAS.
- L. Ge, C. Han, J. Liu and Y. Li, Appl. Catal., A, 2011, 409, 215–222 CrossRef.
- G. Zhang, G. Li and X. Wang, ChemCatChem, 2015, 7, 2864–2870 CrossRef CAS.
- Y. Hou, Y. Zhu, Y. Xu and X. Wang, Appl. Catal., B, 2014, 156, 122–127 CrossRef.
- M. Akple, J. Low, S. Wageh, A. Al-Ghamdi, J. Yu and J. Zhang, Appl. Surf. Sci., 2015, 358, 196–203 CrossRef CAS.
- J. Yu, S. Wang, B. Cheng, Z. Lin and F. Huang, Catal. Sci. Technol., 2013, 3, 1782–1789 CAS.
- J. Yan, H. Wu, H. Chen, L. Pang, Y. Zhang, R. Jiang, L. Li and S. Liu, Appl. Catal., B, 2016, 194, 74–83 CrossRef CAS.
- A. Indra, P. Menezes, K. Kailasam, D. Hollmann, M. Schröder, A. Thomas, A. Brückner and M. Driess, Chem. Commun., 2016, 52, 104–107 RSC.
- S. Yi, J. Yan, B. Wulan, S. Li, K. Liu and Q. Jiang, Appl. Catal., B, 2017, 200, 477–483 CrossRef CAS.
- J. Zou, L. Wang, J. Luo, Y. Nie, Q. Xing, X. Luo, H. Du, S. Luo and S. Sui, Appl. Catal., B, 2016, 193, 103–109 CrossRef CAS.
- Q. Han, B. Wang, J. Gao and L. Qu, Angew. Chem., Int. Ed., 2016, 55, 10849–10853 CrossRef CAS PubMed.
- Y. Shiraishi, Y. Kofuji, S. Kanazawa, H. Sakamoto, S. Ichikaw, S. Tanaka and T. Hirai, Chem. Commun., 2014, 50, 15255–15258 RSC.
- L. Yin, Y. Yuan, S. Cao, Z. Zhang and C. Xue, RSC Adv., 2014, 4, 6127–6132 RSC.
- J. Hong, Y. Wang, Y. Wang, W. Zhang and R. Xu, ChemSusChem, 2013, 6, 2263–2268 CrossRef CAS PubMed.
- K. Chang, X. Hai and J. Ye, Adv. Energy Mater., 2016, 6, 1502555 CrossRef.
- Y. Zhu, Y. Xu, Y. Hou, Z. Ding and X. Wang, Int. J. Hydrogen Energy, 2014, 39, 11873–11879 CrossRef CAS.
- A. Kluwer, R. Kapre, F. Hartl, M. Lutz, A. Spek, A. Brouwer, P. van Leeuwen and J. Reek, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10460–10465 CrossRef CAS PubMed.
- P. Zhang, M. Wang, C. Li, X. Li, J. Dong and L. Sun, Chem. Commun., 2010, 46, 8806–8808 RSC.
- X. Hao, Z. Jin, S. Min and G. Lu, RSC Adv., 2016, 6, 23709–23717 RSC.
- J. Dong, M. Wang, X. Li, L. Chen, Y. He and L. Sun, ChemSusChem, 2012, 5, 2133–2138 CrossRef CAS PubMed.
- C. Caputo, M. Gross, V. Lau, C. Cavazza, B. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 53, 11538–11542 CrossRef CAS PubMed.
- H. Yu, R. Shi, Y. Zhao, G. Waterhouse, L. Wu, C. Tung and T. Zhang, Adv. Mater., 2016, 28, 9454–9477 CrossRef CAS PubMed.
- M. Kanan and D. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- D. Zhong and D. Gamelin, J. Am. Chem. Soc., 2010, 132, 4202–4207 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841–1844 CrossRef CAS PubMed.
- X. Deng and H. Tüysüz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- K. Maeda, K. Ishimaki, Y. Tokunaga, D. Lu and M. Eguchi, Angew. Chem., Int. Ed., 2016, 55, 8309–8313 CrossRef CAS PubMed.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941–947 CrossRef CAS.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675–678 CAS.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- G. Zhang, C. Huang and X. Wang, Small, 2015, 11, 1215–1221 CrossRef CAS PubMed.
- G. Zhang, S. Zang, L. Lin, Z. Lan, G. Li and X. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 2287–2296 CAS.
- G. Zhang, S. Zang, Z. Lan, C. Huang, G. Li and X. Wang, J. Mater. Chem. A, 2015, 3, 17946–17950 CAS.
- M. Zhang, Z. Luo, M. Zhou, C. Huang and X. Wang, Sci. China Mater., 2015, 58, 867–876 CrossRef CAS.
- G. Zhang, Z. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- Z. Pan, Y. Zheng, F. Guo, P. Niu and X. Wang, ChemSusChem, 2017, 10, 87–90 CrossRef CAS PubMed.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867–4871 CrossRef CAS PubMed.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- R. Kuriki, O. Ishitani and K. Maeda, ACS Appl. Mater. Interfaces, 2016, 8, 6011–6018 CAS.
- K. Maeda, R. Kuriki and O. Ishitani, Chem. Lett., 2016, 45, 182–184 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |