
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical control of membrane tethering and interorganellar communication at nanoscales†
Lian
He‡
a,
Ji
Jing‡
 a,
Lei
Zhu‡
b,
Peng
Tan
a,
Guolin
Ma
a,
Qian
Zhang
ac,
Nhung T.
Nguyen
a,
Junfeng
Wang
*b,
Yubin
Zhou
*a and
Yun
Huang
*d
a,
Lei
Zhu‡
b,
Peng
Tan
a,
Guolin
Ma
a,
Qian
Zhang
ac,
Nhung T.
Nguyen
a,
Junfeng
Wang
*b,
Yubin
Zhou
*a and
Yun
Huang
*d
aCenter for Translational Cancer Research, Institute of Biosciences and Technology, Department of Medical Physiology, College of Medicine, Texas A&M University, Houston, TX 77030, USA. E-mail: yzhou@ibt.tamhsc.edu
bKey Laboratory of High Magnetic Field and Ion Beam Physical Biology, Chinese Academy of Sciences, Hefei 230031, Anhui, China. E-mail: junfeng@hmfl.ac.cn
cDepartment of Infectious Diseases, Renmin Hospital of Wuhan University, Wuhan, Hubei 430060, China
dCenter for Epigenetics and Disease Prevention, Institute of Biosciences and Technology, Department of Medical Physiology, College of Medicine, Texas A&M University, Houston, TX 77030, USA. E-mail: yun.huang@ibt.tamhsc.edu
First published on 31st May 2017
Abstract
Endoplasmic reticulum (ER) forms an extensive intracellular membranous network in eukaryotes that dynamically connects and communicates with diverse subcellular compartments such as plasma membrane (PM) through membrane contact sites (MCSs), with the inter-membrane gaps separated by a distance of 10–40 nm. Phosphoinositides (PI) constitute an important class of cell membrane phospholipids shared by many MCSs to regulate a myriad of cellular events, including membrane trafficking, calcium homeostasis and lipid metabolism. By installing photosensitivity into a series of engineered PI-binding domains with minimal sizes, we have created an optogenetic toolkit (designated as ‘OptoPB’) to enable rapid and reversible control of protein translocation and inter-membrane tethering at MCSs. These genetically-encoded, single-component tools can be used as scaffolds for grafting lipid-binding domains to dissect molecular determinants that govern protein–lipid interactions in living cells. Furthermore, we have demonstrated the use of OptoPB as a versatile fusion tag to photomanipulate protein translocation toward PM for reprogramming of PI metabolism. When tethered to the ER membrane with the insertion of flexible spacers, OptoPB can be applied to reversibly photo-tune the gap distances at nanometer scales between the two organellar membranes at MCSs, and to gauge the distance requirement for the free diffusion of protein complexes into MCSs. Our modular optical tools will find broad applications in non-invasive and remote control of protein subcellular localization and interorganellar contact sites that are critical for cell signaling.
Introduction
Phosphoinositide (PI) lipids are indispensable components of cellular membranes and play cardinal roles in controlling cell signalling and membrane trafficking in eukaryotic cells.1 Different PI species with unique functions are localized at various organellar membranes. For example, PI(4,5)P2 is found predominantly in the PM, which can be hydrolysed by phospholipase C (PLC) into two important second messengers, inositol trisphosphate and diacylglycerol, for mediating downstream signal transduction.2 By contrast, PI(3)P is mostly enriched at the endosomal compartments and is closely involved in regulating membrane trafficking and autophagy.3,4PI-dependent membrane recruitment of proteins is predominantly mediated by lipid-binding domains. For example, PIs with two adjacent phosphates in their inositol headgroup can be specifically recognized by proteins harbouring the pleckstrin homology (PH) domains.5 PH domains fused with fluorescent tags have been commonly used as genetically-encoded PI sensors to monitor the dynamic changes of PIs in intact cells.6,7 However, the constitutive expression of PH domains might compete with endogenous PI-binding proteins, and therefore tend to introduce undesired artefacts into the host system by perturbing PI-associated cell signalling. Indeed, the overexpression of PH domains under some circumstances has proven to be toxic to mammalian cells.6,8 In addition to PH domains, polybasic (PB) domains with clusters of positively charged residues can interact with acidic PIs through electrostatic interactions. PB domains are often found at the C-terminal tails of proteins and typically contain four or more Lys or Arg residues.9,10 Many small GTPases, such as Rit and Rin, contain PB domains that bind PI(4,5)P2 and/or PI(3,4,5)P3, two PI species that are abundantly distributed in the inner leaflet of the plasma membrane.10 Other notable examples include the ER-resident Ca2+ sensor proteins, stromal interaction molecules (STIM1 and STIM2), which undergo cytosol-to-PM translocation upon ER Ca2+ depletion to physically interact with and gate ORAI1 Ca2+ channels on the PM. In the absence of the PB domain, STIM1 fails to translocate toward PM to mediate store-operated Ca2+ entry (SOCE).11–14 Compared to PH domains that span nearly 150 residues, a typical PB domain only contains 20–30 residues, thus making it a more attractive target for optogenetic engineering.
In the current study, we aim to devise a series of light-controllable PB domains (designated as ‘OptoPB’). Once expressed in the cytosol, OptoPB can be exploited as a single-component modular scaffold to deliver proteins of interest toward PM upon light stimulation. When tethered to a subcellular compartment, such as ER, we envision that OptoPB may further enable light-inducible assembly of membrane contact sites (MCSs), a subcellular structure that is critical for maintaining lipid biosynthesis and Ca2+ homeostasis.15,16
Results
To achieve optical control over protein–phospholipid interactions with least perturbation to the host cells, we set out to fuse phospholipid-binding PB domains to a small genetically-encoded photoswitch, the LOV2 domain (Light, Oxygen or Voltage-sensing domain, residues 404–546) of Avena sativa phototropin1 (Fig. 1a). AsLOV2 has been successfully used for photoactivatable control of cell motility,17 calcium influx,18–20 protein nuclear translocation21,22 and protein degradation.23 In the dark, the C-terminal Jα helix tightly docks to the LOV2 core body and can thus introduce steric hindrance to the PB domain fused downstream of Jα. Following blue light illumination, photoexcitation generates a covalent photoadduct between the cofactor flavin mononucleotide (FMN) and a cysteine residue (C450) within LOV2 to cause the unwinding and undocking of the Jα helix from the core body,24,25 thereby exposing the C-terminal PB domain to restore its PI-binding function (Fig. 1a).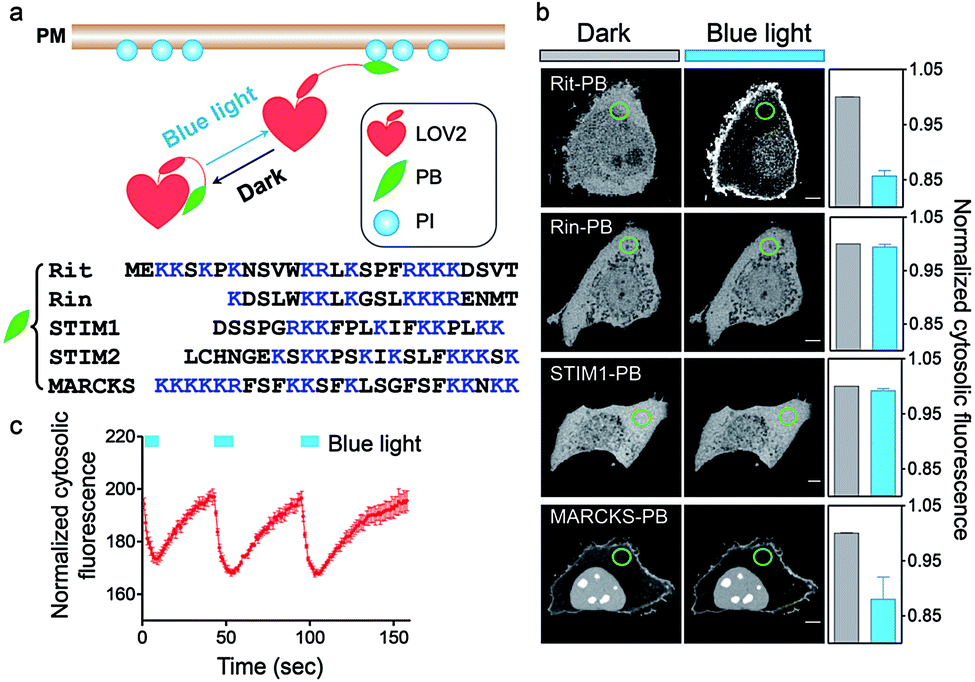 | ||
| Fig. 1 OptoPB as a single-component optogenetic device to photo-control protein translocation toward PM. (a) Design of LOV2-PB chimeras. (b) Confocal images at the middle layer of HeLa cells expressing the indicated mCherry (mCh)-tagged LOV2-PB chimeras before and after blue light illumination (4 mW cm−2 for 1 min). The normalized intensities of cytosolic mCh-OptoPB within the circled areas before (grey) and after light stimulation (blue) were plotted on the right as bar graphs. Scale bar, 5 μm. (c) Reversible cytosol-to-PM translocation of OptoPB. The cells transfected with mCh-LOV2-Rit-PB chimera shown in panel (b) were subjected to three repeated light–dark cycles of stimulation (each time: 10 s, 4 mW cm−2). Data were plotted as normalized cytosolic fluorescence and shown as mean ± s.e.m. (n = 5). | ||
We first generated over two dozen LOV2-PB chimeras (Fig. 1b) by varying the fused PB domains (derived from GTPases Rit and Rin, STIM1 or myristoylated alanine-rich protein kinase C substrate (MARCKS)). By using light-dependent cytosol-to-PM translocation as a straightforward readout, we identified the best performing construct (termed OptoPB) composed of Rit-PB and LOV2404-546. Although the LOV2-MARCKS-PB fusion protein exhibited a moderate degree of cytosol-to-PM translocation, we did not pursue further with it because of notable preactivation and accumulation in the nuclei (Fig. 1b). mCherry (mCh)-tagged OptoPB, which showed an even cytosolic distribution in the dark, rapidly translocated toward PM upon blue light illumination (470 nm at a power density of 4 mW cm−2). The light-induced accumulation of OptoPB did not seem to significantly alter the overall distribution of a PIP2 sensor (constitutive localization at PM given the abundance of PM-resident PIP2; Fig. S1a†) or the PIP3 sensor (cytosolic distribution under starvation conditions; Fig. S1b†), thus ruling out the possibility that OptoPB might adversely affect PI-mediated cell signaling. More importantly, no significant cytotoxicity was detected in HeLa cells overexpressing OptoPB (Fig. S1c†). The translocation of OptoPB between PM and the cytosol can be manipulated by switching on and off the light source for repeated cycles (t1/2, ON = 3 ± 1 s; t1/2, OFF = 17 ± 2 s; Fig. 1c, S2a; Movie 1†).
We further characterized the physiochemical properties of recombinant OptoPB in vitro. OptoPB underwent blue light dependent conformational changes, as reflected by a shift in the size exclusion chromatography (Fig. 2a), the appearance of three characteristic absorbance peaks in the UV-visible spectra (Fig. 2b), and gain of negative circular dichroism signals when recovering from the lit state to the dark state (Fig. 2c and d). More importantly, the recombinant OptoPB showed light-dependent interactions with phospholipids (particularly PI(4,5)P2) immobilized on a hydrophobic membrane (Fig. 2e). We further performed structure homology-modeling and molecular dynamics simulations to predict how the Rit-PB fused after the C-terminus of Jα helix might interplay with LOV2 (Fig. 2f). Electrostatic potential calculations on the surface of the model structure indicated that the poly-basic Rit-PB docked toward two negatively charged pockets within LOV2 (Fig. 2g). Collectively, we have demonstrated the light-controllable protein–phosphoinositides interactions both in vitro and in cellulo.
 | ||
| Fig. 2 In vitro characterization of LOV2-Rit-PB (OptoPB). (a) Gel filtration profiles of recombinant OptoPB before (black) and after (red) light stimulation. Arrow, purified recombinant OptoPB resolved on SDS-PAGE. (b) UV-Vis spectra showing the absorbance changes of OptoPB (120 μM) when recovering from the lit state (blue) to the dark state (black). Signals were recorded every 25 s. (c) Circular dichroism (CD) used to monitor the secondary structural changes of OptoPB (20 μM) before and after light simulation. (d) The time course showing the recovery of CD signals (at 222 nm) to the dark upon 1 min blue light illumination (4 mW cm−2). Two irradiation cycles were applied. Signals during the photostimulation periods (blue bar) were not recorded because the blue light illumination interfered with CD signal acquisition. (e) Schematic representation of lipid species on a membrane lipid strip (upper panel). Lipid strips incubated with the OptoPB (0.5 μg ml−1 LOV2-Rit-PB) recombinant protein with or without blue light (2 h, 4 mW cm−2; lower panel). (f) The modelled structure of OptoPB (LOV2-Rit-PB chimera) in the dark state. Rit-PB, shown in magenta, partially docked to the LOV2 in the dark. (g) Electrostatic potential surface representation of LOV2 (with surface rendering) fused with Rit-PB (magenta, cartoon representation). The positively-charged Rit-PB was docked toward two clusters of negatively charged residues within LOV2 (Pocket I: E409, D501, E541, and E545; Pocket II: E443 and E444). The electrostatic potential is measured in eV, with the ranges shown in the colour bar at the bottom. | ||
Aside from mediating protein–lipids/membrane interactions, the phosphorylated forms of phosphoinositides, polyphosphoinositides (PPIs), play crucial roles in regulating signal transduction occurring at membranes.26 As one of the most abundant PM-embedded PPIs, PI(4,5)P2 can be phosphorylated by Class I phosphoinositide 3-kinases (PI3K) to produce PI(3,4,5)P3, which recruits the nodal kinase Akt and subsequently triggers signaling cascades to regulate cell proliferation, growth, motility and survival.27 To examine whether the OptoPB could be utilized for the metabolic reprogramming of PPIs, we fused mCh-tagged inter-SH2 domain of the PI3K p85α regulatory subunit (iSH2-mCh) with OptoPB to recruit the endogenous PI3K catalytic subunit, p110 (Fig. 3a). Upon blue light stimulation, we observed the translocation of cytosolic iSH2-mCh-OptoPB toward the PM to trigger the conversion of PIP2 into PIP3, as visualized by increased decoration of GFP-tagged PIP3 sensor (PHAkt-GFP) around the plasma membrane (Fig. 3b). By optimizing the lengths of Jα and the linker region between Jα and Rit-PB, we obtained the best design of iSH2-OptoPB fusion for optogenetic control of PIP3 production on PM with a single-component system (V1.3 in Fig. S2b–d†).
 | ||
| Fig. 3 iSH2-OptoPB for reprogramming of PI metabolism in the plasma membrane. (a) Schematic illustrating the design of iSH2-OptoPB for photo-inducible catalysis of PIP3 production on PM. OptoPB fused inter-SH2 (iSH2) domain of p85α was used to recruit the endogenous catalytic p110α-subunit of PI3-kinase. (b) Confocal images showing blue light-induced translocation of iSH2-mCh-OptoPB to the PM, with subsequent enrichment of the PIP3 reporter, GFP-tagged Akt PH domain (PHAkt-GFP), on PM. HeLa cells were co-transfected with mch-iSH2-OptoPB and PHAkt-GFP. Prior to imaging, cells were pre-incubated in DMEM supplemented with 0.5% FBS for 4 h. Scale bar, 5 μm. Data were shown as mean ± s.e.m. (n = 5). | ||
Next, we tethered OptoPB toward the cytosolic side of ER membrane and set out to exploit the idea of further engineering OptoPB to reversibly label and manipulate the formation of MCSs, which are defined as specialized zones of close appositions (or junctions) between two membranous organelles that are separated by a narrow gap without membrane fusion.15 At ER-PM junctions, the two membranes are in close apposition with a gap distance of approximately 10–40 nm.16,28 The junctions function as platforms for lipid transfer and metabolism between the organelles, as well as for cell signaling, particularly SOCE mediated by STIM1 and ORAI1.16,29,30 Upon ER Ca2+ store depletion, the ER-resident Ca2+ sensor STIM1 undergoes oligomerization and subsequently translocates toward PM to form puncta, which traps diffusible ORAI1 in the PM through physical interactions, thereby directly gating ORAI1 Ca2+ channels to mediate Ca2+ influx.14,30–33 This physiological process is facilitated by the C-terminal polybasic domain of STIM1.14,31 We reason that, by keeping the ER-targeting sequences from an ER-resident protein (i.e., the signal peptide and the single transmembrane domain from STIM1) while replacing the entire cytosolic region with LOV2-PB chimeras, we could anchor OptoPB to ER and manipulate the assembly of ER-PM membrane contacts by light (Fig. 4a; Movie 2†).
 | ||
| Fig. 4 ER-tethered OptoPB (OptoPBer) for reversible labelling of ER-PM contact sites and for probing protein-PI interactions in living cells. Blue light-induced manipulation of membrane contact sites between the endoplasmic reticulum (ER) and the plasma membrane (PM) controls lipid metabolism and cell signalling. (a) Schematic illustrating the design of OptoPBer and light-inducible formation of ER-PM contact sites. (b) Confocal images showing the distribution of mRuby2-OptoPBer at the footprint (bottom layer) of HeLa cells before and after blue light stimulation (4 mW cm−2 for 1 min; also see Movie 2†). Areas within the white boxes were enlarged and shown below the whole cell image. (c) Qualification of OptoPBer clustering with two light–dark cycles. Scale bar, 10 μm. (d) Schematic showing the molecular architecture of ER-tethered LOV2-Jα fused with WT or mutant STIM1 PB. (e and f) TIRF images (e) and quantification of fluorescence intensities (f) of the indicated OptoPBer chimeras expressed in HeLa cells before (red) and after (blue) blue light stimulation. Data were shown as mean ± s.e.m. (n = 5). (g) Representative TIRF images of HeLa cells expressing WT STIM1 or STIM1 harboring the indicated mutations. Store depletion was induced by the addition of 1 μM thapsigargin (TG). Scale bar, 5 μm. | ||
Given the best photoresponsive performance of LOV2-Rit-PB in the cytosol, we first tethered this protein toward ER and monitored its subcellular localization before and after light stimulation. As shown in Fig. S3,† although we observed photo-induced increase in puncta formation, a small portion of ER-tethered LOV2-Rit-PB accumulated at ER-PM junctions even in the absence of blue light (Fig. S3b; Movie 3†). To eliminate such ‘dark activity’, we further screened over a dozen constructs (Fig. 4 and S4†) and identified ER-resident LOV2-STIM1-PB (named as ‘OptoPBer’) as the best candidate, which exhibited the highest dynamic range with no noticeable dark activity (Movie 3†). Interestingly, the LOV2-STIM1-PB as a cytosolic protein failed to respond to light to translocate toward PM (Fig. 1b), probably owing to its relatively weak affinity toward PIs. When anchored to the ER membrane, the increased local density of LOV2-STIM1-PB seemed to enhance its capability to associate with PM-resident PIs. In HeLa cells expressing mRuby2-tagged OptoPBer, we observed the immediate formation of OptoPBer puncta at ER-PM junctions following blue light illumination (t1/2, ON = 14 ± 2 s; Fig. 4b and c; Movies 2†). In the absence of light, the OptoPBer puncta were smoothly dispersed back into the cytoplasm (t1/2, OFF = 25 ± 3 s; Fig. 4c; Movies 2†). OptoPBer's distribution at resting and photo-activated conditions was further confirmed by its colocalization with a well-known ER marker (YFP-Sec61β; Fig. S5a†) or a recently-reported ER-resident protein located at ER-PM junctions28,34 (GFP-OSBPL5; Fig. S5b†).
The development of OptoPBer opened new possibilities of ‘grafting’ a PB domain derived from any protein of interest into the ER-tethered LOV2 scaffold, thus allowing us to conveniently identify molecular determinants required for PB-lipid/membrane interactions at real time in living cells. We decided to use STIM1-PB as an initial test case for validating this grafting approach given its well-characterized role in facilitating STIM1 translocation toward PM.14,31,35 Although the deletion of the entire STIM1-PB (aa 672-685) has been shown to inhibit STIM1 puncta formation at ER-PM junctions despite of intact self-oligomerization of STIM1 in ER,11,14,31 the key residues that directly mediate STIM1-phospholipids interactions remain to be defined in cellulo. To address this, we introduced single or double alanine mutations into STIM1-PB to replace lysine residues (Fig. 4d), and assessed how the neutralization of positive charges in STIM1-PB affects the formation of OptoPBer puncta at ER-PM junctions. We found that single Lys-to-Ala mutation within STIM1-PB led to a moderate reduction in the formation of puncta. By contrast, replacement of two adjacent lysines by alanines (K672A/K673A, K680A/K681A, or K684A/K685A) in STIM1-PB almost completely abrogated light-triggered puncta formation at ER-PM junctions (Fig. 4d–f), clearly attesting to the critical role of these lysine residues in driving the movement of STIM1 toward ER-PM junctions. We further validated this finding in the context of full-length STIM1 at physiological conditions (Fig. 4g). Introduction of the K684A/K685A double mutations prevented store depletion-induced puncta formation in HeLa cells expressing WT STIM1 (Fig. 4g, left), and also abolished the spontaneous puncta formation (Fig. 4g, right) in cells expressing the constitutively-active STIM1-D76A mutant, which by itself loses the ER luminal Ca2+-sensing capability and is thus anticipated to form puncta at ER-PM junctions even in the absence of thapsigargin-induced store depletion.36,37 By extending this grafting strategy to other proteins bearing PB domains, we identified key lysine residues in Rit-PB (K205/K208 and K214/K215) and STIM2-PB (K743/K744) that are required to mediate protein–lipid/membrane associations (Fig. S4†). Our site-directed mutagenesis studies have demonstrated that the photo-inducible assembly of ER-PM junctions is mainly driven by the electrostatic interactions between OptoPBer and phospholipids in the PM.
In our prototypical design, OptoPBer is estimated to span a distance of less than 10 nm in space by taking into account the dimension of LOV2 (∼2.4 nm × 4 nm) and the cytosolic linker regions (20 residues; approximately 5 nm in length if totally disordered) in the engineered protein. In store-operated CRAC channels, ORAI1 might form a teardrop-shaped structure with the intracellular regions projecting into the cytoplasm and require a minimum distance of 10 nm between the membranes of ER and PM.38 Therefore, the narrow gap formed by OptoPBer would be theoretically insufficient for accommodating even the ORAI channel itself, let alone the ORAI1-STIM1 complex that needs additional space for STIM1. Indeed, after scrutinizing the distribution of YFP-ORAI1 at the bottom layer (footprint) of HeLa cells before and after photostimulation, we observed remarkable exclusion of YFP-ORAI1 from ER-PM contact sites marked by mRuby2-OptoPBer after blue light illumination (Fig. 5a–c). In line with this observation, we found that HeLa cells expressing this optogenetic tether showed a substantially reduced calcium influx in response to thapsigargin-induced store depletion (Fig. S6†). This motivated us to develop OptoPBer variants that would bridge a flexible range of gaps at membrane contact sites. To achieve this, we constructed a series of ER-tethered OptoPB variants with the insertion of two to eight alpha-helical spacers composed of (EAAAR)4, with each spacer spanning an estimated distance of 3 nm.39 The blue light-induced exclusion of YFP-ORAI1 from the OptoPBer-labelled membrane contact sites was gradually alleviated after the insertion of two, four, or eight helical spacers (Fig. 5a, right), with the calculated intermembrane separation distances in the range of ∼15–35 nm. When the distance was increased to over 30 nm with the insertion of 8× spacers, we did not observe significant exclusion of YFP-ORAI1 distribution at ER-PM junctions before and after photostimulation (Fig. 5d and e). Accordingly, we did not observe a significant inhibition of SOCE in HeLa cells expressing OptoPBer with 8× helical spacers (Fig. S6†). Collectively, with these constructs, we have successfully gauged the minimal distance requirement on the inter-membrane gap (at least 10–15 nm) to enable free diffusion of ORAI1 into the membrane contact sites to mediate efficient Ca2+ influx.
 | ||
| Fig. 5 Use of OptoPBer to photo-tune the gap distances at ER-PM contact sites to constrain the diffusion of PM-resident proteins such as ORAI1. (a) Schematic demonstrating the use of OptoPBer with varying lengths of spacers to manipulate the inter-membrane distances at ER-PM junctions (left). Each spacer is estimated to span a distance of 3 nm. After introduction of 0, 2, 4 or 8 alpha-helical spacers made of (EAAAR)4 repeats, we measured the fluorescence intensity of YFP-ORAI1 at ER-PM junctional sites labeled by OptoPBer before (red) and after (blue) light stimulation (right panel). Data were shown as mean ± s.e.m. (n = 5). (b–d) Representative images of COS-7 cells co-expressing YFP-ORAI1 and mRuby2-OptoPBer with 0× spacer (b) or 8× spacers (d) before and after light stimulation. Areas highlighted in the yellow boxes were enlarged and shown next to the whole cell image. Surface profiles were presented to aid the visualization of subcellular distribution of mRuby-OptoPBer puncta (red, b) and YFP-ORAI1 (green; panel b). The exclusion of YFP-ORAI1 from mRuby2-OptoPBer puncta was indicated by arrows in panel (b). This scenario was not observed in cells expressing mRuby2-OptoPBer with 8 spacers (panel d). The YFP (green) and mRuby2 (red) fluorescence intensities across the arrowhead lines were plotted in panels (c) and (e) to indicate the degree of signal overlap. Scale bar, 5 μm. | ||
Discussions and conclusions
The phospholipids have fundamental roles in eukaryotes and they are often regarded as codes of organelle membranes for labelling of different membranous compartments and membrane subdomains.6 Building upon naturally-evolved PI-interacting PB domains, we have developed a set of optogenetic tools to rapidly and reversibly label PM-resident phospholipids in mammalian cells. Interestingly, PB domains derived from different proteins exhibit differential binding toward PM. The strength of PB-lipid/PM interactions seems to be correlated with the relative positioning and numbers of positively-charged residues. For instance, MARCKS-PB, which contains one ‘KKKKKR’ motif and three ‘KK’ repeats in the primary sequence, constitutively associates with PM, likely owing to its strong affinity for PIs. The Rit-PB, containing one ‘RKKK’ and multiple ‘KK’ or ‘KR’ combinations with a moderate affinity toward PM-resident PIs, shows light-inducible accumulation at PM (Fig. 1a). By contrast, STIM1-PB harbors three “KK” repeats but fails to show overt interaction with PM, probably owing to its relatively weaker affinity for PIs when expressed as a cytosolic protein. However, once anchored to the ER membrane, presumably with increased local concentrations, ER-resident LOV2-STIM1-PB fusion protein (OptoPBer) shows light-inducible accumulation at ER-PM junctions. All the observed interactions seem to require the presence of at least two consecutive positively charged residues (‘KK’ or ‘KR’). Replacement of a single lysine with alanine in such dyad motifs could lead to pronounced reduction in puncta formation; whereas KK-to-AA substitutions often abrogate the PB-lipid/PM associations (Fig. 4 and S4†). Further structural studies on the interaction between PB domains and PIs will likely provide a definitive answer by pinpointing the exact molecular determinants.When expressed as a cytosolic protein, OptoPB can act as a single-component optical device to photo-trigger the translocation and accumulation of proteins of interest toward PM to achieve tailored functions (e.g., reprogramming of phospholipids metabolism). The photo-controllable PM translocation of OptoPB by itself does not seem to cause overt cytotoxicity or significant perturbation over the distribution of major PM-resident PIs. Compared to other two-component optogenetic tools, our modular system requires less engineering efforts and only needs express a single construct in living cells. Single-component optogenetic tools have been recently developed by multiple groups to enable optical control of calcium influx in mammalian cells.18–20,40 Once tethered to the ER network, OptoPBer can be conveniently applied to photo-manipulate ER-PM membrane contact sites in living cells. Although similar operations have been done previously by building upon the chemically inducible dimerization technique,41–43 OptoPB and OptoPBer allow us to remotely control membrane tethering with higher spatial and temporal resolution because of the unparalleled speed and accuracy of light stimulation. Depending on the lengths of linkers, one can fine tune the inter-membrane gap distances at MCSs to control protein behaviors with light. Admittedly, the gap distances at ER-PM junctions are calculated by estimating the size of the engineered molecular tether and by assuming that the rigid helical spacers made of ‘EAAAR’ repeats will project toward PM at a right angle without tilting between ER and PM. Therefore, we might have overestimated the gap distance. It is our future goal to further accurately determine the inter-membrane gap distances by electron microscopy44 in mammalian cells expressing OptoPBer variants before and after photostimulation. Regardless of these complications, such manipulation has been demonstrated to limit the diffusion of PM-resident ORAI1 proteins, and functionally, to regulate store depletion-induced calcium influx at ER-PM junctions. We anticipate that, following a similar approach, lipid metabolism at ER-PM contact sites can likewise be fine-tuned by light.
Together, our study provides an optogenetic toolkit that can be widely applied to aid the study of protein–lipid interactions in cellulo, to expedite the mechanistic dissection of membrane contact sites in situ, and to enable remote control of inter-membrane communications occurring at these specialized subcellular structures. The similar engineering strategies can be broadly extended to photo-manipulate other types of inter-organellar contact sites, such as the ER-endosome/lysosome, ER-peroxisome, and ER-mitochondria MCSs, thus exerting remote control over cellular functions in a light-dependent manner.
Acknowledgements
We are grateful for Qiancheng Zhao and Dr Bailong Xiao at Tsinghua University for sharing the mRuby2-related construct with us. We thank the financial supports from the National Institutes of Health grant (R01GM112003 to YZ), the Welch Foundation (BE-1913 to YZ), the American Cancer Society (RSG-16-215-01 TBE to YZ), the Cancer Prevention and Research Institute of Texas (RR140053 to YH), the Innovation Award from American Heart Association (16IRG27250155 to YH), the Natural Science Foundation of China (U1532269 to JW), Ministry of Science and Technology of China (2016YFA0400901 to JW), and the China Scholarship Council (to JJ), and by an allocation from the Texas A&M University Health Science Center Startup Fund.Notes and references
- G. Di Paolo and P. De Camilli, Nature, 2006, 443, 651–657 CrossRef CAS PubMed.
- M. J. Rebecchi and S. N. Pentyala, Physiol. Rev., 2000, 80, 1291–1335 CAS.
- A. Simonsen, R. Lippe, S. Christoforidis, J. M. Gaullier, A. Brech, J. Callaghan, B. H. Toh, C. Murphy, M. Zerial and H. Stenmark, Nature, 1998, 394, 494–498 CrossRef CAS PubMed.
- H. M. McBride, V. Rybin, C. Murphy, A. Giner, R. Teasdale and M. Zerial, Cell, 1999, 98, 377–386 CrossRef CAS PubMed.
- J. E. Harlan, P. J. Hajduk, H. S. Yoon and S. W. Fesik, Nature, 1994, 371, 168 CrossRef CAS PubMed.
- T. Balla, J. Physiol., 2007, 582, 927–937 CrossRef CAS PubMed.
- X. Li, X. Wang, X. Zhang, M. Zhao, W. L. Tsang, Y. Zhang, R. G. Yau, L. S. Weisman and H. Xu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 21165–21170 CrossRef CAS PubMed.
- D. Raucher, T. Stauffer, W. Chen, K. Shen, S. Guo, J. D. York, M. P. Sheetz and T. Meyer, Cell, 2000, 100, 221–228 CrossRef CAS PubMed.
- T. Yeung, G. E. Gilbert, J. Shi, J. Silvius, A. Kapus and S. Grinstein, Science, 2008, 319, 210–213 CrossRef CAS PubMed.
- W. D. Heo, T. Inoue, W. S. Park, M. L. Kim, B. O. Park, T. J. Wandless and T. Meyer, Science, 2006, 314, 1458–1461 CrossRef CAS PubMed.
- J. Liou, M. Fivaz, T. Inoue and T. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9301–9306 CrossRef CAS PubMed.
- C. M. Walsh, M. Chvanov, L. P. Haynes, O. H. Petersen, A. V. Tepikin and R. D. Burgoyne, Biochem. J., 2009, 425, 159–168 CrossRef PubMed.
- E. Ercan, F. Momburg, U. Engel, K. Temmerman, W. Nickel and M. Seedorf, Traffic, 2009, 10, 1802–1818 CrossRef CAS PubMed.
- C. Y. Park, P. J. Hoover, F. M. Mullins, P. Bachhawat, E. D. Covington, S. Raunser, T. Walz, K. C. Garcia, R. E. Dolmetsch and R. S. Lewis, Cell, 2009, 136, 876–890 CrossRef CAS PubMed.
- C. J. Stefan, A. G. Manford and S. D. Emr, Curr. Opin. Cell Biol., 2013, 25, 434–442 CrossRef CAS PubMed.
- S. Carrasco and T. Meyer, Annu. Rev. Biochem., 2011, 80, 973–1000 CrossRef CAS PubMed.
- Y. I. Wu, D. Frey, O. I. Lungu, A. Jaehrig, I. Schlichting, B. Kuhlman and K. M. Hahn, Nature, 2009, 461, 104–108 CrossRef CAS PubMed.
- L. He, Y. Zhang, G. Ma, P. Tan, Z. Li, S. Zang, X. Wu, J. Jing, S. Fang, L. Zhou, Y. Wang, Y. Huang, P. G. Hogan, G. Han and Y. Zhou, eLife, 2015, 4, e10024 Search PubMed.
- G. Ma, S. Wen, L. He, Y. Huang, Y. Wang and Y. Zhou, Cell Calcium, 2017, 64, 36–46 CrossRef CAS PubMed.
- P. Tan, L. He, G. Han and Y. Zhou, Trends Biotechnol., 2017, 35, 215–226 CrossRef CAS PubMed.
- H. Yumerefendi, A. M. Lerner, S. P. Zimmerman, K. Hahn, J. E. Bear, B. D. Strahl and B. Kuhlman, Nat. Chem. Biol., 2016, 12, 399–401 CrossRef CAS PubMed.
- H. Yumerefendi, D. J. Dickinson, H. Wang, S. P. Zimmerman, J. E. Bear, B. Goldstein, K. Hahn and B. Kuhlman, PLoS One, 2015, 10, e0128443 Search PubMed.
- C. Renicke, D. Schuster, S. Usherenko, L. O. Essen and C. Taxis, Chem. Biol., 2013, 20, 619–626 CrossRef CAS PubMed.
- J. M. Christie, M. Salomon, K. Nozue, M. Wada and W. R. Briggs, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8779–8783 CrossRef CAS.
- S. M. Harper, L. C. Neil and K. H. Gardner, Science, 2003, 301, 1541–1544 CrossRef CAS PubMed.
- T. Balla, Physiol. Rev., 2013, 93, 1019–1137 CrossRef CAS PubMed.
- I. Vivanco and C. L. Sawyers, Nat. Rev. Cancer, 2002, 2, 489–501 CrossRef CAS PubMed.
- Y. Saheki and P. De Camilli, Annu. Rev. Biochem., 2017, 86, 6.1–6.26 CrossRef PubMed.
- M. Prakriya and R. S. Lewis, Physiol. Rev., 2015, 95, 1383–1436 CrossRef CAS PubMed.
- J. Soboloff, B. S. Rothberg, M. Madesh and D. L. Gill, Nat. Rev. Mol. Cell Biol., 2012, 13, 549–565 CrossRef CAS PubMed.
- J. P. Yuan, W. Zeng, M. R. Dorwart, Y. J. Choi, P. F. Worley and S. Muallem, Nat. Cell Biol., 2009, 11, 337–343 CrossRef CAS PubMed.
- Y. Zhou, P. Meraner, H. T. Kwon, D. Machnes, M. Oh-hora, J. Zimmer, Y. Huang, A. Stura, A. Rao and P. G. Hogan, Nat. Struct. Mol. Biol., 2010, 17, 112–116 CAS.
- A. Gudlur, Y. Zhou and P. G. Hogan, Curr. Top. Membr., 2013, 71, 33–58 CAS.
- J. Chung, F. Torta, K. Masai, L. Lucast, H. Czapla, L. B. Tanner, P. Narayanaswamy, M. R. Wenk, F. Nakatsu and P. De Camilli, Science, 2015, 349, 428–432 CrossRef CAS PubMed.
- Y. Zhou, P. Srinivasan, S. Razavi, S. Seymour, P. Meraner, A. Gudlur, P. B. Stathopulos, M. Ikura, A. Rao and P. G. Hogan, Nat. Struct. Mol. Biol., 2013, 20, 973–981 CAS.
- J. Liou, M. L. Kim, W. D. Heo, J. T. Jones, J. W. Myers, J. E. Ferrell Jr and T. Meyer, Curr. Biol., 2005, 15, 1235–1241 CrossRef CAS PubMed.
- G. S. Bird, W. I. DeHaven, J. T. Smyth and J. W. Putney Jr, Methods, 2008, 46, 204–212 CrossRef CAS PubMed.
- Y. Maruyama, T. Ogura, K. Mio, K. Kato, T. Kaneko, S. Kiyonaka, Y. Mori and C. Sato, J. Biol. Chem., 2009, 284, 13676–13685 CrossRef CAS PubMed.
- G. Merutka, W. Shalongo and E. Stellwagen, Biochemistry, 1991, 30, 4245–4248 CrossRef CAS PubMed.
- T. Kyung, S. Lee, J. E. Kim, T. Cho, H. Park, Y. M. Jeong, D. Kim, A. Shin, S. Kim, J. Baek, J. Kim, N. Y. Kim, D. Woo, S. Chae, C. H. Kim, H. S. Shin, Y. M. Han and W. D. Heo, Nat. Biotechnol., 2015, 33, 1092–1096 CrossRef CAS PubMed.
- B. C. Suh, T. Inoue, T. Meyer and B. Hille, Science, 2006, 314, 1454–1457 CrossRef CAS PubMed.
- T. Komatsu, I. Kukelyansky, J. M. McCaffery, T. Ueno, L. C. Varela and T. Inoue, Nat. Methods, 2010, 7, 206–208 CrossRef CAS PubMed.
- P. Varnai, B. Toth, D. J. Toth, L. Hunyady and T. Balla, J. Biol. Chem., 2007, 282, 29678–29690 CrossRef CAS PubMed.
- M. M. Wu, J. Buchanan, R. M. Luik and R. S. Lewis, J. Cell Biol., 2006, 174, 803–813 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplemental sequences, figures referred in the text, supplemental movies. See DOI: 10.1039/c7sc01115f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |