
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments in the use of aza-Heck cyclizations for the synthesis of chiral N-heterocycles
Nicholas J.
Race
 ,
Ian R.
Hazelden
,
Adele
Faulkner
and
John F.
Bower
*
,
Ian R.
Hazelden
,
Adele
Faulkner
and
John F.
Bower
*
School of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: john.bower@bris.ac.uk
First published on 20th June 2017
Abstract
Aza-Heck cyclizations are an emerging method for the construction of chiral N-heterocyclic systems. In these processes, an activated N–O bond replaces the C–X bond (X = halide, OTf) used in conventional Heck reactions, with the associated aza-Pd(II)-intermediate engaging pendant alkenes in a Heck-like manner. This perspective article commences with an historical overview of the area, which stems from Narasaka's seminal studies using oxime esters as the initiating motif. The scope and mechanism of associated chiral N-heterocyclic methodologies are then outlined, including cascade processes that enable diverse alkene 1,2-carboaminations. The recent emergence of new N–O donors and the realization of highly enantioselective aza-Heck cyclizations are then discussed. Collectively, these studies suggest that the aza-Heck approach can underpin a broad family of redox-neutral and enantioselective C–N bond forming processes.
 Nicholas J. Race | Nicholas Race received his undergraduate MChem degree (First Class) from Oxford University in 2010. He worked with Professor Tim Donohoe during his final year where he developed methodology involving a cross metathesis approach to substituted pyrroles. He then moved to Bristol for his graduate studies with Dr John Bower. At Bristol he developed aza-Heck methodology for the synthesis of chiral nitrogen heterocycles. After receiving his PhD in 2015 he moved to Salt Lake City, Utah where he is currently a postdoctoral research fellow with Professor Matthew Sigman. |
 Ian R. Hazelden | Ian Hazelden graduated from the University of Bristol in 2014 with a First Class MSci degree in chemistry. He completed his final year research project in the laboratory of Professor Tim Gallagher. He stayed in Bristol for his PhD studies under the supervision of Dr John Bower. His current research focuses on the development of different activated N–O bonds as initiating motifs for aza-Heck reactions. |
 Adele Faulkner | Adele Faulkner received her undergraduate MSci degree (First Class) from the University of Bristol in 2011. Her PhD studies were also conducted in Bristol with Dr John Bower. Her research focused on metal-catalyzed cyclizations for the synthesis of chiral nitrogen heterocycles. After completing her PhD in 2015, she was awarded a Leverhulme Trust scholarship to carry out postdoctoral research in the laboratory of Professor Ben Feringa. She is currently a senior medicinal chemist at Evotec in Abingdon, UK. |
 John F. Bower | John F. Bower obtained his MSci degree in 2003 from the University of Bristol, where he remained to study for his PhD degree (2007) under the guidance of Professor Timothy Gallagher. He then undertook postdoctoral appointments with Professor Michael Krische at the University of Texas at Austin (2007–2008) and Professor Timothy Donohoe at the University of Oxford (2008–2010). In 2010, he was awarded a Royal Society University Research Fellowship and commenced his independent career at the University of Bristol. Bower's research has been recognized by a number of awards, including the 2013 Royal Society of Chemistry Harrison-Meldola Memorial Prize, the 2015 Royal Society of Chemistry Hickinbottom Award and a 2016 Philip Leverhulme Prize. |
Introduction
Nitrogen-containing molecules hold a privileged position in drug discovery. In 2013, eighty percent of the top twenty best-selling brand name drugs incorporated nitrogen, with two thirds of these compounds containing an N-heterocycle.1 In particular, chiral pyrrolidine and piperidine derivatives are common in pharmaceuticals and alkaloid natural products.2 With these considerations in mind, the development of efficient and versatile routes to complex molecular structures, particularly those containing C(sp3)–N bonds, is of paramount importance.3In this perspective, we highlight the emerging area of aza-Heck reactions for the synthesis of chiral nitrogen heterocycles. Here, an activated N–O bond replaces the C–X bond (X = halide, OTf) used in conventional Heck reactions, with the resulting aza-Pd(II) intermediates mimicking the reactivity of their aryl-Pd(II) counterparts (Scheme 1). In general terms, aza-Heck reactions can be considered complementary to more well-established aza-Wacker processes that require an external oxidant.4 However, as will be seen, the use of an N–O bond as the internal oxidant can confer several synthetic and mechanistic advantages. The following discussion highlights key historical developments in aza-Heck chemistry before focussing on the scope and mechanism of methodologies that provide chiral heterocyclic systems.
 | ||
| Scheme 1 Synthesis of chiral N-heterocycles by aza-Heck cyclization. | ||
The Narasaka–Heck reaction and early heteroaromatic methodologies
The prototypical aza-Heck process was reported in 1999 by Narasaka and co-workers. In this seminal work, exposure of γ,δ-unsaturated pentafluorobenzoyl oxime ester 1 (FBz = pentafluorobenzoyl) to 10 mol% Pd(PPh3)4 effected cyclization to pyrrole 2 in 81% yield (Scheme 2).5 A variety of other systems were shown to cyclize efficiently, providing a direct and flexible entry to differentially substituted pyrroles. The proposed mechanism involves elementary steps that are analogous to the conventional Heck reaction,6 with the process later becoming known as the ‘Narasaka–Heck reaction’.7 Specifically, oxidative addition of Pd(0) into the N–O bond of 1 generates key imino-Pd(II) intermediate 3; this initiation step replaces the C–X oxidative addition step of the conventional Heck reaction. From 3, migratory insertion of the alkene into the Pd–N bond (5-exo imino-palladation)8 is followed by β-hydride elimination and alkene isomerization to deliver the pyrrole target. Importantly, the geometry of the oxime ester moiety of 1 is inconsequential, such that both isomers cyclize with comparable efficiencies. Presumably, E/Z isomerization occurs at the stage of imino-Pd(II) intermediate 3, although the mechanism of this process is not well understood. Imino-Pd(II) intermediates related to 3 have been characterized crystallographically by the groups of Hartwig9 and Stahl,10 providing unambiguous confirmation of the N–O oxidative addition event. | ||
| Scheme 2 The Narasaka–Heck reaction. | ||
Following Narasaka's initial report, a variety of related heteroaromatic methodologies emerged (Scheme 3). A key feature of all of these approaches is that the oxime ester substrates are readily available from the corresponding ketones, which, in turn, can be accessed using well-established carbonyl chemistry (Scheme 3A). In most cases, O-pentafluorobenzoyl oxime esters are required, although processes that exploit oxidative addition into the N–O bond of O-phosphinyl oxime esters11 or the N–N bond of N,N,N-trimethylhydrazonium salts have also been developed.12N-Chloroamines have also been suggested as suitable initiating motifs, although subsequent studies suggest that cyclization of such systems proceeds via a radical-based pathway.13
 | ||
| Scheme 3 Heterocycle synthesis via the Narasaka–Heck reaction. | ||
A representative outline of heteroaromatic compounds accessible via the Narasaka–Heck reaction is presented in Scheme 3B. Pyridines 5 can be accessed by 6-endo cyclization of oxime esters 4; here, chloride additives such as LiCl and n-Bu4NCl were found to play a crucial role.14 Abell and co-workers showed that amidoxime esters 6 cyclize to afford imidazoles 7, thereby demonstrating the viability of N–O oxidative addition beyond oxime ester substrates.15 In conformationally constrained cases, 6-exo cyclizations are feasible; for example, isoquinolines 9 can be accessed by aza-Heck cyclization of precursors 8.16 Other classes of acceptor can also be exploited. For example, cyclization onto pendant cycloheptatrienes generates aza-azulenes after in situ oxidation of the initial aza-Heck adduct (10 to 11).17
The processes discussed so far use the Narasaka–Heck reaction to generate new C(sp2)–N bonds,18 rendering these approaches ideally suited to the synthesis of N-heteroaromatic compounds. Given the increasing importance of chiral N-heterocycles in the pharmaceutical industry,2 generalization of aza-Heck manifolds to the formation of (stereodefined) C(sp3)–N bonds is particularly appealing. The remainder of this Perspective highlights recent aza-Heck methodologies that use N–O bonds to generate chiral nitrogen heterocycles.19 As will be seen, processes that initiate at the N–O bond of oxime esters, as used in the original Narasaka process, have received the most attention. However, recent reports have exploited other redox active N–O donors, such that N-(pentafluorobenzoyloxy)sulfonamide20 and O-phenyl hydroxamates21 are now viable initiating motifs for aza-Heck cyclizations (Scheme 4).
 | ||
| Scheme 4 Historical overview of key developments in aza-Heck reactions. | ||
Chiral N-heterocycles via Narasaka–Heck cyclizations: mechanism and reaction scope
In 2012, the first systematic efforts to exploit aza-Heck cyclizations of oxime esters for the synthesis of C(sp3)–N bonds, and therefore chiral N-heterocycles, were reported by our group. Initial studies explored cyclizations onto cyclic alkenes because the stereoelectronic requirements of syn-iminopalladation and syn-β-hydride elimination were expected to result in the formation of C(sp3)–N bonds.22 Indeed, an example of this type of cyclization had been reported in 2005 by Fürstner and co-workers in the context of a natural product synthesis (Scheme 5).23 In this process, a symmetrical bis-olefinic acceptor unit was specifically incorporated into 12 to offset inherent inefficiencies associated with imino-palladation. Even using this biasing factor, high loadings of Pd catalyst were required to achieve a relatively modest yield of target 13. At the time, these issues were reflective of wider difficulties in promoting efficient aza-Heck cyclizations over diverse substrate classes. | ||
| Scheme 5 Fürstner's use of an aza-Heck cyclization onto a cyclic alkene. | ||
With the aim of developing a general aza-Heck methodology involving cyclic alkenes, oxime ester 14 was used as a model substrate (Scheme 6A).22 Extensive investigations revealed that efficient cyclization to 15 was possible, but only when electron-deficient phosphine ligands were used. By far the most effective subset of ligands were fluorinated triarylphosphines, with P(3,5-(CF3)2C6H3)3 emerging as the optimal system (Scheme 6B). This ligand allowed cyclization of 14 to proceed in 93% yield at 60 °C with 5 mol% Pd loading. Highly electron-deficient phosphine ligands, such as P(C6F5)3 were not effective and low conversions were observed, likely due to slow N–O oxidative addition. However, within the effective ligand range outlined in Scheme 6B, oxidative addition does not appear to be rate limiting such that the choice of ligand is dictated by the efficiency of steps later in the cycle. Interestingly, bulky electron-rich phosphine ligands, such as those typically employed in conventional Heck reactions (where oxidative addition is often rate limiting), are not effective and lead to a different mechanistic pathway (vide infra).
 | ||
| Scheme 6 Key features of the aza-Heck cyclization of O-pentafluorobenzoyl oxime esters. | ||
The beneficial effects of P(3,5-(CF3)2C6H3)3 are consistent with stoichiometric studies by Hartwig and co-workers, who demonstrated that migratory insertion of alkenes into Pd-NPh2 bonds is facilitated by bulky and/or electron-deficient ligands.24 An additional key factor for the conversion 14 to 15 resides in suppression of protodepalladation of imino-Pd intermediate 16. Electron-deficient phosphine ligands are expected to increase σ-donation from the imino moiety, which, in turn, should suppress competing protonation of this unit. Indeed, under unoptimized conditions, protodepalladation dominates, leading to ketone by-product 19, likely via the intermediacy of hydrolytically sensitive NH imine 18.
Also of considerable importance for the cyclization of 14 to 15 is the pentafluorobenzoate leaving group. This activating group was introduced in Narasaka's seminal publication as a result of empirically driven studies;5 substrates of this type were found to offer the best balance of starting material stability and catalytic activity. With respect to the former aspect, the key issue is suppression of competing Beckmann rearrangement of the starting material, which becomes problematic when more polarizing activating groups (e.g. O-sulfonyl groups) are used. Nevertheless, the requirement of a pentafluorobenzoate leaving group for the cyclization of 14 was striking given that Uemura and co-workers have shown that facile oxidative addition of Pd(0) into the N–O bond of less polarized O-benzoyl oximes can be used to trigger β-carbon eliminations.25 Note that the Uemura process does not involve alkene imino-palladation, suggesting a critical role for the leaving group on the efficiency of this step in aza-Heck reactions.
Investigations aimed at understanding the efficiencies of different leaving groups (in particular pentafluorobenzoate vs. benzoate) revealed that, for the cyclization of 14 to 15, addition of exogenous C6F5CO2H or C6H5CO2H decreases both the reaction rate and yield.26 Furthermore, halide additives, such as n-Bu4NCl, completely suppress aza-Heck cyclization. These studies suggest that, for efficient cyclization, the leaving group must dissociate after oxidative addition to provide cationic imino-Pd(II) intermediate 17 (Scheme 6A). Relatively electron-rich carboxylates, such as benzoate, do not dissociate readily, resulting in less efficient cyclization of 14 and increased levels of protodepalladation at the stage of imino-Pd(II) intermediate 16. 19F NMR studies revealed a surprising additional benefit of the pentafluorobenzoate leaving group. It was found the interaction of this by-product with protonated triethylamine, which is generated by deprotonation of the Pd-hydride species released upon β-hydride elimination, results in facile protodecarboxylation to C6F5H. This process removes otherwise inhibitory pentafluorobenzoate from the reaction system, such that equilibrium access to cationic imino-Pd(II) intermediate 17 is maintained in subsequent cycles. In favorable cases, the protodecarboxylation process allows substoichiometric (catalytic) quantities of Et3N to be used. Thus, efficient aza-Heck cyclization is dependent on both a pentafluorobenzoate leaving group and an electron-deficient phosphine. A complete mechanistic picture of the aza-Heck cyclization of 14 to 15, as discussed above, is outlined in Scheme 7.
 | ||
| Scheme 7 Detailed catalytic cycle of the aza-Heck cyclization of oxime ester 14. | ||
The conditions optimized for 14 proved to be generally applicable for 5-exo aza-Heck cyclizations onto cyclic alkenes (Scheme 8).22 Indeed, a variety of aryl-, alkyl-, and heteroaryl-substituted oxime esters underwent efficient aza-Heck reaction to give the corresponding cis-fused bicyclic N-heterocycles in high yield (Scheme 8A). Cyclic alkenes of different ring sizes were also tolerated, as well as systems with substitution at C2 of the oxime ester. Interestingly, oxime ester 20a, which possesses a non-defined α-stereocenter, underwent stereoconvergent cyclization to generate bicyclic system 21a in high diastereopurity; a similar result was observed for 21b (Scheme 8B). Presumably, initial cyclization of both diastereomers is followed by equilibration (via the enamine) to the thermodynamically favored product, wherein the R2-substituent resides on the less hindered face of the dihydropyrrole ring.
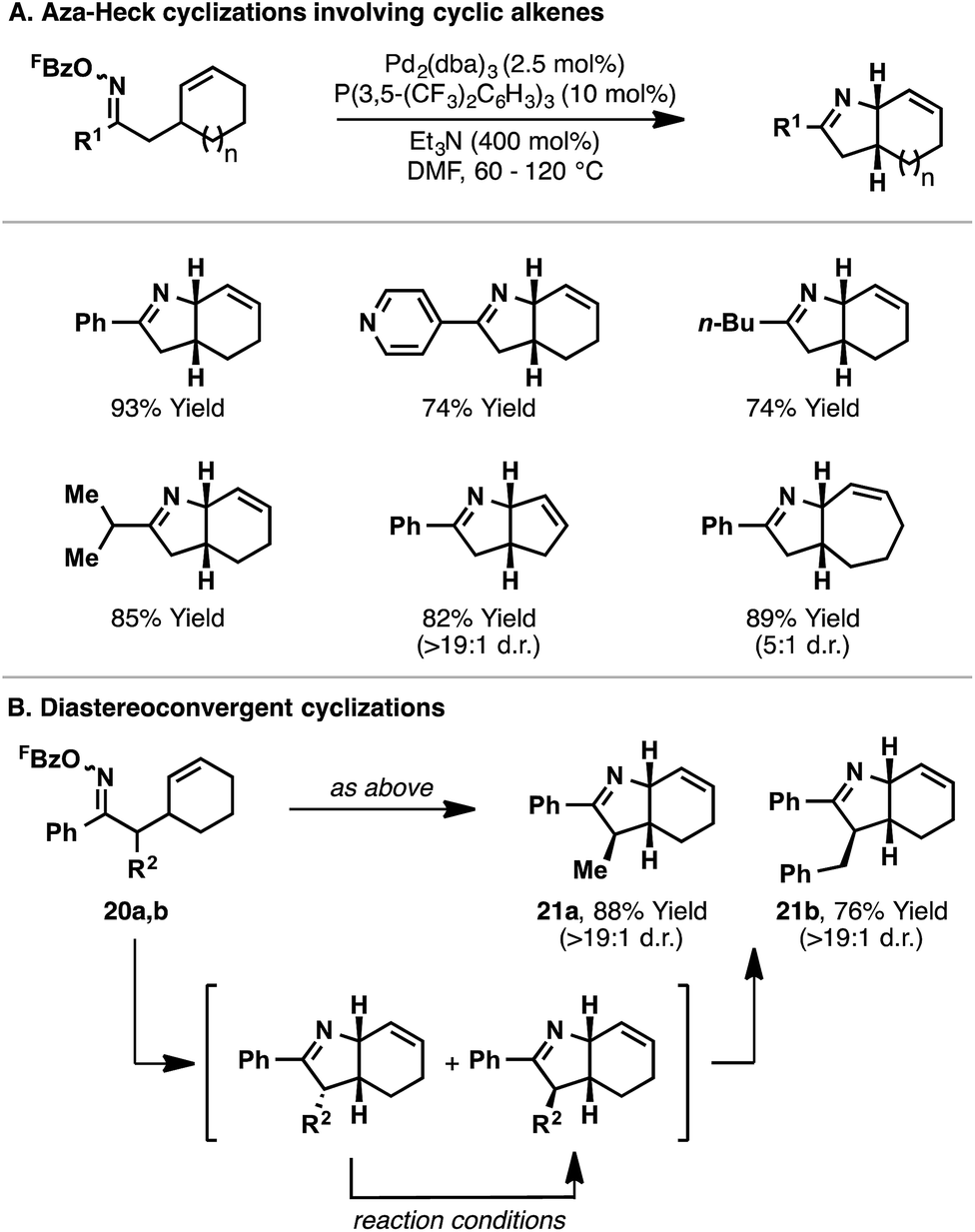 | ||
| Scheme 8 Aza-Heck cyclizations of oxime esters with cyclic alkenes. | ||
An inherent consequence of using the Narasaka–Heck cyclization to generate C(sp3)–N bonds is that the product contains imine and alkene units for further derivatization. Oxime ester (R)-14 (93% e.e.) underwent diastereoselective aza-Heck cyclization to afford N-heterocycle (3aR,7aS)-15 without loss of enantiomeric purity (Scheme 9). Further manipulation of this product via the imine and alkene afforded complex bicyclic system 22 with high levels of control for all five stereocenters. The sequence in Scheme 9 demonstrates the power of aza-Heck methodology for the formation of complex, chiral N-heterocycles.
 | ||
| Scheme 9 Synthesis of a complex, chiral N-heterocycle using aza-Heck methodology. | ||
Further studies sought to investigate the scope of the Pd2(dba)3/P(3,5-(CF3)2C6H3)3 catalyst system with respect to other classes of alkene acceptor. It was found that cyclizations of oxime esters with 1,1-disubstituted alkenes (Scheme 10) provides a general entry to α,α-disubstituted systems 24.27 Here, 5-exo cyclization of 23 generates alkyl-Pd(II) intermediate 25, which can only undergo β-hydride elimination via C7–H to deliver chiral product 24. The process tolerates sterically and electronically diverse substituents at R1 and R2, providing an efficient approach to challenging tetra-substituted nitrogen-bearing stereocentres. Ultimately, this study served as the basis for the development of enantioselective variants, as described later.
 | ||
| Scheme 10 Aza-Heck cyclization of oxime esters with 1,1-disubstituted alkenes. | ||
In the studies outlined above, which involve cyclic or 1,1-disubstituted alkenes, the nature of the alkene acceptor enforces the formation of chiral products (new C(sp3)–N bond). For processes involving 1,2-disubstituted alkenes, 5-exo cyclization generates alkyl-Pd(II) intermediate 27, from which β-hydride elimination can occur via either Ha or Hb (Scheme 11). The former pathway leads to pyrrole products; Narasaka has shown that cyclisation of 26 using Pd(PPh3)4 affords pyrrole 28 with 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity over dihydropyrrole 29 (73% combined yield).5
:1 selectivity over dihydropyrrole 29 (73% combined yield).5
 | ||
| Scheme 11 β-Hydride elimination pathways in aza-Heck cyclizations with 1,2-disubstituted alkenes. | ||
To provide selectivity for chiral products in processes involving 1,2-disubstituted alkenes, strategic installation of an aryl ring at R2 was explored (Scheme 12).28,29 This approach proved effective, such that a variety of dihydropyrroles 31a–e were generated from oxime esters 30a–e with high selectivity over the corresponding pyrroles 32a–e. For systems with substitution at C4 (e.g.31e) good levels of 1,2-diastereocontrol were observed, whereas C3-methyl substituted product 31d was formed in low diastereoselectivity. Gratifyingly, extension of the method to systems where R2 = alkyl also resulted in selective formation of chiral products (e.g.30f to 31f). Here, studies on the effects of different ligands revealed that the relatively wide cone angle of P(3,5-(CF3)2C6H3)3 is responsible for preferential β-hydride elimination via Hb.
 | ||
| Scheme 12 Aza-Heck cyclizations of oxime esters with 1,2-disubstituted alkenes; the ratio of dihydropyrrole 31:pyrrole 32 is shown in brackets below the isolated yield of pure 31. | ||
The dihydropyrrole products accessed in Scheme 12 are ideally suited to diastereoselective manipulations (Scheme 13). Indeed, hydrogenation of the imine of 31a afforded pyrrolidine 33 in 95% yield and 10:1 d.r. Similarly, allyl addition (allylMgCl) to 31a occurred from the less hindered face of the imine to provide 34 in high diastereoselectivity (>20:1 d.r.). Bicyclic systems common to many indolizidine alkaloids can also be constructed. For example, DIBAL-H reduction of the imine of 31a was followed by N-butenylation and ring-closing metathesis to afford bicyclic system 35 in high diastereopurity. More stereochemically complex aza-Heck products can also be manipulated, with DIBAL-H reduction of 31e occurring from the face opposite the C2-substituent to provide complex pyrrolidine 36 in >20:1 d.r.
 | ||
| Scheme 13 Chiral N-heterocycles accessible from dihydropyrrole product. | ||
Imino-palladation versus iminyl radical cyclization in Narasaka–Heck reactions and a copper-catalyzed protocol
The studies described so far highlight the importance of electron-deficient ligands for efficient aza-Heck cyclization. In part, this may be due to an enhancement of alkene migratory insertion at the stage of the imino-Pd(II) intermediate. However, another key benefit resides in the mechanistic control that such ligands offer for the N–O oxidative addition step. In early studies, we found that when common electron-rich ligand systems (e.g. 1,1′-bis(di-tert-butylphosphino)ferrocene (dt-bpf), P(t-Bu)3, PCy3etc.) were used for cyclization of oxime ester 14 or pivaloyl variant 37, aza-Heck product 15 was not generated and instead saturated system 40 formed (Scheme 14).30 Although 40 is formally a reductive aza-Heck product, a series of mechanistic experiments support an alternate radical-based reaction pathway. Indeed, the yield of 40 is enhanced by the inclusion of common hydrogen atom donors, such as γ-terpinene, whereas hydride donors offer no appreciable benefit. Our collective studies suggest that single-electron transfer from the electron-rich Pd(0)-catalyst to 14 or 37 forms iminyl radical 38, with concomitant generation of Pd(I) and carboxylate anion. Fast 5-exo cyclization then affords alkyl radical 39, which can abstract a hydrogen atom from γ-terpinene to generate ‘reductive’ aza-Heck product 40 and bis-allylic radical 41. Hydrogen-atom transfer from 41 to Pd(I) affords p-cymene and a Pd(II)-hydride, which undergoes base-promoted reductive elimination to regenerate the Pd(0)-catalyst. The process outlined in Scheme 14A is generally applicable, with the unusual hybrid organometallic-radical protocol31 providing an appealing entry to iminyl radical chemistry.32 Indeed, products 40a–c were all accessed efficiently using this approach (Scheme 14B). | ||
| Scheme 14 Reductive aza-Heck reaction via iminyl radical cyclization. | ||
Overall, the ligand effects revealed during our studies highlight important factors that may impact other processes involving oxidative addition of Pd(0) into the N–O bond of oxime esters. Specifically, it appears that electron-neutral or electron-poor Pd(0)-systems undergo N–O oxidative addition via a concerted two-electron redox process, whereas electron-rich Pd-systems can deviate into a SET pathway. For the process in Scheme 14, this pathway is interrupted by fast cyclization of the iminyl radical onto the pendant alkene. In systems without an alkene acceptor, stoichiometric studies from the groups of Hartwig9 and Stahl10 have shown that electron-rich Pd-systems (e.g. Pd(0)/PCy3) lead to imino-Pd(II) intermediates. In these cases, it is possible that SET oxidative addition is operative. The mechanistic divergence observed for different phosphine ligands during N–O oxidative addition is reminiscent of the substrate dependent mechanistic divergence often observed for oxidative addition of Pd(0) into aryl iodides vs. alkyl iodides (concerted vs. SET).31
In Scheme 14B, iminyl radical cyclization leads to products lacking an alkene. However, under copper-catalyzed conditions we have found that Heck-like products (i.e. those with an alkene) can be accessed using iminyl radical-based cyclizations (Scheme 15).13b,33 This finding is significant as it allows the replacement of a rare-earth transition metal catalyst with an isoelectronic first-row system. Our studies in this area were inspired, in part, by reports from Liebeskind and co-workers, who harnessed copper-based systems to activate the N–O bonds of oxime esters.34 Under optimized conditions, Cu(2-ethylhexanoate)2-catalyzed cyclization of a range of pivaloyl oxime esters 42 occurs in good to excellent yields (Scheme 15A). The reaction tolerates cyclic, 1,1-disubstituted and 1,2-disubstituted alkenes, demonstrating similar scope to the aforementioned Pd-catalyzed protocols. For comparative purposes, the yields obtained for Pd-catalyzed aza-Heck cyclization of the corresponding O-pentafluorobenzoyl oxime ester are included in Scheme 15A. Mechanistic studies suggest that Cu(I)-carboxylate (generated in situ) induces N–O homolysis of oxime ester 42a, with the resulting iminyl radical cyclizing to alkyl radical 43 (Scheme 15B). Cu(II)-carboxylate then traps 43 to generate alkyl-Cu(III) intermediate 44. As supported by studies from Kochi and co-workers,35 alkyl-Cu(III) intermediate 44 then undergoes oxidative elimination, via transition state 45, to afford product 29 with concomitant regeneration of the active Cu(I)-catalyst.
 | ||
| Scheme 15 Copper-catalyzed aza-Heck-like cyclization via an iminyl radical. | ||
New N–O donors: aza-Heck cyclizations of N-(pentafluorobenzoyloxy)sulfonamides and O-phenyl hydroxamates
A key limitation of the Narasaka–Heck protocol is that, in general, aldoxime esters are not suitable substrates. For example, in the attempted aza-Heck cyclization of 46, target product 48 was not observed with nitrile side product 47 forming instead (Scheme 16A). Two key factors contribute to preferential formation of 47: (a) access to reactive (Z)-imino-Pd(II) intermediate 49 is not enforced by the small hydrogen substituent of aldoxime 46, and (b) deleterious β-hydride elimination from sterically favored (E)-imino-Pd(II) intermediate 50 is driven by the formation of the relatively stable nitrile 47. To facilitate cyclizations of systems possessing hydrogen substituents adjacent to nitrogen, aza-Heck cyclizations of non-oxime derived N–O donors were sought (Scheme 16B). Specifically, cyclizations of N-pentafluorobenzoyloxysulfonamides 51 (to bicycle 53) were considered appealing, because, at the stage of aza-Pd(II) intermediate 52, β-hydride elimination generates a less thermodynamically stable imine (54), such that this detrimental pathway was expected to be slower.20 A further benefit of the approach is that the substrates can be prepared directly by Mitsunobu reaction of the appropriate alcohol with N-(pentafluorobenzoyloxy)sulfonamide reagents, which we have found easy to prepare on multi-gram scale. As such, the overall sequence allows the two-step conversion of bis-homoallylic alcohols to chiral pyrrolidine targets. | ||
| Scheme 16 Development of alternate N–O bond donors for aza-Heck cyclizations. | ||
In the event, the approach outlined in Scheme 16B was successful, providing efficient access to a diverse array of heterocyclic ring systems (Scheme 17). The optimized catalyst system is the same as that described earlier, with fine-tuning of reaction solvent required on a case-by-case basis. Notable features of the process include: (a) selective β-hydride elimination away from nitrogen for cyclizations involving 1,2-disubstituted alkenes (e.g.57a–b), (b) excellent diastereoselectivities for cyclizations of substrates bearing substituents α to nitrogen (e.g.57c–d) and (c) the ability to promote challenging 6-exo cyclizations (e.g.57f). Furthermore, transannular cyclizations are also possible; cyclization of 58 provide tropane ring system 59 in high yield. Mechanistic studies support a reaction pathway analogous to the Narasaka–Heck reaction (see Scheme 7), with protodecarboxylation of the pentafluorobenzoate leaving group once again playing a key role.
 | ||
| Scheme 17 Aza-Heck cyclizations of N-(pentafluorobenzoyloxy)sulfonamides. | ||
Conceptually, the approach outlined in Scheme 16B is of broader interest because it uses sulfonamides 55 as bifunctional amino reagents. Specifically, the Mitsunobu step exploits the nucleophilic character of 55, whereas the Pd-catalyzed aza-Heck step harnesses the electrophilic reactivity of the N–O bond. As such, further nucleophilic–electrophilic sequences could be envisaged with 55, perhaps leading to a diverse array of methodologies. Studies towards this broad goal are ongoing.
Recently, Watson and co-workers reported a third distinct class of aza-Heck reaction, based on cyclizations of O-phenyl hydroxamates 60 (Scheme 18).21 The choice of an O-phenyl leaving group was crucial for efficient cyclization, as it suppressed competing Lossen rearrangement of 60. Optimized conditions employ a Pd-system modified with the highly electron deficient phosphite P(OCH2CF3)3. Using this system, 5-exo cyclizations involving a wide range of di-, tri- and tetra-substituted alkenes were all effective, providing the targets in moderate to excellent yields. 6-Exo cyclizations are also achievable but require conformationally predisposed substrates (e.g.61f). Note that a secondary O-phenyl hydroxamate is required (R(CO)NHOPh), and N-alkylated systems are not tolerated. The proposed mechanism closely follows those outlined already, with oxidative addition of Pd(0) into the N–O bond of 60 occurring in advance of syn-stereospecific alkene amino-palladation; a series of experiments supported this latter proposition. In contrast to the processes described earlier, it is unclear whether or not this class of aza-Heck cyclization occurs via a cationic aza-Pd intermediate. Indeed, compared to pentafluorobenzoate, phenoxide is expected to dissociate much less readily, such that in the Watson process cyclization likely occurs in “neutral mode”.
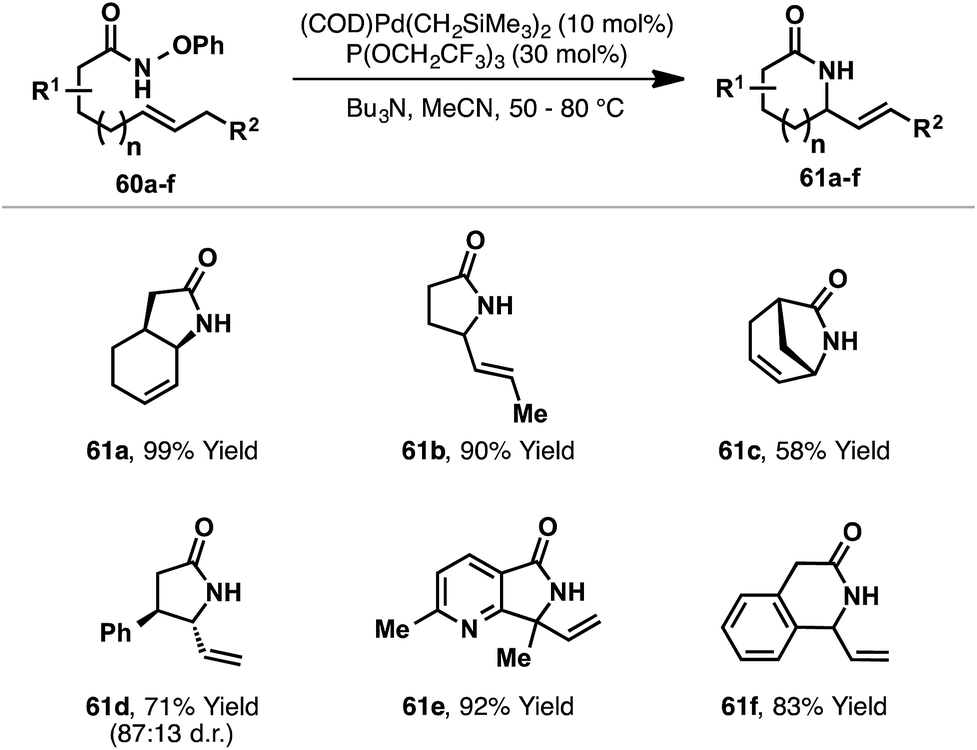 | ||
| Scheme 18 Aza-Heck cyclizations of O-phenyl hydroxamates. | ||
The processes outlined in Schemes 17 and 18 could also potentially be achieved using an aza-Wacker approach, wherein the allylic amine targets are generated by Pd(II)-catalyzed cyclization of an NH nucleophile with an alkene under oxidative conditions.4 Although often efficient, this approach suffers from several limitations. For example, few cyclizations of primary amides are known under aza-Wacker conditions (cf.Scheme 18),36 highlighting an inherent limitation of the method. With respect to aza-Wacker cyclizations involving NH-sulfonamides (cf.Scheme 17): large α-substituents are not tolerated (cf.57d), hindered acyclic olefins do not participate (cf.57d) and electron-deficient alkenes cannot be used due to competing conjugate addition (cf.57e).37 Furthermore, NH-sulfonamides required for aza-Wacker cyclization are not prepared directly from the alcohol because the requisite primary sulfonamides do not engage efficiently in conventional Mitsunobu reactions (cf.55 to 51). Additional advantages of the aza-Heck approach include predictable syn-migratory insertion of the alkene37c and the ability to use tunable phosphine ligands (because oxidative conditions are avoided).
Alkene 1,2-carboaminations via aza-Heck cascades
In all of the aza-Heck processes discussed so far, the alkyl-Pd(II) intermediate formed after alkene migratory insertion undergoes β-hydride elimination to generate a new alkene. Further synthetic flexibility is potentially enabled by harnessing this intermediate for processes other than β-hydride elimination.38,39 Such an approach provides an appealing mechanistic framework for the development of synthetically valuable alkene 1,2-carboamination reactions.40It has been shown that cascade aza-Heck reactions are possible by using specifically designed substrates, bearing multiple alkenes. In 2001, Narasaka reported a series of processes leading to spirocyclic imines (Scheme 19A).41 In one example, aza-Heck–Heck cyclization of oxime ester 62 generated spirocyclic system 64 in 82% yield. Initial imino-palladation of the 1,1-disubstituted alkene affords intermediate 63, where β-hydride elimination is not possible. As such, a second migratory insertion can occur, this time involving the mono-substituted alkene, to deliver ultimately spirocycle 64. Similar cascade reactions have been reported using more recently developed N–O donors (Scheme 19B and C).20,21
 | ||
| Scheme 19 Synthesis of spirocyclic N-heterocycles via cascade aza-Heck cyclizations. | ||
The processes in Scheme 19 involve intramolecular capture of the alkyl-Pd(II) intermediate. Related intermolecular processes offer further flexibility. For example, our group has demonstrated carbonylative cascades involving a range of C-based or alcohol nucleophiles.26 Under a CO atmosphere, exposure of oxime ester 65 and Ph4B·NHEt3 to a P(3,5-(CF3)2C6H3)3 ligated Pd(0)-catalyst effected cyclization to target 66 in 62% yield. The proposed mechanism involves cyclization and carbonylation to afford cationic acyl-Pd(II) intermediate 67, which is intercepted by the pre-activated tetraarylborate (to give 68), leading to product 66. The choice of a tetraarylborate with a triethylammonium counterion was critical, with this latter component providing the proton required to trigger protodecarboxylation of the otherwise inhibitory pentafluorobenzoate leaving group (cf.Scheme 7). Note that in the oxime ester-based processes described earlier, the necessary proton is generated via the β-hydride elimination step. The carboamination protocol could be extended to other systems (e.g.69a–e), and alcohols and phenols were also found to be competent nucleophiles, leading to ester products (e.g.69f).26,42
Related carbonylative cascades involving alkynyl or vinyl boronates provide ynone and enones targets 70 and 71, respectively (Scheme 21A). Here, the addition of a protic additive was not required, which suggests that the pentafluorobenzoate leaving group is sequestered as FBzO-BPin, via Suzuki-like transmetallation from neutral acyl-Pd(II) intermediate 72. Similarly, under non-carbonylative conditions, aryl, vinyl or alkynyl boronates participated to provide 1,2-carboamination products 74a–f from cyclization of oxime esters 73 (Scheme 21B).
The processes in Schemes 20 and 21 demonstrate that N–O oxidative addition can serve as the basis for a diverse suite of alkene 1,2-carboamination reactions. Specifically, 1,2-amino-acylation, -carboxylation, -vinylation, -arylation and -alkynylation reactions are all achieved within a unified mechanistic framework. Other redox-neutral Pd-catalyzed methods for alkene 1,2-aminoarylation exploit oxidative addition into the incoming C-based fragment,40b,c,e,f and cannot access carbonylative products such as 66. Conversely, carbonylative aza-Wacker cyclizations, which are oxidative processes, provide 1,2-aminocarboxylation products (cf.69f), but do not tolerate organometallic nucleophiles because these effect reduction of the active Pd(II)-catalyst.40d Accordingly, this manifold does not provide access to, for example, 1,2-aminoarylation products. Given the new aza-Heck N–O donors that have recently been developed (vide supra), it is likely that further variants of the “umpoled” alkene 1,2-carboamination strategy summarized here will emerge in the near future.
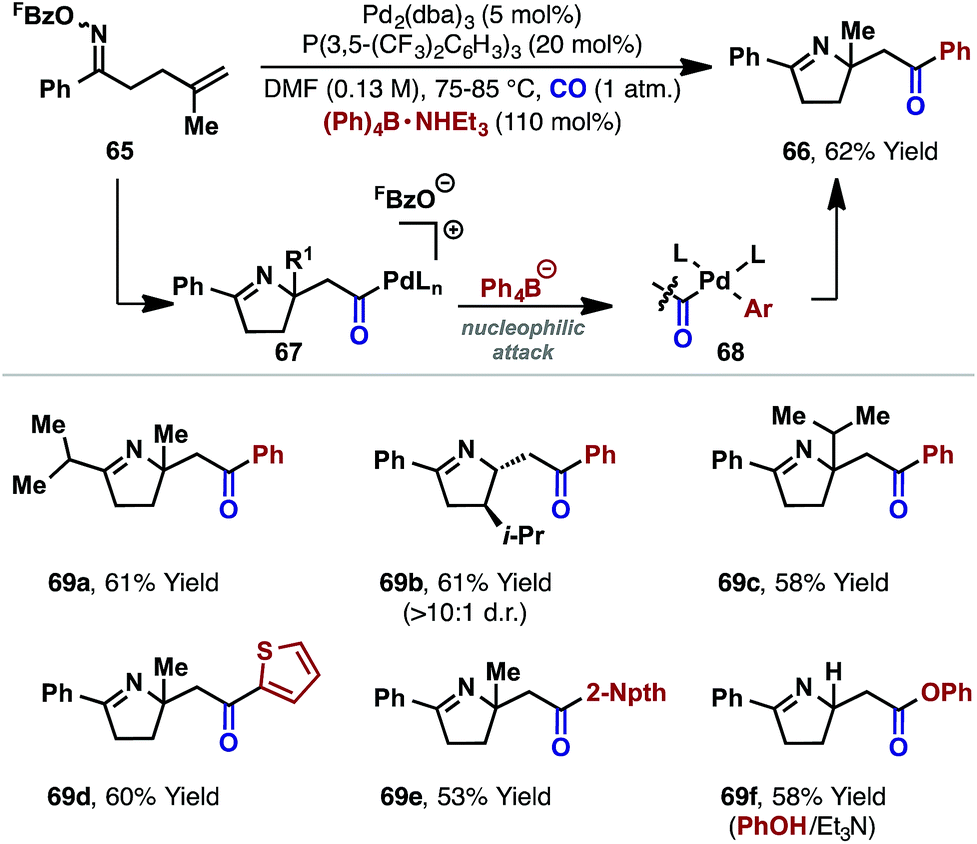 | ||
| Scheme 20 Carbonylative alkene 1,2-aminoacylation reactions. | ||
 | ||
| Scheme 21 Further classes of 1,2-carboamination reaction. | ||
Enantioselective aza-Heck reactions
From the discussion so far, it is clear that aza-Heck reactions provide convenient and flexible access to a diverse range of chiral N-heterocycles. Given the prevalence of stereodefined C–N bonds in natural products and pharmaceuticals, the development of enantioselective aza-Heck cyclizations became a key focus of our group. As highlighted above, aza-Heck cyclizations have relatively strict ligand requirements, such that the identification of effective chiral ligands was challenging. After extensive investigations, we found that SPINOL-derived P,N-ligand (Sa,S)-75 (Fig. 1) could promote highly enantioselective aza-Heck cyclizations of oxime esters with 1,1-disubstituted alkenes (Scheme 22).43,44 Presumably, the weakly donating oxazoline moiety is able to mimic the electronic effects of achiral electron-deficient triaryl phosphines used earlier (see Scheme 6B). | ||
| Fig. 1 SPINOL-derived P,N-ligand (Sa,S)-75 for enantioselective aza-Heck cyclizations. | ||
 | ||
| Scheme 22 An enantioselective aza-Heck reaction. | ||
Using ligand (Sa,S)-75, we found that enantioselective aza-Heck cyclization is efficient with a range of sterically diverse 1,1-disubstituted alkenes, generating targets 24a–f in 68–86% yield and 91:9–95:5 e.r. (Scheme 22A). Notably, systems with large or small groups at R2 cyclized with similar levels of efficiency (e.g.25f, R2 = Me, 94:6 e.r. vs24f, R2 = iPr, 93:7 e.r.). A stereochemical model that accounts for this observation is outlined in Scheme 22B. The imino group resides trans to the oxazoline and the alkene coordinates cis to this unit. In this arrangement, the alkene favors an orientation where the –CH2R3 substituent is positioned away from the phenyl substituent of the oxazoline. In this model, there are minimal steric interactions between the R2 substituent and the chiral ligand, which accounts for the similar enantioselectivities observed for 24d, 24e and 24f.
The enantioenriched dihydropyrrole products are suitable for derivatization via either the imine or alkene moieties (Scheme 22C). Exhaustive hydrogenation of 24d provided chiral α-tertiary amine 76 in 97% yield, whereas DIBAL-H reduction of imine 24f generated pyrrolidine 77 with good levels of diastereocontrol. The cyclizations in Scheme 22A are the first examples of highly enantioselective aza-Heck reactions, adding to a rare yet powerful class of processes that involve enantioselective migratory insertion of alkenes into Pd–N bonds.4,8,45 The development of further enantioselective aza-Heck cyclizations, including processes that use other N–O donors, is the focus of ongoing efforts.
Conclusions
This perspective summarizes recent progress in the use of aza-Heck cyclizations for the synthesis of chiral nitrogen heterocycles. Building on Narasaka's seminal report, and aided by a detailed interrogation of reaction mechanism, highly efficient cyclizations of oxime esters with cyclic, 1,1- and 1,2-disubstituted alkenes have been developed. Our collective studies have highlighted key ligand requirements and provided a rationalization for the privileged role of pentafluorobenzoate leaving groups in such processes. This information guided the design of alkene 1,2-carboamination processes that are initiated by N–O oxidative addition of Pd(0)-catalysts. This fundamental mechanistic step is key to the work described here, and Narasaka's observation of this unusual process heralded the development of a wide range of N–O based processes outside the immediate area of aza-Heck chemistry. Indeed, imino-Pd(II) intermediates now underpin a diverse array of redox-neutral processes including alkene 1,2-carboaminations,26 aryl C–H aminations,9 alkene aziridinations,46 alkene 1,2-iodoaminations,47 aryne aminofunctionalizations,38 and C–C bond activations.25 Exciting recent developments in aza-Heck chemistry have extended N–O oxidative addition to other N–O donors; this is an area that has great potential. Indeed, recent examples of highly enantioselective aza-Heck processes set the stage for the development of a family of redox-neutral enantioselective C–N bond forming processes that are initiated by oxidative addition at electrophilic nitrogen sources.Acknowledgements
We thank the EPSRC (EP/J007455/1) for financial support and Ph.D. studentships (to A. F. and I. R. H). A. F. and I. R. H. thank AstraZeneca for partial support, and N. J. R. thanks the Bristol Chemical Synthesis Doctoral Training Centre, funded by the EPSRC (EP/G036764/1), for a Ph.D. studentship. N. J. R. also thanks the SCI for a postgraduate scholarship. J. F. B. is indebted to the Royal Society for a University Research Fellowship.Notes and references
- http://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/TopUSPharmaceuticalProductsof2013.pdf, accessed on 28th March 2017.
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (b) A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS PubMed; (c) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- (a) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed; (b) N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trindade, Chem. Rev., 2009, 109, 2703 CrossRef CAS PubMed.
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed.
- (a) H. Tsutsui and K. Narasaka, Chem. Lett., 1999, 28, 45 CrossRef; (b) H. Tsutsui, M. Kitamura and K. Narasaka, Bull. Chem. Soc. Jpn., 2002, 75, 1451 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed.
- (a) M. Kitamura and K. Narasaka, Chem. Rec., 2002, 2, 268 CrossRef CAS PubMed; (b) K. Narasaka and M. Kitamura, Eur. J. Org. Chem., 2005, 4505 CrossRef CAS.
- For a review that encompasses processes involving migratory insertion into Pd–N bonds, see: P. S. Hanley and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 8510 CrossRef CAS PubMed.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef CAS PubMed.
- W. P. Hong, A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 13664 CrossRef CAS PubMed.
- (a) J.-L. Zhu and Y.-H. Chan, Synlett, 2008, 1250 CAS; (b) J.-L. Zhu, Y.-L. Su, Y.-H. Chan, I.-C. Chen and C.-C. Liao, Heterocycles, 2009, 78, 369 CrossRef CAS; (c) S.-W. Tsao and J.-L. Zhu, Heterocycles, 2012, 85, 383 CrossRef CAS.
- M. Kitamura, H. Yanagisawa, M. Yamane and K. Narasaka, Heterocycles, 2005, 65, 273 CrossRef CAS.
- (a) J. Helaja and R. Göttlich, Chem. Commun., 2002, 720 RSC; (b) A. Faulkner, N. J. Race, J. S. Scott and J. F. Bower, Chem. Sci., 2014, 5, 2416 RSC.
- M. Kitamura, D. Kudo and K. Narasaka, ARKIVOC, 2006, 3, 148 Search PubMed.
- S. Zaman, K. Mitsuru and A. D. Abell, Org. Lett., 2005, 7, 609 CrossRef CAS PubMed.
- H. Tsutsui and K. Narasaka, Chem. Lett., 2001, 30, 526 CrossRef.
- (a) M. Kitamura, S. Chiba, O. Saku and K. Narasaka, Chem. Lett., 2002, 31, 606 CrossRef; (b) S. Chiba, M. Kitamura, O. Saku and K. Narasaka, Bull. Chem. Soc. Jpn., 2004, 77, 785 CrossRef CAS.
- (a) J. Ichikawa, K. Sakoda, J. Mihara and N. Ito, J. Fluorine Chem., 2006, 127, 489 CrossRef CAS; (b) J. Ichikawa, R. Nadano and N. Ito, Chem. Commun., 2006, 4425 RSC; (c) K. Sakoda, J. Mihara and J. Ichikawa, Chem. Commun., 2005, 4684 RSC.
- For a review on the use of electrophilic nitrogen sources in transition metal cross-coupling reactions, see: T. J. Barker and E. R. Jarvo, Synthesis, 2011, 3954 CAS.
- I. R. Hazelden, X. Ma, T. Langer and J. F. Bower, Angew. Chem., Int. Ed., 2016, 55, 11198 CrossRef CAS PubMed.
- S. A. Shuler, G. Yin, S. B. Krause, C. M. Vesper and D. A. Watson, J. Am. Chem. Soc., 2016, 138, 13830 CrossRef CAS PubMed.
- A. Faulkner and J. F. Bower, Angew. Chem., Int. Ed., 2012, 51, 1675 CrossRef CAS PubMed.
- (a) A. Fürstner, K. Radkowski, H. Peters, G. Seidel, C. Wirtz, R. Mynott and C. W. Lehmann, Chem.–Eur. J., 2007, 13, 1929 CrossRef PubMed; (b) A. Fürstner, K. Radkowski and H. Peters, Angew. Chem., Int. Ed., 2005, 44, 2777 CrossRef PubMed.
- (a) P. S. Hanley and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 15661 CrossRef CAS PubMed; (b) P. S. Hanley, D. Marković and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 6302 CrossRef CAS PubMed . See also ref. 8.
- (a) T. Nishimura, Y. Nishiguchi, Y. Maeda and S. Uemura, J. Org. Chem., 2004, 69, 5342 CrossRef CAS PubMed; (b) T. Nishimura and S. Uemura, J. Am. Chem. Soc., 2000, 122, 12049 CrossRef CAS . See also, ref. 9 and 10.
- A. Faulkner, J. S. Scott and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 7224 CrossRef CAS PubMed.
- A. Faulkner, J. S. Scott and J. F. Bower, Chem. Commun., 2013, 49, 1521 RSC.
- N. J. Race and J. F. Bower, Org. Lett., 2013, 15, 4616 CrossRef CAS PubMed.
- For studies that delineate factors responsible for β-hydride elimination selectivity from alkyl-Pd(ii) intermediates, see: (a) E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 9692 CrossRef CAS PubMed; (b) E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 13981 CrossRef CAS PubMed.
- N. J. Race, A. Faulkner, M. H. Shaw and J. F. Bower, Chem. Sci., 2016, 7, 1508 RSC.
- For a comprehensive review, see: (a) U. Jahn, Top. Curr. Chem., 2012, 320, 323 CrossRef CAS PubMed. For selected recent examples of Pd-catalyzed hybrid organometallic radical processes that employ alkyl iodides: (b) K. S. Bloome and E. J. Alexanian, J. Am. Chem. Soc., 2010, 132, 12823 CrossRef CAS PubMed; (c) H. Liu, Z. Qiao and X. Jiang, Org. Biomol. Chem., 2012, 10, 7274 RSC; (d) B. M. Monks and S. P. Cook, Angew. Chem., Int. Ed., 2013, 52, 14214 CrossRef CAS PubMed; (e) E. R. Fruchey, B. M. Monks, A. M. Patterson and S. P. Cook, Org. Lett., 2013, 15, 4362 CrossRef CAS PubMed; (f) C. M. McMahon and E. J. Alexanian, Angew. Chem., Int. Ed., 2014, 53, 5974 CrossRef CAS PubMed; (g) A. R. O. Venning, P. T. Bohan and E. J. Alexanian, J. Am. Chem. Soc., 2015, 137, 3731 CrossRef CAS PubMed.
- (a) J. C. Walton, Acc. Chem. Res., 2014, 47, 1406 CrossRef CAS PubMed; (b) S. Z. Zard, Synlett, 1996, 1148 CrossRef CAS; (c) K. Narasaka and M. Kitamura, ARKIVOC, 2006, 7, 245 Search PubMed.
- For Cu-catalyzed radical cyclization of activated hydroxysulfonamides with alkenes, see: (a) W.-M. Liu, Z.-H. Liu, W.-W. Cheong, L.-Y. T. Priscilla, Y. Li and K. Narasaka, Bull. Korean Chem. Soc., 2010, 31, 563 CrossRef CAS. For an aza-Heck type reaction of oxime esters promoted by stoichiometric Cu(OAc)2, see: (b) M. Bingham, C. Moutrille and S. Z. Zard, Heterocycles, 2014, 88, 953 CrossRef CAS.
- (a) S. Liu, Y. Yu and L. S. Liebeskind, Org. Lett., 2007, 9, 1947 CrossRef CAS PubMed; (b) S. Liu and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 6918 CrossRef CAS PubMed.
- (a) J. K. Kochi, J. Am. Chem. Soc., 1962, 84, 3271 CrossRef CAS; (b) J. K. Kochi, A. Bemis and C. L. Jenkins, J. Am. Chem. Soc., 1968, 90, 4616 CrossRef CAS; (c) C. L. Jenkins and J. K. Kochi, J. Am. Chem. Soc., 1972, 94, 843 CrossRef CAS; (d) C. L. Jenkins and J. K. Kochi, J. Am. Chem. Soc., 1972, 94, 856 CrossRef CAS.
- Q. Lui, E. M. Ferreira and B. M. Stoltz, J. Org. Chem., 2007, 72, 7352 CrossRef PubMed.
- For selected methods, see: (a) M. Rönn, J.-E. Backväll and P. G. Andersson, Tetrahedron Lett., 1995, 36, 7749 CrossRef; (b) R. I. McDonald, P. B. White, A. B. Weinstein, C. P. Tam and S. S. Stahl, Org. Lett., 2011, 13, 2830 CrossRef CAS PubMed; (c) Aza-Wacker cyclizations proceed via condition-dependent syn- or anti-amino-palladation pathways, see: ; (d) G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2007, 129, 6328 CrossRef CAS PubMed . For a review encompassing enantioselective aza-Wacker cyclizations, see ref. 4.
- T. Gerfaud, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 572 CrossRef CAS PubMed.
- M. Kitamura, Y. Moriyasu and T. Okauchi, Synlett, 2011, 643 CrossRef CAS.
- For recent reviews that encompass Pd-catalyzed alkene 1,2-carboamination and related processes, see: (a) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed; (b) J. P. Wolfe, Synlett, 2008, 2913 CrossRef CAS; (c) D. M. Schultz and J. P. Wolfe, Synthesis, 2012, 44, 351 CrossRef CAS PubMed; 1,2-aminoacylation: (d) T. A. Cernak and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 3124 CrossRef CAS PubMed; 1,2-aminoarylation: (e) C. F. Rosewall, P. A. Sibbald, D. V. Liskin and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 9488 CrossRef CAS PubMed; (f) P. A. Sibbald, C. F. Rosewall, R. D. Swartz and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 15945 CrossRef CAS PubMed; 1,2-aminoarylation: (g) B. A. Hopkins and J. P. Wolfe, Chem. Sci., 2014, 5, 4840 RSC, and references cited therein; (h) S. Hayashi, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2009, 48, 7224 CrossRef CAS PubMed; (i) L. Bagnoli, S. Cacchi, G. Favrizi, A. Goggiomani, C. Scarponi and M. Tiecco, J. Org. Chem., 2010, 75, 2134 CrossRef CAS PubMed; 1,2-aminoalkynylation: (j) S. Nicolai, R. Sedigh-Zadeh and J. Waser, J. Org. Chem., 2013, 78, 3783 CrossRef CAS PubMed. For an intermolecular oxidative Pd-catalyzed 1,2-aminocarboxylation reaction, see: (k) J. Cheng, X. Qi, P. Chen and G. Liu, J. Am. Chem. Soc., 2015, 137, 2480 CrossRef CAS PubMed.
- (a) M. Kitamura, S. Zaman and K. Narasaka, Synlett, 2001, 974 CrossRef CAS; (b) S. Zaman, M. Kitamura and K. Narasaka, Bull. Chem. Soc. Jpn., 2003, 76, 1055 CrossRef CAS.
- S. Zaman, M. Kitamura and A. D. Abell, Aust. J. Chem., 2007, 60, 624 CrossRef CAS.
- N. J. Race, A. Faulkner, G. Fumagalli, T. Yamauchi, J. S. Scott, M. Rydén-Landergren, H. A. Sparkes and J. F. Bower, Chem. Sci., 2017, 8, 1981 RSC.
- This ligand class is effective for Ir-catalyzed enantioselective hydrogenation, see: (a) C. Guo, D.-W. Sun, S. Yang, S.-J. Mao, X.-H. Xu, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2015, 137, 90 CrossRef CAS PubMed; (b) S.-F. Zhu, J.-B. Xie, Y.-Z. Zhang, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886 CrossRef CAS PubMed; (c) S. Li, S.-F. Zhu, C.-M. Zhang, S. Song and Q.-L. Zhou, J. Am. Chem. Soc., 2008, 130, 8584 CrossRef CAS PubMed.
- For examples involving enantioselective redox-neutral 1,2-carboamination of alkenes, see: (a) D. Y. Mai and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 12157 CrossRef CAS PubMed; (b) B. A. Hopkins and J. P. Wolfe, Angew. Chem., Int. Ed., 2012, 51, 9886 CrossRef CAS PubMed; (c) Y. Miyazaki, N. Ohta, K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 3732 CrossRef CAS PubMed. For examples involving enantioselective oxidative 1,2-carboaminations of alkenes, see: (d) K.-T. Yip, M. Yang, K.-L. Law, N.-Y. Zhu and D. Yang, J. Am. Chem. Soc., 2006, 128, 3130 CrossRef CAS PubMed; (e) W. He, K.-T. Yip, N.-Y. Zhu and D. Yang, Org. Lett., 2009, 11, 5625 Search PubMed. For examples involving enantioselective 1,2-diaminations of dienes and trienes, see: (f) H. Du, W. Yuan, B. Zhao and Y. Shi, J. Am. Chem. Soc., 2007, 129, 11688 CrossRef CAS PubMed; (g) L. Xu and Y. Shi, J. Org. Chem., 2008, 73, 749 CrossRef CAS PubMed. For examples involving enantioselective aza-Wacker cyclizations, see: (h) A. B. Weinstein and S. S. Stahl, Angew. Chem., Int. Ed., 2012, 51, 11505 CrossRef CAS PubMed and ref. 37b. For enantioselective Cu-catalyzed 1,2-carboaminations of alkenes, see: (i) T. W. Liwosz and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 2020 CrossRef CAS PubMed.
- K. Okamoto, T. Oda, S. Kohigashi and K. Ohe, Angew. Chem., Int. Ed., 2011, 40, 11470 CrossRef PubMed.
- C. Chen, L. Hou, M. Cheng, J. Su and X. Tong, Angew. Chem., Int. Ed., 2015, 54, 3092 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |