
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimizing side chains for crystal growth from water: a case study of aromatic amide foldamers†
Xiaobo
Hu
 ,
Simon J.
Dawson
,
Pradeep K.
Mandal
,
Xavier
de Hatten
,
Benoit
Baptiste‡
and
Ivan
Huc
*
,
Simon J.
Dawson
,
Pradeep K.
Mandal
,
Xavier
de Hatten
,
Benoit
Baptiste‡
and
Ivan
Huc
*
Université de Bordeaux, CNRS, IPB, CBMN, UMR 5248, Institut Européen de Chimie et Biologie, 2 Rue Escarpit, 33600 Pessac, France. E-mail: i.huc@iecb.u-bordeaux.fr
First published on 8th March 2017
Abstract
The growth of crystals of aromatic compounds from water much depends on the nature of the water solubilizing functions that they carry. Rationalizing crystallization from water, and structure elucidation, of aromatic molecular and supramolecular systems is of general value across various fields of chemistry. Taking helical aromatic foldamers as a test case, we have validated several short polar side chains as efficient substituents to provide both solubility in, and crystal growth ability from, water. New 8-amino-2-quinolinecarboxylic acids bearing charged or neutral aminomethyl, carboxymethyl, sulfonic acid, or bis(hydroxymethyl)-methoxy side chains in position 4 or 5, were prepared on a multi gram scale. Fmoc protection of the main chain amine and suitable protections of the side chains ensured compatibility with solid phase synthesis. One tetrameric and five octameric oligoamides displaying these side chains were synthesized and shown to be soluble in water. In all cases but one, crystals were obtained using the hanging drop method, thus validating the initial design principle to combine polarity and rigidity. The only case that resisted crystallization appeared to be due to exceedingly high water solubility endowed by eight sulfonic acid functions. The neutral side chain did provide crystal growth ability from water but contributed poorly to solubility.
Introduction
Structure elucidation is a necessary step to understand structure–property relationships and the design of functions at the molecular level. Among other techniques, X-ray crystallographic analysis provides unsurpassed accuracy and stands out as a direct way to gather structural information and to make further design possible. The first and main difficulty of crystallographic analysis is crystal growth. In particular, there is a perception – possibly a misperception – by organic chemists that growing crystals from water is difficult, despite the availability of multiple methods, commercial kits and additives that have been developed to crystallize complex biomacromolecules1 and metallo-organic structures.2 Water is a most relevant medium in which to elicit molecular and supramolecular functions. Indeed, there is a notable trend in supramolecular chemistry and host–guest chemistry to shift from traditional organic solvents to protic media including water.3 If the main structure elucidation method was complicated in water due to difficult crystallogenesis, this would constitute a true bottle neck. In the following, we report our own difficulties in growing crystals of aromatic amide foldamers suitable for X-ray crystallographic analysis from water and how we successfully overcame these difficulties through the design of short and polar side chains that promote both high solubility in water and high crystal growth ability. This work constitutes a case study focused on aromatic helical foldamers, but it certainly has a general value for the crystallization from water of innumerable aromatic molecular and supramolecular systems across various fields of chemistry.In the past two decades, aromatic foldamers and in particular aromatic amide foldamers have emerged as a new class of folded molecular architectures distinct from peptidic and nucleotidic structures.4 Aromatic amide foldamers have been designed to adopt a variety of shapes, including helices,5 macrocycles,6 sheets,7 linear and zig-zag8 ribbons, as well as bundle-like architectures.9 They give access to structures beyond the reach of biopolymers and find applications in, for example, molecular recognition.10 Folding in these systems is driven firstly by polar interactions such as hydrogen bonds and local electrostatic repulsions, and secondly by interactions associated with aromatic stacking. By and large, aromatic amide foldamer structures have been characterized in organic solvents from which they crystallize well. The crystal growth ability of these systems, including some of the largest non-biological molecules ever crystallized,9,11 is assigned to their high conformational stability in organic solvents and to the use of appropriate side chains. In particular, the isobutoxy side chain has proven extremely useful in that it endows foldamers with both high solubility in organic medium and a high crystal growth ability.5b,5d–g,6d,7,9,10a,11 This observation can be related to the numerous crystal structures of lipophilic peptides and pseudo peptides that often contain short branched side chains such as leucine or valine residues.12
Aromatic amide foldamers may be composed of diverse units including benzene, pyridine or anthracene based monomers. Oligoamides of 8-amino-2-quinolinecarboxylic acids (Q) such as those shown in Fig. 1 form very stable helices spanning two turns every five units that have been characterized in solution13 and in the solid state.5b Intrigued by the potential use of these medium-sized objects in a biological context, we and others have started to explore their interactions with nucleic acids14 and proteins15 as well as their cell penetration properties.16 In parallel, other groups have focused on rod-like architectures for protein surface recognition.17 A first step was to endow Qn oligomers, a priori very hydrophobic molecules, with high (millimolar range) solubility in water. This was successfully achieved by introducing cationic (QOrn) or anionic side chains (QAsp),16,18 as well as neutral oligoethylene glycol side chains (QTeg).19 Remarkably, we found that the helical conformations were considerably more stable in methanol than in non-protic organic solvents, and were further stabilized upon adding water presumably due to the hydrophobic component associated with aromatic stacking,19a to eventually result in kinetic inertness of some P and M helices in pure water.19c This property should have been an advantage for growing crystals, but a considerable number of attempts all proved unsuccessful. Regardless of their length, neither anionic, cationic or zwitterionic helical sequences yielded crystals suitable for X-ray analysis. This was despite the use of multiple screens and a great deal of experience including success at solving difficult cases.20 In the best scenarios, crystals were obtained that could be shown to be of the foldamers but that did not diffract sufficiently or were polycrystalline. Eventually, crystal structures of the helical foldamers were obtained from water when these were bound to the surface of a protein or a nucleic acid.14b,15a In the case of linear aromatic foldamers, crystallization from water has been achieved through the assistance of transition metal coordination.21 A few other structures of water soluble oligomers have been reported.22
 | ||
| Fig. 1 Structures of (a) QXxx and (b) Q5Xxx amino acid monomers, and (c) a summary of aromatic amide foldamer sequences 1–6. New side chains developed in this study are shown in red. The terminal 8-nitro group of oligomer 3 replaces the terminal NH function. | ||
We hypothesized that the reason for these failures might be the relative flexibility of the side chains of QAsp and QOrn. Flexible groups create an entropic barrier to crystallization. They give rise to disorder within the crystal lattice and may perturb long range order. This phenomenon is known to protein crystallographers who have developed surface entropy reduction mutagenesis, targeting mainly side chains such as lysines and glutamates.23 In the following, we describe the design and synthesis of new QXxx monomers bearing short and more rigid water solubilizing side chains, their incorporation into octameric or tetrameric helical sequences, and the successful growth of crystals suitable for X-ray diffraction analysis. We find that the removal of a single atom from each side chain may lead to dramatic changes in crystal growth behaviour.
Results and discussion
Monomer design and synthesis
Four new side chains for Q monomers were considered in position 4 and, for two of them, in position 5 as well. For the purpose of the subsequent solid supported synthesis (SPS)18 of the oligoamide sequences, each new monomer was produced as an Fmoc-protected amine having a free carboxylic acid function. Furthermore, in anticipation that the targeted monomers would serve both the purpose of solubility and crystal growth, and thus that their incorporation in foldamer sequences could become frequent, multi gram scale syntheses were developed.Sulfonic acid was identified as a desirable functional group due to the rich background of crystal structures afforded from aryl sulfonates2d,24 including in complex with proteins.25 It is short, rigid, very polar, and anionic over a wide pH range. Among several possible synthetic approaches, two appeared to be suitable to introduce sulfonic acid on 8-amino-2-quinoline carboxylic monomers: thiol oxidation and direct sulfonation. The synthesis was first successful with the latter route thanks to a short and simple synthetic protocol (Scheme 1a). Only three steps were needed from commercially available methyl 8-aminoquinoline-2-carboxylate to monomer 9 which has a sulfonic acid group in position 5 (overall yield 60%). Sulfonation is made easy by the donor effect of the amine in the para position. Unfortunately, this eventually turned into a disadvantage: the reciprocal electron withdrawing effect of the sulfonic acid on the amine dramatically reduced its reactivity. Even under harsh coupling conditions (60 °C, DBU, 9 equiv. of acid chloride, repeated couplings), chain extension from the amine was too inefficient for SPS and the use of 9 was limited to that of an N-terminal monomer.
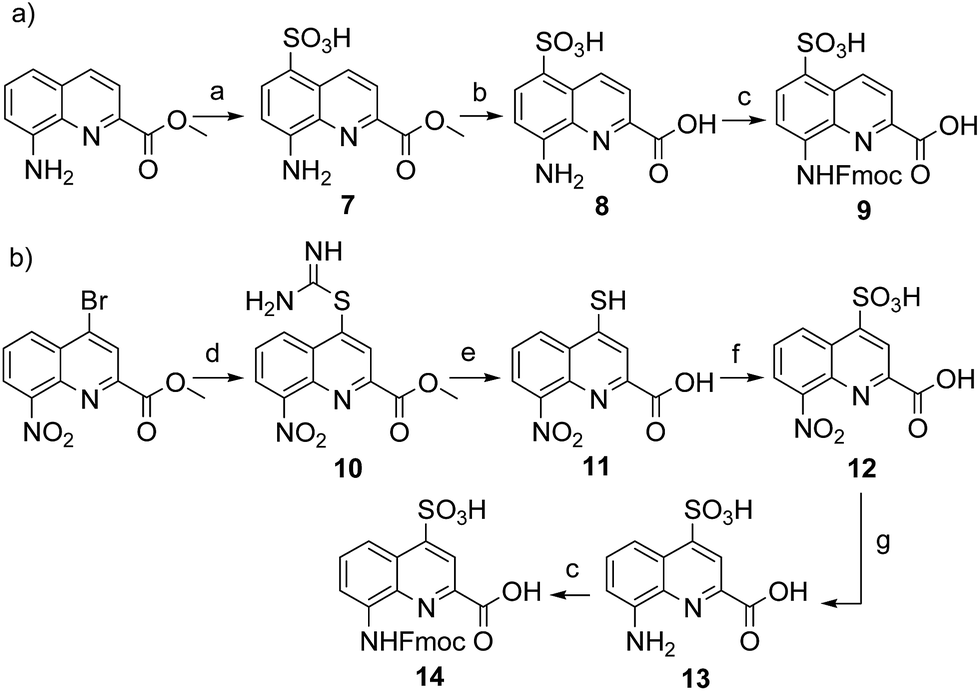 | ||
| Scheme 1 Synthesis of (a) Fmoc–Q5SO3H–OH (9) and (b) Fmoc–QSO3H–OH (14). Reagents and conditions: (a) ClSO3TMS, dioxane, 100 °C, overnight; (b) NaOH, THF/H2O, r.t., 1 h; (c) Fmoc–Cl, dioxane/H2O, NaHCO3 (10% w/vol), 0 °C to r.t., overnight; (d) thiourea, acetone, reflux, overnight; (e) NaOH, MeOH/THF, r.t., 3 h; (f) 30% H2O2, formic acid, 0 °C, 2 h; (g) H2, Pd/C, MeOH, r.t., 1 d. | ||
The synthesis of monomer 14 was then planned (Scheme 1b) with the expectation that a sulfonic acid in position 4 should not alter much the reactivity of the amine function. The starting 4-bromo-quinoline15a was first substituted by thiourea. Subsequent hydrolysis yielded quinoline mercaptan 11. After oxidation of the thiol and hydrogenation of the nitro group, Fmoc protection was installed to afford monomer 14. Particular care must be taken to control temperature during the oxidation step with hydrogen peroxide because of the inherent risk of explosion associated with this reagent, in particular on a large scale. All reactions proceeded efficiently, yet the overall yield was not as high as expected. This was mainly because of the high polarity and water solubility of the sulfonic acid intermediates, which impeded the use of classical work-ups such as extraction or column chromatography. For example, water solubility was surprisingly high for the Fmoc-protected final product 14 which proved difficult to extract and even harder to isolate using chromatography. Isolation was eventually achieved by direct precipitation from the reaction mixture but, by this method, high purity was possible only by collecting part of the material.
We initially thought that sulfonic acid protection would be necessary during SPS, but this proved not to be the case. This was fortunate because our trials to make sulfonate esters were all unsuccessful. Thus, the activation of 14 into a carbonyl chloride and its coupling to amines proceeded normally. The SPS procedure nevertheless required some slight changes: once one or more monomer 14 molecules have been added to a sequence, the sulfonic acid may retain some of the piperidine used for subsequent Fmoc deprotections, even after several solvent washes. Being a secondary amine, piperidine may consume part of the acid chlorides added in the next steps, causing a drop in yield that we found to be disastrous when multiple sulfonic acids were present. To address this problem, an additional DIPEA wash after Fmoc deprotection was introduced in the SPS protocol to exchange piperidine with DIPEA.
The extraordinary success of the isobutoxy group at promoting both solubility in, and crystallization from organic solvents,5b,5d–5g,6d,7,9,10a,11 guided the design of monomer 19 (Scheme 2). It was hoped that the branching along with two hydroxyl functions of a side chain derived from 2-hydroxymethyl-1,3-propanediol may result in similar properties in aqueous media. The identification of a protecting group suitable for SPS proved to be delicate. Acetals were found to be too acid labile (cleavage during acid chloride activation with Ghosez’s reagent18) or not enough (low lability in presence of TFA). Eventually, the di-tert-butylsilylene (DTBS) group was found to be stable during monomer synthesis and under SPS conditions. It resists TFA treatment normally used for resin cleavage but can conveniently be deprotected in the presence of TBAF. The preparation of 19 entailed usual protocols for side chain introduction.5b,15a,16a,18a Mitsunobu reaction, saponification, hydrogenation and Fmoc installation provided 19 in an overall yield of 45%. The SPS proceeded normally (see below).
 | ||
| Scheme 2 Synthesis of Fmoc–Qdiol(DTBS)–OH (19). Reagents and conditions: (a) DTBS ditriflate, anhydrous pyridine, anhydrous THF, −78 °C to r.t., 4 h; (b) 15, DIAD, PPh3, anhydrous THF, 0 °C to 50 °C, 5 h; (c) LiOH·H2O, THF/H2O, r.t., 1 h; (d) H2, Pd/C, THF, r.t., overnight; (e) Fmoc–Cl, dioxane/H2O, NaHCO3 (10% w/vol), 0 °C to r.t., overnight. | ||
The series of polar side chain functionality was further expanded from sulfonic acid (14) and diol (19) to ammonium (25, Scheme 3) and carboxylic acid (31 and 36, Scheme 4). The purpose was to cover a range of functionalities and to provide side chains resembling those of QAsp and QOrn which have proven to be useful in nucleic acid recognition14 and protein recognition,15c,d yet being shorter and therefore more rigid. A novel feature in this chemistry shared by the aminomethyl chain of 25 and the carboxymethyl chains of 31 and 36 is that they are connected to the quinoline ring by a carbon–carbon bond. In the context of biological applications, this linkage may confer a better metabolic stability than hydrolytically cleavable ethers, but the validation of this hypothesis is beyond the scope of the present study.
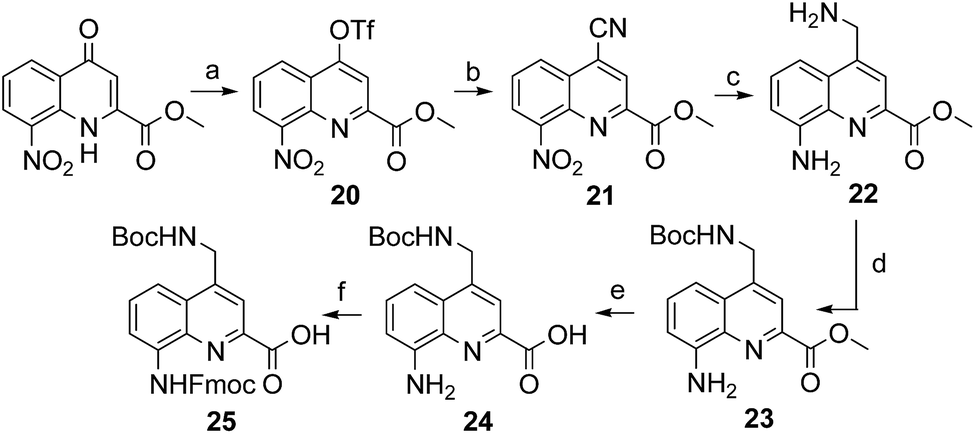 | ||
| Scheme 3 Synthesis of Fmoc–QNH(Boc)–OH (25). Reagents and conditions: (a) Tf2O, anhydrous pyridine, anhydrous DCM, r.t., overnight; (b) KCN, Pd(PPh3)4, anhydrous toluene/DMF, 100 °C, 2 h; (c) H2, Pd/C, AcOH/THF, r.t., overnight; (d) Boc2O, DIPEA, DCM/CH3CN, r.t., overnight; (e) LiOH·H2O, THF/H2O, r.t., 1 h; (f) Fmoc–Cl, dioxane/H2O, NaHCO3 (10% w/vol), 0 °C to r.t., overnight. | ||
 | ||
| Scheme 4 Synthesis of (a) Fmoc–Q5CO2(tBu)–OH (31) and (b) Fmoc–QCO2(tBu)–OH (36). Reagents and conditions: (a) ethylvinylether, H2SO4, AcOH, 70 °C to 120 °C, 30 min; (b) SeO2, pyridine, 80 °C, 2 d; (c) benzyl bromide, K2CO3, DMF, r.t., 12 h; (d) 2-tert-butoxy-2-oxoethyl zinc(II) bromide, bis(dibenzylidene acetone)-palladium [0], 1,2,3,4,5-pentaphenyl-1′-(di-tert-butylphosphino)ferrocene, THF, 70 °C, 12 h; (e) H2, Pd/C, THF, r.t., 1 d; (f) Fmoc–Cl, dioxane/H2O, NaHCO3 (10% w/vol), 0 °C to r.t., overnight; (g) LiOH·H2O, THF/H2O, r.t., 1 h; (h) 2-tert-butoxy-2-oxoethyl zinc(II) bromide, bis(dibenzylidene acetone)-palladium [0], 1,2,3,4,5-pentaphenyl-1′-(di-tert-butylphosphino)ferrocene, anhydrous THF, r.t., 4 h. (c) Possible decarboxylation mechanism. | ||
The synthesis of 25, equipped with a Boc-protected side chain amine and an Fmoc-protected main chain amine, is straightforward (Scheme 3). The yield of each step was above 90% on a multi gram scale. It is noteworthy that the protocol required no column chromatography which should make scale up easy. All six steps of the synthesis and final purification can be achieved in one week with an overall yield as high as 75%.
In contrast, the syntheses of 31 and 36, which possess a carboxymethyl side chain protected as an acid labile tert-butyl ester, was less easy. The key step is the aryl–alkyl carbon–carbon bond formation via a Pd-catalyzed α-arylation of the zinc enolate of tert-butyl acetate.26 The synthetic route and conditions were first developed and optimized for monomer 31 (Scheme 4a). In order to install the side chain in position 5, the 5-bromo quinoline precursor 28 was prepared in three steps from commercial 5-bromo-2-nitroaniline. The substitution of the bromide by freshly prepared Reformatsky reagent 2-tert-butoxy-2-oxoethyl zinc(II) bromide was delicate. The key to success was to strictly avoid oxygen. Operating this reaction in a glove box helps ensure a high and reproducible yield and avoids wasting costly catalyst and precursor. The purity of product 29 was found to be crucial to obtain a final Fmoc monomer sufficiently pure (>97%) for multi-step SPS coupling. Indeed, amino acid 30 and final product 31 are both difficult to purify. When all necessary care is taken, monomer 31 can be prepared on a 10 g scale with a yield of 7% over six steps.
The introduction of the same carboxymethyl side chain in position 4 was achieved starting from methyl 4-bromo-8-nitroquinoline-2-carboxylate which was prepared following previously published protocols.15a Synthesis was initially considered from the methyl ester via its subsequent saponification (LiOH) or demethylation (LiI). However, in both cases, purification of the final product 36 was problematic, possibly due to some tert-Bu ester cleavage. Instead, the two-step introduction of a benzyl ester proved to be rewarding (Scheme 4b). The subsequent aryl–alkyl cross-coupling, combined benzyl ester hydrogenolysis and nitro group hydrogenation, and final Fmoc introduction all worked as for compound 31. Monomer 36 was obtained in good purity (RP-HPLC purity > 97%) and good yield (overall 31% after 5 steps) without any column chromatography purification. The synthesis of sequences composed of this monomer was successful (see below). However, we found that decarboxylation of the side chain may occur under relatively mild conditions for the C-terminal monomer, for example in pure DMSO at room temperature.27 We suppose that this decarboxylation is assisted by the neighbour main chain terminal carboxylic acid function (Scheme 4c). The use of this monomer is thus fine within an oligoamide sequence but it should be avoided as a C-terminal unit. In principle, such decarboxylation may also occur under UV irradiation.28 Although we did not encounter any degradation under normal laboratory conditions, protection from UV light is advised.
Oligomer synthesis and crystallography
In order to test the effect of these new side chains on the solubility and crystal growth ability of oligomers in water, oligoamides 1–6 (Fig. 1) were synthesized from Fmoc acid monomers 14, 19, 25, 31, 36 and Fmoc–QOMe–OH using the previously reported SPS methods18 (see ESI†). As discussed above, the amino group of monomer 9 was too unreactive to elongate an oligoamide sequence. This monomer is only suitable as an N-terminal unit which greatly limits the scope of its applications. Also, as mentioned above, we have observed decarboxylation of the carboxymethyl side chain in position 4 when placed at the C-terminus of the sequence (as in monomer 36, see Scheme 4c). This degradation is faster in DMSO and in aqueous acidic medium but it does not occur during TFA mediated resin cleavage and side chain deprotection. Fortunately, it is very slow under slightly basic conditions (pH > 8). According to LC-MS monitoring, sequence 6 had undergone less than 5% degradation after six months. The recommendation is nevertheless to avoid placing this monomer at the C-terminus. Eventually, all oligomers but sequence 1 produced crystals from aqueous media by a standard hanging drop technique (see ESI† for crystallization conditions).Tetraamide 3 derived from the branched neutral diol side chain was synthesized first. SPS as well as side chain deprotection and RP-HPLC purification proceeded smoothly (Fig. 2b and ESI†). The product, however, had a disappointingly low solubility in water, barely reaching 1 mM. This was enough to record an NMR spectrum, but we inferred that this side chain, while compatible with water, does not confer sufficient water solubility to help dissolve sequences that would also contain hydrophobic residues. Nevertheless, crystals of 3 grew, though not from pure water but from an acetonitrile/aqueous ammonium sulfate mixture. The design that led to propose a branched di-hydroxyl side chain by analogy with the isobutyl group thus proved to be valid. The structure could be solved and is shown in Fig. 2c. It is centrosymmetric and thus contains both right-handed (P) and left-handed (M) helices. Helices of each handedness pile-up in head-to-tail infinite stacks. Side chains are projected away from the helices. Inter-helix hydrogen bonds occur directly. Some inter-helix contacts also appear to be mediated by hydrophobic patches of the main chain and side chains (Fig. 2c bottom).
 | ||
| Fig. 2 Formula (a), RP-HPLC chromatogram (b) and crystal structure (c) of 3. Carbon, nitrogen and oxygen atoms are shown in green, blue and red, respectively. Hydrogen atoms have been omitted for clarity. C- and N-termini and helix handedness are indicated in cartoons. | ||
Several features of the structure of 3 were found to be recurrent in other crystals: the centro-symmetrical nature of the lattice which is common in racemic foldamer helices10a,29 and the tendency of the helices to form cylindrical stacks that hide the hydrophobic aromatic cross-sections.15a,b,30 For oligomers 4–6, crystals grew easily. Disappointingly, none of the datasets recorded for crystals of cationic octaamide 4 could be solved, even though crystal growth was the fastest for this sequence (3 days) and two differently shaped crystals were obtained (Fig. 3c). The resolution (at best 1.48 Å) remained too low to solve the structure using ab initio methods and molecular replacement was never successful in our hands despite the availability of high quality models. The diffraction frames (Fig. 3b) with intense diffraction near 3.5 Å and unit cell dimensions unambiguously indicated the presence of the oligoamide in the lattice. We considered this to be sufficient evidence to validate our design approach, and therefore we did not proceed further with efforts to solve the structure of this particular sequence.
 | ||
| Fig. 3 (a) Formulae of octamers 4–6. Diffraction pattern (b) and crystals (c) of 4. Crystal structures of 5 (d) and 6 (e and f). In (d), some side chains have been omitted for clarity. Hydrogen bonds (dashed grey lines) connect two stacked helices of 6 at their C-termini (f). Carbon, nitrogen, and oxygen atoms are shown in green, blue, and red, respectively. Calcium is shown as purple balls. Hydrogen atoms have been omitted for clarity. C- and N-termini and helix handedness are indicated in cartoons. | ||
Single crystals of 5 suitable for X-ray crystallographic analysis were obtained from a Ca(OAc)2 solution. This time, the arrangement of the helices within the cylindrical stacks were head-to-head, thus creating two distinct interfaces: N-terminus to N-terminus, and C-terminus to C-terminus (Fig. 3d). This arrangement is held together by calcium bridges involving side chain carboxylates of different helices and a main chain C-terminal carboxylate. In fact, calcium chelates to almost all carboxylate functional groups in the structure, bridging the side chains to other helices or to amide carbonyl groups of the same helix.
X-ray quality crystals of 6 were obtained under conditions different from those that yielded crystals of 5, despite the related nature of their side chains (see ESI†). This structure was resolved as shown in Fig. 3e. In the absence of calcium bridges, hydrogen bonds between helices seem to be a prevalent interaction in the crystal lattice, including at the C-terminus/C-terminus aromatic interface within the stacks where hydrogen bonds form between carboxylic acid and quinoline functions (Fig. 3f). Other differences between the structures of 5 and 6 include the packing of the helices, which is pseudo-hexagonal in the structure of 6 (Fig. S3†), whereas it is square for 5.
The difference in crystallization behaviour between oligomers 5 and 6, and oligomers containing the QAsp monomer18 is noteworthy. The side chains of QAsp are only a single oxygen atom longer than those of 5 and 6. In absolute terms, the side chain of QAsp is not very flexible, for such aryl–alkyl ethers, rotation about the aryl–oxygen bond is restricted and the first carbon of the side chain is generally found in the plane of the quinoline ring (see structure 3 in Fig. 2). In addition, the side chain of QAsp is isosteric to an isobutoxy group that provides crystal growth ability from organic solvents. The different effects of –OCH2CH(CH3)2 in organic solvents and –OCH2CO2− in water may reflect the involvement of solvent or counter ion effects in addition to the inherent flexibility of the side chain. In water, shortening –OCH2CO2− to –CH2CO2− is found to greatly facilitate crystallization.
Another feature worthy of comment is the potential role of the diethyleneglycol tail in 4–6. Its intended purpose was not to enhance solubility in water. A single short tail has little influence on the solubility of such large molecules and even multiple ethylene glycol side chains do not provide solubility in pure water if they are not long enough.19a,b The intended purpose was to attempt to decrease aggregation in solution through the stacking of the aromatic cross-sections of the helices that we have observed to occur in water,15 in particular to obtain sharp NMR spectra in water. The effect of ethylene glycol chains on crystallization is presumed not to be favourable, as opposed to terminal nitro groups (as in 3) that are present in the crystal structures of many organic soluble aromatic foldamers. The crystallization of 4–6 thus occurred despite the presence of a diethylene glycol tail. Also, the structures of 5 and 6 reveal that the tail is not effective at preventing the stacking of the aromatic cross-sections of the helices in the solid state.
In the sulfonic acid series, octaamides 1 and 2 bearing eight and four sulfonic acid groups, respectively, were synthesized from monomer 14 and Fmoc–QOMe–OH. Octamer 1 was synthesized first and, quite disappointingly, did not yield any crystal. Instead, crystals of additives (e.g. salts) recurrently grew from the hanging drops. This observation hinted at the possible high solubility of 1 which we endeavoured to rigorously assess (see ESI†). Remarkably, solubility in basic and neutral aqueous solutions was so high that we failed to reach saturation with the limited amount of material available. We then measured a maximum solubility of 1 of 108 mM in 0.1 M aqueous HCl. The absence of crystals of 1 was thus not due to direct detrimental effects of sulfonic acid residues but due to enhanced solubility. Oligomer 2 was thus designed so that four sulfonic acid side chains have been replaced by four hydrophobic side chains (–OMe). Oligomer 2 was still very soluble in water, but it eventually crystallized from a CaCl2 aqueous solution after three weeks. Its crystal structure could be solved as shown in Fig. 4 and 5. The packing somewhat resembles the structure of 6 (head-to-head stacks of helices having the same handedness in a pseudo-hexagonal arrangement). Inter-helix interactions are mediated by: (i) aromatic stacking between helix cross-sections; (ii) some hydrophobic contacts between methoxy groups which form a linear array in the crystal lattice (Fig. 4 and S4†); and (iii) intermolecular calcium ions and water mediated bridges between sulfonic acids and main chain amide carbonyl groups (Fig. S4d†).
 | ||
| Fig. 4 Crystal structure of 2. Carbon, nitrogen, and oxygen atoms are shown in green, blue, and red, respectively. Calcium is shown as purple balls. Hydrogen atoms have been omitted for clarity. C- and N-termini and helix handedness are indicated in cartoons. | ||
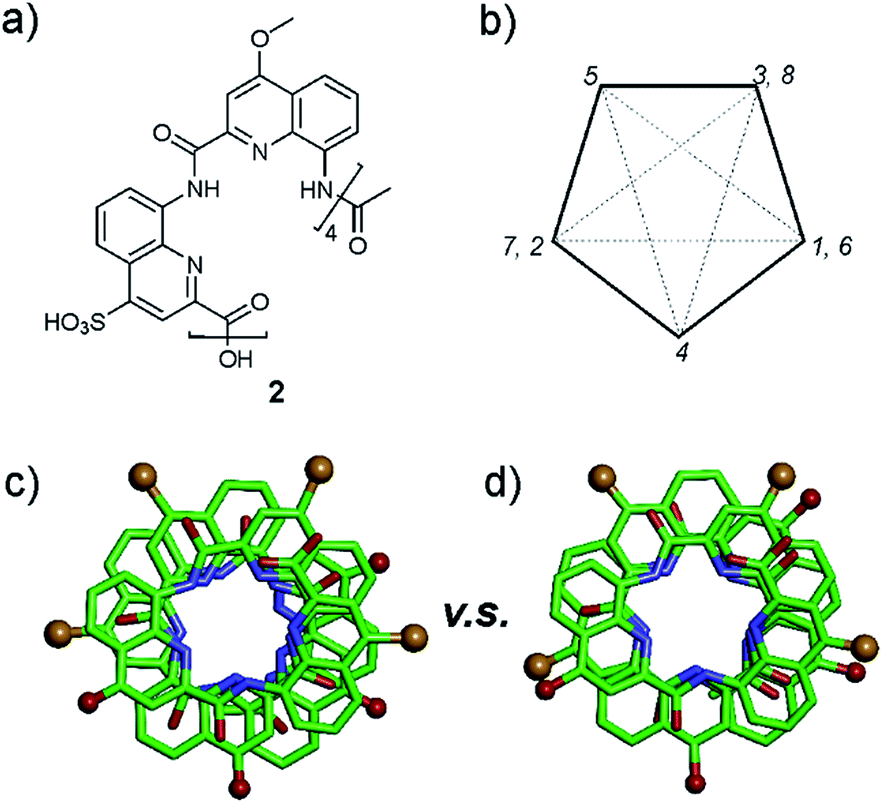 | ||
| Fig. 5 (a) Formula of octamer 2. (b) Five-pointed star showing the usual positions of side chains in quinoline-based helices counting monomers from 1 to 8. Top views of the crystal structure of 2 (c), and of 6 (d) in which carboxylic acid side chains have been replaced by the side chains of 2. All side chains are shown as golden (sulfonic acid) or red balls (methoxy). | ||
A remarkable aspect of the structure of 2 was its curvature. Oligoamides of 8-amino-2-quinolinecarboxylic acids normally form helices comprised of almost exactly five units per two turns (Fig. 5b). This is recurrently observed in crystals grown from organic solvents5b and has also been shown in solution.13 The solid state structures of water soluble 3, 5 and 6 described above make no exception. In contrast, the structure of 2 deviates from this pattern and possesses a slightly larger curvature – i.e. fewer units per turn – revealing a certain degree of flexibility for these helices (Fig. 5c and d). It is not clear whether this is the result of the packing interactions mentioned above or a direct outcome of intramolecular interactions between side chains and the main chain (a related effect was observed upon the introduction of bromine substituents in position 5 (ref. 11)).
Conclusions
In summary, synthetic protocols have been developed to prepare quinoline monomers bearing new short polar – neutral, anionic or cationic – side chains on multi gram scales. Side chain protections (or their absence) were made to be compatible with SPS. One tetrameric and five octameric oligoamides displaying these side chains were synthesized and shown to be soluble in water. In all cases but one, crystals were obtained using the hanging drop method, thus validating the initial design principle to combine polarity and rigidity. The only case that resisted crystallization appeared to be due to exceedingly high water solubility endowed by eight sulfonic acid functional groups. The neutral side chain did provide crystal growth ability from water but contributed poorly to solubility. The difference in crystal growth ability between oligomers 5 and 6 on one hand, and oligomers of QAsp (Fig. 1) on the other hand, is striking, considering that the side chains of the latter are just one atom longer than those of the former.The results presented here in the case of helical aromatic amide foldamers may certainly be extrapolated to other aromatic molecular and supramolecular systems. Extrapolation to other short and rigid polar functional groups may also be considered. These developments will be particularly useful to elucidate structures in projects where remarkable properties of foldamers have been observed in aqueous media, including endomolecular recognition and double helix formation.31 Along this line, an investigation of helix bundles constructed from monomers bearing the new side chains is in progress in our laboratory.
Acknowledgements
The authors thank Dr Brice Kauffmann for help during crystal structure determination and refinement. This work has benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform at IECB, CNRS UMS 3033, INSERM US001, Université de Bordeaux. We also thank staff on Proxima-1 beamline (at Synchrotron SOLEIL, Paris), ID29 beamline and BM30A beamline (at ESRF, Grenoble) for beam time and help during data collection. Preliminary contributions by Dr Marine Stupfel for the synthesis of 21 and by Dr Victor Dos Santos for the synthesis of 31 are gratefully acknowledged. This work was supported by the China Scholarship Council (PhD scholarship to X. H.) and the European Research Council (Grant Agreement Number ERC-2012-AdG-320892, postdoctoral fellowship to S. J. D. and P. K. M.).Notes and references
- (a) A. McPherson, Introduction to Macromolecular Crystallography, Wiley-Blackwell, Hoboken, 2nd edn, 2009 Search PubMed; (b) R. Giegé, B. Lorber and A. Théobald-Dietrich, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 339 CrossRef PubMed; (c) A. Ducruix and R. Giegé, in Crystallization of Nucleic Acids and Proteins. A Practical Approach, ed. A. Ducruix and R. Giegé, Oxford, IRL Press/Oxford Univ. Press, 1992 Search PubMed.
- (a) T. Sawada, M. Yoshizawa, S. Sato and M. Fujita, Nat. Chem., 2009, 1, 53 CrossRef CAS PubMed; (b) C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. Di Pasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802 CrossRef CAS PubMed; (c) J. L. Bolliger, T. K. Ronson, M. Ogawa and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 14545 CrossRef CAS PubMed; (d) J. R. Nitschke, D. Schultz, G. Bernardinelli and D. Gerard, J. Am. Chem. Soc., 2004, 126, 16538 CrossRef CAS PubMed; (e) S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and D. Williams, Science, 1999, 283, 1148 CrossRef CAS PubMed; (f) R. Carballo, B. Covelo, N. Fernandez-Hermida, E. Garcia-Martinez, A. B. Lago, M. Vazquez and E. M. Vazquez-Lopez, Cryst. Growth Des., 2006, 6, 629 CrossRef CAS.
- (a) E. A. Kataev and C. Müller, Tetrahedron, 2014, 70, 137 CrossRef CAS; (b) A. Dalla Cort, G. Forte and L. Schiaffino, J. Org. Chem., 2011, 76, 7569 CrossRef CAS PubMed; (c) A. P. Davis, Nature, 2010, 464, 169 CrossRef CAS PubMed; (d) S. Kubik, Chem. Soc. Rev., 2009, 38, 585 RSC; (e) G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366 CrossRef CAS PubMed; (f) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS PubMed; (g) T. J. Mooibroek, J. M. Casas-Solvas, R. L. Harniman, C. M. Renney, T. S. Carter, M. P. Crump and A. P. Davis, Nat. Chem., 2016, 8, 69 CrossRef CAS PubMed.
- (a) I. Huc, Eur. J. Org. Chem., 2004, 1, 17 CrossRef; (b) D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271 CrossRef CAS PubMed; (c) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC; (d) I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726 RSC.
- (a) J. Zhu, R. D. Parra, H. Zeng, E. Skrzypczak-Jankum, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2000, 122, 4219 CrossRef CAS; (b) H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448 CrossRef CAS PubMed; (c) H.-P. Yi, C. Li, J.-L. Hou, X.-K. Jiang and Z.-T. Li, Tetrahedron, 2005, 61, 7974 CrossRef CAS; (d) Z.-Q. Hu, H.-Y. Hu and C.-F. Chen, J. Org. Chem., 2006, 71, 1131 CrossRef CAS PubMed; (e) B. Baptiste, J. Zhu, D. Haldar, B. Kauffmann, J.-M. Leger and I. Huc, Chem.–Asian J., 2010, 5, 1364 CAS; (f) Y. Ferrand, A. M. Kendhale, J. Garric, B. Kauffmann and I. Huc, Angew. Chem., Int. Ed., 2010, 49, 1778 CrossRef CAS PubMed; (g) Q. Gan, C. Bao, B. Kauffmann, A. Grelard, J. Xiang, S. Liu, I. Huc and H. Jiang, Angew. Chem., Int. Ed., 2008, 47, 1715 CrossRef CAS PubMed.
- (a) S. Ferguson, K. Yamato, R. Liu, L. He, X. C. Zeng and B. Gong, Angew. Chem., Int. Ed., 2009, 48, 3150 CrossRef PubMed; (b) H. Fu, Y. Liu and H. Zeng, Chem. Commun., 2013, 49, 4127 RSC; (c) Y.-Y. Zhu, C. Li, G.-Y. Li, X.-K. Jiang and Z.-T. Li, J. Org. Chem., 2008, 73, 1745 CrossRef CAS PubMed; (d) H. Jiang, J.-M. Léger, P. Guionneau and I. Huc, Org. Lett., 2004, 6, 2985 CrossRef CAS PubMed.
- (a) L. Sebaoun, V. Maurizot, T. Granier, B. Kauffmann and I. Huc, J. Am. Chem. Soc., 2014, 136, 2168 CrossRef CAS PubMed; (b) L. Sebaoun, B. Kauffmann, T. Delclos, V. Maurizot and I. Huc, Org. Lett., 2014, 16, 2326 CrossRef CAS PubMed.
- (a) Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1996, 118, 7529 CrossRef CAS; (b) J. T. Ernst, J. Becerril, H. S. Park, H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2003, 42, 535 CrossRef CAS PubMed; (c) E. Kolomiets, V. Berl, I. Odriozola, A.-M. Stadler, N. Kyritsakas and J.-M. Lehn, Chem. Commun., 2003, 7, 2868 RSC; (d) Z.-Q. Wu, X.-K. Jiang, S.-Z. Zhu and Z.-T. Li, Org. Lett., 2004, 6, 229 CrossRef CAS PubMed.
- (a) N. Delsuc, J.-M. Léger, S. Massip and I. Huc, Angew. Chem., Int. Ed., 2007, 46, 214 CrossRef CAS PubMed; (b) N. Delsuc, S. Massip, J.-M. Léger, B. Kauffmann and I. Huc, J. Am. Chem. Soc., 2011, 133, 3165 CrossRef CAS PubMed.
- (a) N. Chandramouli, Y. Ferrand, G. Lautrette, B. Kauffmann, C. D. Mackereth, M. Laguerre, D. Dubreuil and I. Huc, Nat. Chem., 2015, 7, 334 CrossRef CAS PubMed; (b) Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401 CrossRef CAS PubMed; (c) K.-J. Chang, D. Moon, M. S. Lah and K.-S. Jeong, Angew. Chem., Int. Ed., 2005, 44, 7926 CrossRef CAS PubMed; (d) H. Juwarker, J.-M. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316 RSC.
- X. Li, T. Qi, K. Srinivas, S. Massip, V. Maurizot and I. Huc, Org. Lett., 2016, 18, 1044 CrossRef CAS PubMed.
- (a) G. W. Collie, K. Pulka-Ziach and G. Guichard, Chem. Sci., 2016, 7, 3377 RSC; (b) K. Basuroy, B. Dinesh, N. Shamala and P. Balaram, Angew. Chem., Int. Ed., 2013, 52, 3136 CrossRef CAS PubMed; (c) K. Basuroy, B. Dinesh, N. Shamala and P. Balaram, Angew. Chem., Int. Ed., 2012, 51, 8736 CrossRef CAS PubMed.
- C. Dolain, A. Grélard, M. Laguerre, H. Jiang, V. Maurizot and I. Huc, Chem.–Eur. J., 2005, 11, 6135 CrossRef CAS PubMed.
- (a) S. Muller, K. Laxmi-Reddy, P. V. Jena, B. Baptiste, Z. Dong, F. Godde, T. Ha, R. Rodriguez, S. Balasubramanian and I. Huc, ChemBioChem, 2014, 15, 2563 CrossRef PubMed; (b) P. K. Mandal, B. Baptiste, B. L. d’Estaintot, B. Kauffmann and I. Huc, ChemBioChem, 2016, 17, 1911 CrossRef CAS PubMed.
- (a) J. Buratto, C. Colombo, M. Stupfel, S. J. Dawson, C. Dolain, B. L. d’Estaintot, L. Fischer, T. Granier, M. Laguerre, B. Gallois and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 883 CrossRef CAS PubMed; (b) M. Jewginski, L. Fischer, C. Colombo, I. Huc and C. D. Mackereth, ChemBioChem, 2016, 17, 727 CrossRef CAS PubMed; (c) M. Jewginski, T. Granier, B. Langlois d’Estaintot, L. Fischer, C. D. Mackereth and I. Huc, J. Am. Chem. Soc., 2017, 139, 2928 CrossRef CAS PubMed; (d) S. Kumar and A. D. Miranker, Chem. Commun., 2013, 49, 4749 RSC; (e) S. Kumar, M. Birol, D. E. Schlamadinger, S. P. Wojcik, E. Rhoades and A. D. Miranker, Nat. Commun., 2016, 7, 11412 CrossRef CAS PubMed.
- (a) E. R. Gillies, F. Deiss, C. Staedel, J.-M. Schmitter and I. Huc, Angew. Chem., Int. Ed., 2007, 46, 4081 CrossRef CAS PubMed; (b) J. Iriondo-Alberdi, K. Laxmi-Reddy, B. Bougueme, C. Staedel and I. Huc, ChemBioChem, 2010, 11, 1679 CrossRef CAS PubMed.
- (a) J. A. Hebda, I. Saraogi, M. Magzoub, A. D. Hamilton and A. D. Miranker, Chem. Biol., 2009, 16, 943 CrossRef CAS PubMed; (b) I. Saraogi, J. A. Hebda, J. Becerril, L. A. Estroff, A. D. Miranker and A. D. Hamilton, Angew. Chem., Int. Ed., 2010, 49, 736 CrossRef CAS PubMed; (c) S. Kumar, D. E. Schlamadinger, M. A. Brown, J. M. Dunn, B. Mercado, J. A. Hebda, I. Saraogi, E. Rhoades, A. D. Hamilton and A. D. Miranker, Chem. Biol., 2015, 22, 369 CrossRef CAS PubMed; (d) B. P. Orner, J. T. Ernst and A. D. Hamilton, J. Am. Chem. Soc., 2001, 123, 5382 CrossRef CAS PubMed; (e) H. Yin, G. Lee, K. A. Sedey, O. Kutzki, H. S. Park, B. P. Orner, J. T. Ernst, H.-G. Wang, S. M. Sebti and A. D. Hamilton, J. Am. Chem. Soc., 2005, 127, 10191 CrossRef CAS PubMed; (f) H. Yin, G. Lee, H. S. Park, G. A. Payne, J. M. Rodriguez, S. M. Sebti and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 2704 CrossRef CAS PubMed; (g) J. M. Rodriguez and A. D. Hamilton, Angew. Chem., Int. Ed., 2007, 46, 8614 CrossRef CAS PubMed; (h) J. P. Plante, T. Burnley, B. Malkova, M. E. Webb, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Commun., 2009, 5091 RSC; (i) J. M. Rodriguez, N. T. Ross, W. P. Katt, D. Dhar, G. Lee and A. D. Hamilton, ChemMedChem, 2009, 4, 649 CrossRef CAS PubMed; (j) W. E. Martucci, J. M. Rodriguez, M. A. Vargo, M. Marr, A. D. Hamilton and K. S. Anderson, MedChemComm, 2013, 4, 1247 RSC; (k) V. Azzarito, J. A. Miles, J. Fisher, T. A. Edwards, S. L. Warriner and A. J. Wilson, Chem. Sci., 2015, 6, 2434 RSC.
- (a) B. Baptiste, C. Douat-Casassus, K. Laxmi-Reddy, F. Godde and I. Huc, J. Org. Chem., 2010, 75, 7175 CrossRef CAS PubMed; (b) S. J. Dawson, X. Hu, S. Claerhout and I. Huc, Methods Enzymol., 2016, 580, 279 CAS.
- (a) T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337 RSC; (b) C. Tsiamantas, S. J. Dawson and I. Huc, C. R. Chim., 2016, 19, 132 CrossRef CAS; (c) S. J. Dawson, Á. Mészáros, L. Petho, C. Colombo, M. Csékei, A. Kotschy and I. Huc, Eur. J. Org. Chem., 2014, 4265 CrossRef CAS.
- P. K. Mandal, B. Kauffmann, H. Destecroix, Y. Ferrand, A. P. Davis and I. Huc, Chem. Commun., 2016, 52, 9355 RSC.
- V. Pavone, S.-Q. Zhang, A. Merlino, A. Lombardi, Y. Wu and W. F. DeGrado, Nat. Commun., 2014, 5, 3581 Search PubMed.
- (a) C. G. Cummings and A. D. Hamilton, Tetrahedron, 2013, 69, 1663 CrossRef CAS; (b) L. A. Estroff, C. D. Incarvito and A. D. Hamilton, J. Am. Chem. Soc., 2004, 126, 2 CrossRef CAS PubMed.
- Z. S. Derewenda and P. G. Vekilov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, D62, 116 CrossRef CAS PubMed.
- (a) M. Selkti, A. W. Coleman, I. Nicolis, N. Douteau-Guével, F. Villain, A. Tomas and C. de Rango, Chem. Commun., 2000, 161 RSC; (b) S. J. Dalgarno, M. J. Hardie, M. Makha and C. L. Raston, Chem.–Eur. J., 2003, 9, 2834 CrossRef CAS PubMed; (c) S. J. Dalgarno, M. J. Hardie, J. L. Atwood, J. E. Warren and C. L. Raston, New J. Chem., 2005, 29, 649 RSC; (d) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed.
- (a) R. E. McGovern, A. A. McCaerthy and P. B. Crowley, Chem. Commun., 2014, 50, 10412 RSC; (b) L. Jiao, S. Ouyang, M. Liang, F. Niu, N. Shaw, W. Wu, W. Ding, C. Jin, Y. Peng, Y. Zhu, F. Zhang, T. Wang, C. Li, X. Zuo, C.-H. Luan, D. Li and Z.-J. Liu, J. Virology, 2013, 87, 6829 CrossRef CAS PubMed; (c) A. Schuetz, J. Min, T. Antoshenko, C.-L. Wang, A. Allali-Hassani, A. Dong, P. Loppnau, M. Vedadi, A. Bochkarev, R. Sternglanz and A. N. Plotnikov, Structure, 2007, 15, 377 CrossRef CAS PubMed; (d) H. P. Morgan, I. W. McNae, M. W. Nowicki, W. Zhong, P. A. M. Michels, D. S. Auld, L. A. Fothergill-Gilmore and M. D. Walkinshaw, J. Biol. Chem., 2011, 286, 31232 CrossRef CAS PubMed.
- T. Hama, X. Liu, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 11176 CrossRef CAS PubMed.
- G. R. Jurch and K. C. Ramey, Chem. Commun., 1968, 1211b RSC.
- F. R. Stermitz and W. H. Huang, J. Am. Chem. Soc., 1971, 93, 342 CrossRef.
- (a) G. Lautrette, B. Kauffmann, Y. Ferrand, C. Aube, N. Chandramouli, D. Dubreuil and I. Huc, Angew. Chem., Int. Ed., 2013, 52, 11517 CrossRef CAS PubMed; (b) S. Shin, M. Lee, I. A. Guzei, Y. K. Kang and S. H. Choi, J. Am. Chem. Soc., 2016, 138, 13390 CrossRef CAS PubMed; (c) M. Lee, J. Shim, P. Kang, I. A. Guzei and S. H. Choi, Angew. Chem., Int. Ed., 2013, 52, 12564 CrossRef CAS PubMed; (d) J.-H. Eom, R. Jeong, J. Gong, R. W. Driver and H.-S. Lee, Bull. Korean Chem. Soc., 2015, 36, 2583 CrossRef CAS; (e) D. E. Mortenson, J. D. Steinkruger, D. F. Kreitler, D. V. Perroni, G. P. Sorenson, L. Huang, R. Mittal, H. G. Yun, B. R. Travis, M. K. Mahanthappa, K. T. Forest and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13147 CrossRef PubMed.
- See also: H. Zhao, W. Q. Ong, F. Zhou, X. Fang, X. Chen, S. F. Y. Li, H. Su, N.-J. Cho and H. Zeng, Chem. Sci., 2012, 3, 2042 RSC.
- (a) N. Chandramouli, M. F. El-Behairy, G. Lautrette, Y. Ferrand and I. Huc, Org. Biomol. Chem., 2016, 14, 2466 RSC; (b) J. Shang, Q. Gan, S. J. Dawson, F. Rosu, H. Jiang, Y. Ferrand and I. Huc, Org. Lett., 2014, 16, 4992 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols for synthesis; characterization of new compounds; methods for X-ray crystallography; solubility studies. CCDC 1521878, 1525865, 1525770 and 1523119. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00430c |
| ‡ Present address: Université Pierre et Marie Curie, CNRS, IMPMC (UMR7590), 4 place Jussieu, 75005 Paris, France. |
| This journal is © The Royal Society of Chemistry 2017 |