
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCellular and cell-free studies of catalytic DNA cleavage by ruthenium polypyridyl complexes containing redox-active intercalating ligands†
Cynthia
Griffith
,
Adam S.
Dayoub
,
Thamara
Jaranatne
,
Nagham
Alatrash
,
Ali
Mohamedi
,
Kenneth
Abayan
,
Zachary S.
Breitbach
,
Daniel W.
Armstrong
 and
Frederick M.
MacDonnell‡
*
and
Frederick M.
MacDonnell‡
*
Department of Chemistry and Biochemistry, University of Texas at Arlington, Arlington, TX 76019, USA. E-mail: macdonn@uta.edu
First published on 8th March 2017
Abstract
The ruthenium(II) polypyridyl complexes (RPCs), [(phen)2Ru(tatpp)]2+ (32+) and [(phen)2Ru(tatpp)Ru(phen)2]4+ (44+) are shown to cleave DNA in cell-free studies in the presence of a mild reducing agent, i.e. glutathione (GSH), in a manner that is enhanced upon lowering the [O2]. Reactive oxygen species (ROS) are involved in the cleavage process as hydroxy radical scavengers attenuate the cleavage activity. Cleavage experiments in the presence of superoxide dismutase (SOD) and catalase reveal a central role for H2O2 as the immediate precursor for hydroxy radicals. A mechanism is proposed which explains the inverse [O2] dependence and ROS data and involves redox cycling between three DNA-bound redox isomers of 32+ or 44+. Cultured non-small cell lung cancer cells (H358) are sensitive to 32+ and 44+ with IC50 values of 13 and 15 μM, respectively, and xenograft H358 tumors in nude mice show substantial (∼80%) regression relative to untreated tumors when the mice are treated with enantiopure versions of 32+ and 44+ (Yadav et al. Mol Cancer Res, 2013, 12, 643). Fluorescence microscopy of H358 cells treated with 15 μM 44+ reveals enhanced intracellular ROS production in as little as 2 h post treatment. Detection of phosphorylated ATM via immunofluorescence within 2 h of treatment with 44+ reveals initiation of the DNA damage repair machinery due to the ROS insult and DNA double strand breaks (DSBs) in the nuclei of H358 cells and is confirmed using the γH2AX assay. The cell data for 32+ is less clear but DNA damage occurs. Notably, cells treated with [Ru(diphenylphen)3]2+ (IC50 1.7 μM) show no extra ROS production and no DNA damage by either the pATM or γH2AX even after 22 h. The enhanced DNA cleavage under low [O2] (4 μM) seen in cell-free cleavage assays of 32+ and 44+ is only partially reflected in the cytotoxicity of 32+ and 44+ in H358, HCC2998, HOP-62 and Hs766t under hypoxia (1.1% O2) relative to normoxia (18% O2). Cells treated with RPC 32+ show up to a two-fold enhancement in the IC50 under hypoxia whereas cells treated with RPC 44+ gave the same IC50 whether under hypoxia or normoxia.
Introduction
The use of transition-metal complexes in medicine has enjoyed extensive attention given the tremendous success of cisplatin (cis-Pt(NH3)2Cl2) as a chemotherapeutic agent and the ability of many metal complexes to interact with and damage cellular structures, particularly DNA.1–3 While research in metallopharmaceuticals continues to focus on platinum complexes, ruthenium complexes with labile chloride ligands are also explored as their substitution kinetics are similar to that of platinum. The promise of such compounds being their potential applicability to a wider-range of tumors and less severe toxicity relative to cisplatin. The anti-tumor agents NAMI-A (imidazolium [trans-imidazoledimethylsulfoxide-tetrachlororuthenate(III), KP1019 (indazolium [trans-tetrachlorobis(1H-indazole) ruthenate(III)]), RDC11 ([cis-bis(acetonitrile)-1,10-phenanthroline-2-phenylpyridineruthenium(II)] hexafluorophosphate),4 and ruthenium-aryl-X complexes,5 such as RAPTA-C,6 all contain labile ligands and the loss of these ligands and subsequent binding of the ruthenium to biological substrates is implicated in their biological activity. As many of these complexes contain Ru(III) ions, activation by bioreduction to Ru(II) is known to be essential to their mode of action.7,8 NAMI-A and KP1019 are reported to have advanced to stage I and II clinical trials.9–14Ruthenium polypyridyl complexes (RPCs), which we will constrain to Ru(II) complexes that are coordinatively saturated with polypyridyl ligands, constitute a fundamentally different class of metallo-drugs than those with labile ligands. RPCs have enjoyed the most attention in chemotherapeutic applications as photodynamic therapy (PDT) agents, some of which show demonstrable tumor reduction in vivo.15–17 Photoexcitation of the RPC in the metal to ligand charge transfer (MLCT) region generates long-lived triplet species which can activate O2 to form ROS,18–24 directly oxidize DNA,25,26 or induce ligand loss,27–31 such that the released ligand or resulting complex is damaging to the DNA. Because the metal ion in a RPC is coordinatively saturated and substitutionally inert, it is generally unable to directly form bonds with biological targets, unless activated with light, which is in contrast to complexes such as NAMI-A, RAPTA-C, and KP1019. That said, RPCs do show some interesting biological activity even without light activation.
The homoleptic complexes, such as the trisphenanthroline complex (12+) and the trisbipyridine complex shown in Fig. 1, were extensively studied by the groups of Dwyer and Shulman in the 1950's and 1960's.32 RPCs 12+ and 22+ are modestly cytotoxic (IC50's between 10−3 and 10−4 M) with enhanced cytotoxicity generally observed by increasing the lipophilicity of the complex.33,34 The 3,4,7,8-tetramethyl-1,10-phenanthroline derivative of 12+ was shown to inhibit the growth of dispersed tumor cells (Landshultz ascites) in mice.35 Early studies in which radiolabeled [106Ru(phen)3]2+ was injected intraperitoneally into mice and rats showed that the intact complex cation was the bioactive unit, it was not metabolized in vivo, it did not accumulate in any organ in amounts greater than blood, and nearly all the complex was recovered in the urine.36
 | ||
| Fig. 1 Chemical structures of RPCs and reference numbering scheme. All of these cations are soluble in water as the chloride salts. | ||
More recently, the DNA-binding and molecular-light switch behavior of [Ru(phen)2(dppz)]2+ (62+)37 and [Ru(phen)2(tpphz)]2+ (72+)38,39 has led to a resurgence in this area, with numerous studies of their uses as biological probes40–45 and the inherent (non-light activated) cytotoxicity of RPCs.34,39,43,46–54 Because of the tendency of RPCs to bind DNA, it is often assumed that this is the biological target,55–58 although recent data reveal that other cellular organelles are sometimes targeted, including the mitochondria,46,52,54,59–64 endoplasmic reticulum,42 ribosomes65 and cell membrane.34,66–70 It is not known how these RPCs act on the molecular targets but given their chemical inertness, it is postulated they non-covalently bind at specific sites disrupting important cellular processes.
Given the extensive attention of RPCs binding to DNA, it is somewhat surprising that very few cause observable damage unless they are activated by an external factor, such a light irradiation.17,18,20,66–68 We have shown that the two ruthenium(II)–tatpp complexes, 32+ and 44+ (shown in Fig. 1) are effective DNA cleaving agents upon in situ reduction by common biological reducing agents, such as glutathione.69,70 This is not a light activated process and thus these complexes have potential as systemic chemotherapeutics which can seek out and kill micro-metastases whose presence and location are unknown. In contrast, PDT requires knowing the location of the metastases for effective treatment, although there is now evidence that such treatments can sometimes boost the systemic immune response.71 The DNA cleavage activity of 32+ and 44+ in cell-free assays is enhanced under low oxygen conditions which is both unusual and potentially beneficial as many tumors possess hypoxic regions.69,70 Their anti-tumor efficacy has been demonstrated in mouse models in which nude mice bearing xenograph H358 tumors in their thighs showed an ∼83% regression of tumor growth and more than doubling survival time upon intraperitoneal treatment with enantiopure Δ-32+ and ΔΔ-44+.70 While direct proof was lacking, we postulated that the in vivo activity was also due to DNA damage.
In this paper, we demonstrate that these two RPCs catalytically induce DNA single-strand breaks (SSBs) by activation of O2 through a multi-stage redox-cycling mechanism which generates not only superoxide, but the more potent reactive oxygen species (ROS), H2O2. Significantly, at lower [O2] there is a greater build-up of a doubly-reduced versions of 32+ and 44+, which are competent for direct H2O2 formation upon reaction with O2. Thus a lower [O2] favors enhanced catalytic formation of H2O2 over superoxide, the former being more effectively converted into the highly toxic hydroxyl radical species. Significantly, we can show that ROS is generated in the nuclei of cultured human non-small cell lung carcinoma (NSCLC) cells (H358) within 2 h of treatment with 32+ or 44+. Moreover, multiple DNA double strand breaks (DSBs) are also detected using immunofluorescent techniques to reveal the presence of pATM, an early marker of ROS induced DNA DSB damage, and γH2AX, a downstream marker of DNA DSBs. For cells treated with 44+ this is evident within 2 hours of treatment whereas for 32+, the induction period is considerably longer, on the order of 8 h. To the best of our knowledge, this is the first time that the cell-free DNA cleavage activity of a ruthenium-based drug has been directly correlated with nuclear DNA damage in live human cancer cells.
Experimental
Chemicals
RPCs [1]Cl2,72 [2]Cl2,73 [3]Cl2,74 [4]Cl4,75 [5]Cl4,75 [6]Cl2, [7]Cl2,76,77 [8]Cl4,77 were prepared as described in the literature and were used as the chloride salts. All RPCs were used as racemates or diastereotopic mixtures. Furfural standard was purchased from Sigma Aldrich. 5-Methylene furanone (5MF) was synthesized according to literature78 using the modified procedure described below. The intermediate 3,5-di-O-p-toluoyl-2-deoxy-D-ribono-1,4-lactone was converted to 5 MF using Schlenk line techniques under a N2 atmosphere. The final product structure was confirmed using GCMS for which the MS pattern matched that reported for 5 MF.79 All reagents for the 5 MF synthesis were purchased from Sigma Aldrich.In vitro DNA cleavage assays
All chemicals for buffers and related in vitro DNA experiments were purchased commercially and used without further purification unless otherwise noted. Millipore water was used for all buffers and reactions that required water. All plasmid DNA (pUC18 and pUC19) and DNA ladders were purchased from Bayou Biolabs. Chemicals needed for the DNA electrophoresis assay, ethidium bromide, GSH, trizma base, mono and dibasic phosphates, ethylenediaminetetraacetic acid (EDTA) and agarose were purchased from Sigma Aldrich. Chemicals for the T4 ligase assay, T4 DNA ligase high concentration (HC), T4 DNA ligase 10× buffer, acetylated bovine serum albumin, EcoRI, and buffer HC 10× buffer were purchased from Promega.DNA cleavage experiments were conducted with pUC18 or pUC19 DNA at room temperature in air and low light for all reactions. The concentration of complex used and conditions of the experiment are given in the figure legends. A buffer solution (50 mM phosphate and 10 mM NaCl, pH 7.2) was used to bring the total volume to 40 μL. As a rule, buffer was added first, then the reagents and RPC, and finally the plasmid DNA. Reactions were quenched by placing the samples in an ice bath (dry ice and acetone). Plasmid reaction products were analyzed by addition of 10 μL of 6× loading buffer (30% glycerol in water with 0.1% w/v bromophenol blue) to the sample, vortexing, and loading 10 μL per well. The 1% agarose gels, containing ethidium bromide, were run in TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH 8) at 5 V cm−1 (60 V) for 2 h and imaged using a Bio Rad Gel Doc™ XR+. Experiments in which scavengers, SOD, catalase, or other modifiers were used, the concentration is noted in the figure caption.
DNA scission products assay
Reactions were conducted under the following conditions, (45.5 mL) 700 μM ctDNA, (4.1 mL) 58.3 μM (44+), (19.9 mL) 5.8 mM GSH, (30.5 mL) 50 mM phosphate, 10 mM NaCl buffer at pH 7.4. Samples were digested at room temperature in air overnight, then heated at 90 °C for 1 h in a GC oven. The reaction was quenched with ice bath (dry ice and acetone), extracted with 20 mL dichloromethane (DCM) 3×, dried with MgSO4 and concentrated. Samples were resuspended in pure MeCN for HPLC analysis. The mobile phase of for HPLC was 0.1 TFA/MeCN 90/10, flow rate: 0.1 mL min−1, injection vol: 10 μL, stationary phase: Zorbax Eclipse XDB-C18 4.6 × 150 column. The same method was conducted for 32+.Cultured cells, materials and methods
All solvents were reagent and cell culture grade. All reagents and work environments were maintained sterile. H358, HOP-62 and HCC2998 cells were purchased from The National Cancer Institute (NCI) at Frederick Central Repository. Hs766T cells were purchased from American Type Culture Collection (ATCC). RPMI-1640 and DMEM medium, penicillin/streptomycin, fetal bovine serum (FBS), 100× BME vitamin solution, bovine serum albumin (BSA), para-formaldehyde, methanol, sodium azide, dimethyl sulfoxide (DMSO), 0.04% trypan blue and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma Aldrich. Phosphate buffered saline 10× was purchase from Biorad. The DNA double strand break (DSB) γH2AX monoclonal antibody was purchased from EMD millipore and goat anti-mouse IgG (H + L) secondary antibody Alexa Fluor488 (ABCAM) and Pro-gold anti-fade mounting agent were purchased from Invitrogen.Cell culturing and experimentation
Cell incubation was maintained by a Thermofisher HeriCell CO2 Incubator. Hypoxic incubation was maintained by a New Brunswick Galaxy 14s Dual Channel CO2/N2 incubator. Confocal microscopy was performed using a Zeiss Axio-Plane 540 with mercury lamp and argon laser. Absorbance readings were obtained using a BMG Labtech FLUOstar Omega plate reader.Cell culture
H358, HOP-62 and HCC-2998 cells were grown in RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1.1% penicillin/streptomycin and 1× BME vitamin complex solution. Hs766T cells were grown in DMEM medium supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1.1% penicillin/streptomycin and 1× BME vitamin complex solution. Cells were grown and passaged in T-25 and T-75 Corning culture flasks at 37 °C under 5% CO2 and humidified atmosphere. Cells grown in hypoxic induced stress environments were grown and passaged in T-25 and T-75 Corning culture flasks at 37 °C under 5% CO2, 1.1% O2 and humidified atmosphere.Hypoxic and normoxic cell viability
Cytotoxic effects of complexes 32+ and 44+ were tested on cell growth populations of H358, HCC-2998, HOP-62 and Hs766T. Normoxic [O2] levels were adjusted to 18% in atmosphere and hypoxic [O2] levels were adjusted to 1.1% in atmosphere in two separate incubation environments, the latter using a New Brunswick Galaxy 14s Dual Channel CO2/N2 incubator. Under aerobic conditions, cells were passaged and seeded onto 96 well plates. Plates were then placed in either a normoxic or hypoxic incubator and left to grow for 24 h. At this time, cells were inoculated with complexes 32+ and 44+ at titrating doses and placed back in their respective incubators for 72 h, after which the plates were removed and immediately assayed with MTT (5 mg mL−1) for 3.5 h. Plates were read at 570 nm for absorbance of formazan production in the supernatant.Measurement of intracellular ROS
The generation of ROS in H358 cells was measured using a ROS sensitive fluorescent probe, 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA, ThermoFisher). DCFH-DA can be oxidized to 2′,7′-dichlorofluorescein (DCF) by ROS and exhibits green fluorescence intensity.80 H358 cells were seeded on 25 × 25 mm microscope cover glass slips in BD Falon 60 × 60 mm tissue culture dishes for 72 h. Untreated cells were maintained as the negative controls whereas 10, 20 and 30% H2O2 solution in PBS was administered to cells for 15 minutes as positive controls.54 H358 cells were also dosed with IC50 values of various complex as follows: 44+ (15 μM), 33+ (13 μM) and 22+ (1.7 μM) for 3 time periods of 2, 8, and 22 h. The cells were passaged and washed 3× in ice cold PBS then suspended in 10 mM DCFH-DA in PBS and incubated in the dark for 30 min. The levels of intracellular ROS were examined by confocal microscopy using long pass light filters and a 1.3 airy unit pinhole at 488/529 nm with a Zeiss axio-plane inverted fluorescence microscope.ATM pathway response assay
H358 cells were seeded on 25 × 25 mm microscope cover glass slips in BD Falon 60 × 60 mm tissue culture dishes for 72 h. Cells were then treated with: etoposide (6 μM), 32+ (13 μM) and 44+ (15 μM) at their respective IC50's for 2, 8, and 22 h. The cover slips were removed and washed 3× in ice-cold phosphate-buffered saline (PBS) to remove residual drug. Cells were fixed with 4% para-formaldehyde solution, permeabilized with 0.25% Triton and blocked with 3% BSA, anti-phospho ATM (phospho s1981, ABCAM) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) in 3% BSA/1% sodium azide was administered for 1.5 h in the dark at room temperature. Cells were then washed 3× in ice-cold PBS and goat anti-rabbit IgG (H + L) secondary antibody Alexa Fluor488 (1:2000) in 3% BSA/1% sodium azide was administered for 3 h in the dark at room temperature. Cells were then treated with propidium iodide (5 mg mL−1) for 5 min. After washing 3× with ice-cold PBS, the cells were fixed on microscope slide with Pro-gold antifade reagent. Confocal microscopy was performed using long pass light filters and a 1.3 airy unit pinhole at 488/514 nm. 60× oil immersion objectives were used and digital camera images (DCIM) were captured using ZEN software.
:1000) in 3% BSA/1% sodium azide was administered for 1.5 h in the dark at room temperature. Cells were then washed 3× in ice-cold PBS and goat anti-rabbit IgG (H + L) secondary antibody Alexa Fluor488 (1:2000) in 3% BSA/1% sodium azide was administered for 3 h in the dark at room temperature. Cells were then treated with propidium iodide (5 mg mL−1) for 5 min. After washing 3× with ice-cold PBS, the cells were fixed on microscope slide with Pro-gold antifade reagent. Confocal microscopy was performed using long pass light filters and a 1.3 airy unit pinhole at 488/514 nm. 60× oil immersion objectives were used and digital camera images (DCIM) were captured using ZEN software.
γH2AX double strand break assay
H358 cells were seeded on 25 × 25 mm microscope cover glass slips in BD Falon 60 × 60 mm tissue culture dishes for 72 h. Cells were then treated with complexes: etoposide, 32+ and 44+ at their respective IC50's for 2, 8, and 22 h. The cover slips was removed and washed 3× in ice-cold phosphate-buffered saline (PBS) to remove residual drug. Cells were fixed with 4% para-formaldehyde solution, permeabilized with 0.25% Triton and blocked with 3% BSA, anti-phospho-histone (Ser139) γH2AX (ABCAM) (1:1000) in 3% BSA/1% sodium azide was administered for 1 h in the dark at room temperature. Cells were then washed 3× in ice-cold PBS and goat anti-mouse IgG (H + L) secondary antibody Alexa Fluor488 (1:2000) in 3% BSA/1% sodium azide was administered for 2 h in the dark at room temperature. Cells again were washed 3× in ice-cold PBS and then fixed on microscope slide with Pro-gold antifade reagent. Confocal microscopy was performed using long pass light filters and a 1.3 airy unit pinhole at 488/519 nm. 60× oil immersion objectives were used and digital camera images (DCIM) were captured using ZEN software. Cell sorting and foci count were analyzed with Image J software for an average of 25 cells per image count.
Results
DNA-binding and cleavage activity
RPCs 12+ and 22+ bind predominantly via electrostatics with a binding constant on the order of 103 M−1, the remainder 32+–84+, with large planar aromatic units: dppz(dipyrido[3,2-a:2′,3′-c]phenazine), tpphz(tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine), tatpp(9,11,20,22-tetraazatetrapyrido[3,2-a:2′,3′-c:3′′,2′′-1:2′′′,3′′′-n]-pentacene), tatpq(9,11,20,22-tetraazatetrapyrido[3,2-a:2′,3′-c:3′′,2′′-1:2′′′,3′′′-n]-pentacene-10,21-quinone, bind more tightly due to intercalation and exhibit binding constants in the range of 105 to 108 M−1.81,82RPC binding to DNA does not generally equate with RPCs causing damage. In the absence of deliberate irradiation to access the excited state chemistry of these complexes, the vast majority do not cause any DNA damage after binding. In Fig. 2, we assay the DNA cleaving activity of RPCs 12+, 44+, 54+, 62+, and 84+ using a plasmid cleavage assay under physiologically relevant conditions (50 mM phosphate, 10 mM NaCl, 5.8 mM GSH, pH 7.2, aerobic). In this experiment, we monitored the conversion of supercoiled plasmid DNA (Form I) to nicked, open circular (Form II) and double-strand cleaved linear (Form III) DNA by agarose gel electrophoresis. As seen in Fig. 2, we contrast the cleavage activity of the tatpp complex 44+ and the tatpq complex 54+, with a number of structurally related RPCs. RPC 32+ also cleaves DNA under these conditions.70 Control experiments lacking GSH showed no cleavage activity by any complex. The presence of circular over linear DNA indicates SSBs are prevalent. Hydrolytic cleavage of the DNA was ruled out by treating samples of RPC/GSH cleaved DNA with the T4 ligase repair enzyme to see if the cleavage was reversible; the presumption being that hydrolytic cleavage is reversible whereas DNA damaged by oxidation is not easily relegated. The cleavage induced by RPC 32+, 44+, or 54+/GSH combinations was not reversible (see ESI, Fig. S1 and S2†), supporting oxidative DNA damage.
 | ||
| Fig. 2 Agarose gel showing DNA cleavage products of pUC19 after treatment with RPCs 1–8 in the presence of GSH under aerobic conditions. Lane C, control showing open circular (Form II, top), linear (Form III, middle) and supercoiled (Form I, bottom) plasmid DNA. Lane 1, supercoiled plasmid DNA (144 μM DNA-bp) after 2 h incubation. Lane 2, supercoiled DNA (144 μM DNA-bp) with 120 μM GSH present after 2 h incubation. Lanes 3–7 supercoiled DNA (144 μM DNA-bp) with 12 μM RPC indicated and 120 μM GSH after 2 h incubation. | ||
Activation by reduction is a common mechanism by which transition metal complexes activate O2 to form ROS, but the redox processes typical for RPCs are generally not accessible by common cellular reductants or oxidants. For virtually all RPCs, the first oxidative process is the Ru2+/3+ couple which occurs at about 1.5 V vs. NHE and is far too positive to be accessed in water or more pointedly in vivo (via non-photochemical pathways).83 Reductions in RPCs are generally associated with ligand couples, such as the [RuII(phen)3]2+/[RuII(phen)2(phen˙−)]+ couple in which the electron is localized in one of the low-lying acceptor orbitals on the polypyridyl ligands, usually the LUMO. The potentials for these ‘ligand-based’ redox couples can vary dramatically with ligand structure. Fig. 3 (bottom) shows a line graph of the observed first reduction potential for 12+–84+ as obtained in acetonitrile (see Table S1 in the ESI†). From this data, we observe that the phen and bpy ligands in RPCs 12+ and 22+ are the hardest to reduce at −1.1 V, followed by the dppz and tpphz ligands in 62+, 72+, and 84+ in the −0.76 to −0.5 V potential range, and finally the tatpp and tatpq ligands in 32+, 44+ and 54+ in the respective −0.1 to 0.05 V range (all potential are quoted vs. NHE). It is clear that the RPC's with DNA cleavage activity, which are indicated by the circles filled with black dots (Fig. 3, bottom), possess the most positive reduction potentials (>−0.2 V vs. NHE). With a reduction potential of −0.24 V (vs. NHE, pH 7) for the glutathione disulfide/glutathione couple,84 GSH can only reduce 32+, 44+ and 54+ of all the RPCs examined (12+–84+) and it is no coincidence that these are the only RPCs observed to cleave DNA.
 | ||
| Fig. 3 Top: Relevant redox isomers of 44+ in aqueous solution (pH 7.2). Black ball represents the [Ru(phen)2]2+ fragment. Equivalent redox isomers exist for 32+ with respect to the tatpp ligand. Bottom: Plot of the first reduction potential for the RPCs 12+–84+ in MeCN solvent. Open circles indicate complexes that are inactive for DNA cleavage and partially filled circles indicate complexes which cleave DNA in the presence of GSH. | ||
Cleavage by O2 activation could then be explained by GSH reduction of the RPC to a ligand radical species, shown for 44+ in reaction (1), which then reacts with O2 to form superoxide, as shown in reaction (2). As 44+ is regenerated in reaction (2), it could then redox-cycle much like the related quinone-based anticancer drugs doxorubicin and daunorubicin to generate more ROS.85–87 Superoxide is not potent enough to directly attack DNA but can form more potent ROS including H2O2 and hydroxyl radical through subsequent reductions. The involvement of hydroxyl radical in DNA cleavage was assessed by adding a number of OH˙ scavengers all of which attenuate the cleavage activity. Specifically, addition of sodium benzoate, sodium formate, mannitol, ethanol, or DMSO were all observed to inhibit the cleavage activity in a dose dependent manner for both 32+ and 44+ (see ESI, Fig. S3–7†).88,89 The gels (see ESI† Fig. 3–7) were scanned by densitometry and the data reporting the relative amount of Form II nicked DNA are tabulated in Table 1. In addition to the observation of nicked DNA in agarose gels, large scale DNA cleavage reactions were extracted with dichloromethane and the characteristic small molecule by-products of deoxyribose degradation, 5 MF and furfural, could be identified by HPLC analyses (see ESI Fig. S11†).90,91 5 MF and furfural are characteristic neutral byproducts of hydrogen atom abstraction from the C1 and C5 deoxyribose positions, respectively. Their presence indicates non-specific H atom abstraction and is supporting of hydroxyl radical as the active agent.
| 44+ + ½GSH → 4˙3+ + ½GSSH | (1) |
| 4˙3+ + O2 → O2˙− + 44+ | (2) |
| Inhibitor | % Form II cleavage from 44+ digestion | % Form II cleavage from 32+ digestion | |
|---|---|---|---|
| Benzoate (mM) | 0 | 74 | 74 |
| 2 | 44 | 53 | |
| 4 | 33 | 40 | |
| 6 | 30 | 36 | |
| Formate (mM) | 0 | 62 | 75 |
| 2 | 44 | 57 | |
| 4 | 30 | 43 | |
| 6 | 27 | 39 | |
| Mannitol (mM) | 0 | 67 | 71 |
| 2 | 55 | 53 | |
| 4 | 39 | 49 | |
| 6 | 39 | 36 | |
| EtOH (mM) | 0 | 71 | 79 |
| 2 | 54 | 55 | |
| 4 | 39 | 46 | |
| 6 | 39 | 45 | |
| Pyruvate (mM) | 0 | 74 | 78 |
| 2 | 30 | 34 | |
| 4 | 31 | 28 | |
| 6 | 28 | 23 | |
| DMSO (mM) | 0 | 64 | 74 |
| 2 | 43 | 50 | |
| 4 | 34 | 37 | |
| 6 | 31 | 31 | |
| DEF (mM) | 0 | 65 | 50 |
| 2 | 26 | 19 | |
| 4 | 22 | 17 | |
| 6 | 22 | 17 | |
| SOD (μg mL−1) | 0 | 64 | 77 |
| 15 | 39 | 50 | |
| Catalase (μg mL−1) | 0 | 67 | 75 |
| 15 | 19 | 18 | |
| SOD and catalase (μg mL−1) | 0 | 67 | 75 |
| 15 | 20 | 20 | |
Cleavage reactions in the presence of SOD and/or catalase revealed a central role for H2O2 over superoxide. As shown in Fig. 5, addition of SOD attenuates but does not completely stop the cleavage activity of 44+/GSH or 32+/GSH aerobic mixtures whereas catalase does. The cleavage was also strongly attenuated upon addition of sodium pyruvate (see ESI Fig. S9†), which is a selective scavenger for H2O2, further revealing H2O2 as an integral intermediate in the cleavage mechanism.92,93 As SOD scrubs out superoxide and produces O2 and H2O2, a basal level of cleavage activity would be expected if H2O2 were generated via this pathway and if H2O2 is the primary precursor to hydroxy radical. Catalase, which decomposes H2O2 to water and O2, would completely arrest cleavage if H2O2 were the necessary precursor to hydroxy radical, as is observed.
 | ||
| Fig. 4 In vitro DNA plasmid cleavage assay in which pUC19 DNA (154 μM DNA-bp) was incubated with 44+ (31 μM) in PBS buffer (pH 7.2) and 1.0 mM GSH at varying [O2]. Lane 1: control, no 44+, 220 mM O2. Lane 2–5: DNA, 44+, and varying amounts of O2. Lane 4 also contains 30 μM 3,4-dihydroxybenzoate to show that this does not interfere with the assay; lane 5 contains 30 μM 3,4-dihydroxybenzoate and 5 units of protocatechuate dioxygenase. | ||
 | ||
| Fig. 5 Effect of varying concentrations of SOD and catalase on the DNA cleavage activity of 32+ ((A) top gel) and 44+ ((B) bottom gel). Agarose gel (1%) stained with ethidium bromide of supercoiled pUC18 DNA (154 μM) cleavage products after incubation at 25 °C for 48 h with RPC (12.8 μM), GSH (256 μM) in 50 mM Na3PO4/10 mM buffer (pH 7.2). Lane 1: DNA control; lane 2: GSH and DNA; lane 3: DNA and RPC; lane 4: SOD (15 μg mL−1) DNA; lane 5: catalase (15 μg mL−1) and DNA; lane 6: RPC, GSH and DNA; lane 7: RPC, GSH, SOD (15 μg mL−1) and DNA; lane 8: RPC, GSH, catalase (15 μg mL−1) and DNA; lane 9: RPC, GSH, SOD (15 μg mL−1), catalase (15 μg mL−1) and DNA. All reactions were carried out under aerobic conditions. | ||
Given the requirement for H2O2, reactions (1) and (2) do not explain the observed catalase inhibition, as reaction (2) only yields H2O2 indirectly, by either disproportionation, reaction (3), or by superoxide reduction by some other substrate. This latter reaction is limited, however, by the modest reduction potential of superoxide (+0.36 V at pH 7 and 25 C).94 Disproportionation (rxn 2) should become slow at low [O2] as this would lead to low [O2˙−], but as reported previously, DNA cleavage by 44+ was enhanced as the [O2] was lowered.95 Oxygen concentrations were not quantitated in this previous study, so we examined the DNA cleavage activity of 44+/GSH mixtures at three [O2]. Aerobic solutions had a measured [O2] of 220 μM, as determined by an O2 sensitive electrode. Solutions prepared in a nitrogen-filled glove box measured 4.0 μM [O2] and solutions in the nitrogen glove box and which were internally scrubbed by addition of protocatechuate 3,4-dioxygenase, and its substrate, 3,4-dihydroxybenzoate96 showed no measureable [O2] by the O2 sensitive electrode. As shown in Fig. 4, considerably greater cleavage is observed for samples with 4.0 μM O2 (lanes 3 and 4), compared to that under normoxic conditions (220 μM O2, lane 2). However, no cleavage is seen in the absence of O2 (lane 5). Lane 4 is a control in which 3,4-dihydroxybenzoate is present, and reveals this additive does not affect the DNA cleavage, nor does the protocatechuate 3,4-dioxygenase protein (data not shown).
| O2˙− + H2O2 → HO˙ + HO− + O2 | (3) |
With these data the question becomes, how does low [O2] paradoxically favor enhanced DNA cleavage by ROS and how is H2O2 produced? Both of these questions can be explained by consideration of the multiple accessible and reversible redox states present in RPCs 32+, 44+ and 54+. As shown in Fig. 3 (top), 44+ can undergo a single reduction to form the radical complex 4˙3+, or two reductions, accompanied by protonation at pH 7.2, to yield the diamagnetic complex H244+ (shown as the benzoid tautomer).97 These three isomers are analogous to the quinone, semiquinone radical, and hydroquinone isomers seen in the anthracyclines above, but based on reversible imine/amine couples.
A mechanistic pathway consistent with these results is shown in Scheme 1, which details a multistep pathway by which O2 is activated to form superoxide and hydrogen peroxide upon redox cycling by the RPC 44+ (or 32+). As indicated in the column on the left, RPC 44+ can bind to DNA and be interconverted between three redox states by reaction with GSH and O2. The relative amount of a given redox isomer being dictated by the GSH/O2 ratio under steady-state conditions. At low GSH/O2, the steady-state concentrations are shifted to the more oxidized isomers whereas at low GSH/O2 the opposite occurs. As H244+ (or H232+) can directly produce H2O2 in a single step via a 2-electron, 2-proton transfer to O2 (reaction (4)), circumstances which favor a greater steady state concentration of [H244+] also favor H2O2 production. Now the enhanced DNA cleavage activity under low [O2] can be rationalized in terms of the enhanced efficiency at producing H2O2 relative to superoxide under hypoxia-like conditions. The direct production of H2O2 from the oxidation of hydroquinones,98,99 polyphenols,100 dihydroflavins,101,102 and even dihydropyrazines103 is well established.
| H244+ + O2 → H2O2 + 44+ | (4) |
 | ||
| Scheme 1 | ||
Given the situation in which H2O2 production increases as the [O2] is lowered, enhanced DNA cleavage is explained by the H2O2 activation via Fenton chemistry (reaction (5))104 or reactions with other reductants, such as reactions (6) and (7), to yield the hydroxy radical. Adventitious Fe2+ is frequently observed to play a significant role in the activation of H2O2 (ref. 104) and appears to play a role here. Addition of the iron chelator, deferoxamine,105 to cleavage solutions of 44+ or 32+ and GSH and air mostly quenches the cleavage reaction (see ESI Fig. 10†), suggests that trace Fe2+ is involved in the hydroxy radical production.
| H2O2 + Fe2+ → HO˙ + OH− + Fe3+ | (5) |
| O2˙− + H2O2 → HO˙ + HO− + O2 | (6) |
| 4˙3+ + H2O2 → 44+ + HO˙ + HO− + O2 | (7) |
Reaction (7) shows that the radical 4˙3+ (or 3˙2+) can also activate H2O2 in a manner analogous to the semiquinone radical of anthracyclines.89,106,107 Assuming these RPCs are DNA bound, this would generate the hydroxy radical in the immediate vicinity of the DNA, possibly increasing potency.
It is important to emphasize the difference in this redox activity with that of other known ruthenium-based drugs which are activated by reduction. A number of Ru(III) drugs, such as KP-1019 and NAMI-A, are bioreduced in situ to form the ‘active’ Ru(II) drug.7,8 This reduction is irreversible and the resulting Ru(II) is thought to form adducts with many cellular structures, resulting in apoptosis, however even today the exact cellular targets and mechanism of action are not fully understood.6,108,109 In contrast, the ruthenium(II) center in RPC 32+ and 44+, are essentially inert throughout the process. Instead, the tatpp ligand is the redox-active unit which redox cycles with GSH and O2 to catalytically generate ROS, which is conceptually related to the redox-cycling of Cu(II)phen and Fe–bleomycin.8,110–112 However, again an important distinction remains in that Cu(II)phen and Fe–bleomycin show a directly proportional relationship between the observed DNA cleavage activity and the [O2], whereas RPC 32+ and 44+ show an inverse proportionality. This is due to the presence of two ligand-based redox couples, the doubly-reduced form being increasingly accessed as the GSH/O2 ratio climbs and which can then react with the remaining O2 to directly produce H2O2 with better efficiency. This unusual [O2] dependence could have utility in enhancing the treatment in hypoxic regions of tumors.
As a point of clarification, we have previously reported that the doubly-reduced [H24]Cl4 cleaved DNA in the absence of both GSH and O2.69 Moreover, added 2,2,6,6-tetramethyl-1-piperdinyloxy (TEMPO) attenuated this cleavage while added DMSO did not. TEMPO can quench carbon-based radical species113,114 whereas DMSO is primarily a scavenger for ROS.105,115 At that time, we were using a nitrogen glove box for ‘anerobic‘ work for which we now know leads to solutions with measurable [O2]. Once this was understood, we assumed that ROS were responsible for the observed cleavage, however, this is at odds with the cleavage activity observed in the presence of DMSO. Another possibility, that we still need to demonstrate, is oxidation of H244+ by O2 leads to some DNA-bound 4˙3+. This radical persists for long periods in intimate contact with the DNA (and we know this radical is remarkably stable116,117) and possibly can directly abstract a H-atom from the deoxyribose unit. Such dual cleavage mechanisms (O2 dependent and O2 independent) are precedented in the related antibiotic anthraquinones, daunorubicin and doxorubicin,86,118 and DNA cleaving-dihydropyrazines,103,119–122 both which form DNA bound radical species that can either activate O2 or directly attack the DNA.
ROS production and DNA cleavage activity in cultured human cancer cells
The inhibitory concentrations 50% (IC50) for many of the RPCs in cultured human NSCLC H358 cells are reported in Table 2 and were determined using the MTT assay. In general, RPCs that are not redox-active and which do not induce DNA cleavage in cell-free assays, as described previously, are less cytotoxic. The clear exception being RPC 22+, which is the most potent of all those examined, with an IC50 of 1.7 μM. RPCs 32+ and 44+ were the next most potent at 13–15 μM. We have previously shown that H358 cells treated with 32+ or 44+ (5 μM) for as little as 1 hour have appreciable quantities of ruthenium in both the whole cell and nuclear fractions, as detected by graphite furnace atomic absorption spectroscopy, revealing facile transport into the cells and nucleus.75 RPC 22+ is well-known for its cytotoxic properties and is found to locallize in lysosomes and mitochondria.24 It's cytotoxicity is largely been attributed to mitochondiral poisoning.24| Compound (RPC) | Abbr | H358 IC50 (μM) | Ref. |
|---|---|---|---|
| [Ru(phen)3]2+ | 12+ | 86.7 ± 4.1 | 75 |
| [Ru(Ph2phen)3]2+ | 22+ | 1.7 ± 0.1 | This work |
| [(phen)2Ru(tatpp)]2+ | 32+ | 13.2 ± 1.8 | 75 |
| [(phen)2Ru(tatpp)Ru(phen)2]4+ | 44+ | 15.2 ± 1.8 | 75 |
| [(phen)2Ru(dppz)]2+ | 62+ | 35.1 ± 0.71 | This work |
| [(phen)2Ru(tpphz)]2+ | 72+ | 44.0 ± 3.0 | 75 |
| [(phen)2Ru(tpphz)Ru(phen)2]4+ | 84+ | 41.8 ± 2.7 | 75 |
H358 cells treated with RPC 44+ (15 μM) and to a lesser extent, 32+, (13 μM) show significantly elevated ROS levels in within 2 hours of treatment as observed using a fluorescent ROS-sensitive dye, DCFH-DA,123 and fluorescent microscopy. DCFH-DA is an oxidation sensitive dye that fluoresces brightly and is measured in the green when intracellular ROS is generating in a cell.54,80,124 DCFH-DA was commonly and inaccurately thought to be a H2O2 specific marker but more recently shown to be a more general ROS detection dye, as it is also sensitive to superoxide ion.80,125 As shown in Fig. 6, the green fluorescence image tracks indicate ROS production within the H358 cells treated with H2O2, RPCs 32+, 44+ and 22+, or untreated cells. The negative control shows the basal levels of ROS whereas the positive control shows the dye activity in the presence of H2O2, principly in the cytoplasm. Cells treated with RPC 22+ (1.7 μM) represent a negative control in that this RPC, while quite cytotoxic, does not redox-cycle nor generate ROS unless specifically irradiated with light,17,24,126 which is avoided here. Comparisons of the columns in which RPCs 32+, 44+ and 22+ were used show that RPC 44+ clearly promotes significant ROS production in cells after only 2 h incubation, whereas RPC 32+ does elevate ROS production also, but less dramatically so. At longer incubation periods (22 h), both 32+ and 44+ generate substantial amounts of ROS intracellularly. RPC 22+ does not result ROS production over the basel level at 2 or 8 h. At 22 h, the increase can be largely attributed to indirect pathways to ROS production as the cytotoxic activity results in activation of apoptotic pathways. Most significantly, the ROS activity seen in the gel-shift assays for 32+ and 44+ and the lack of activity for 22+ are clearly mirrored here.
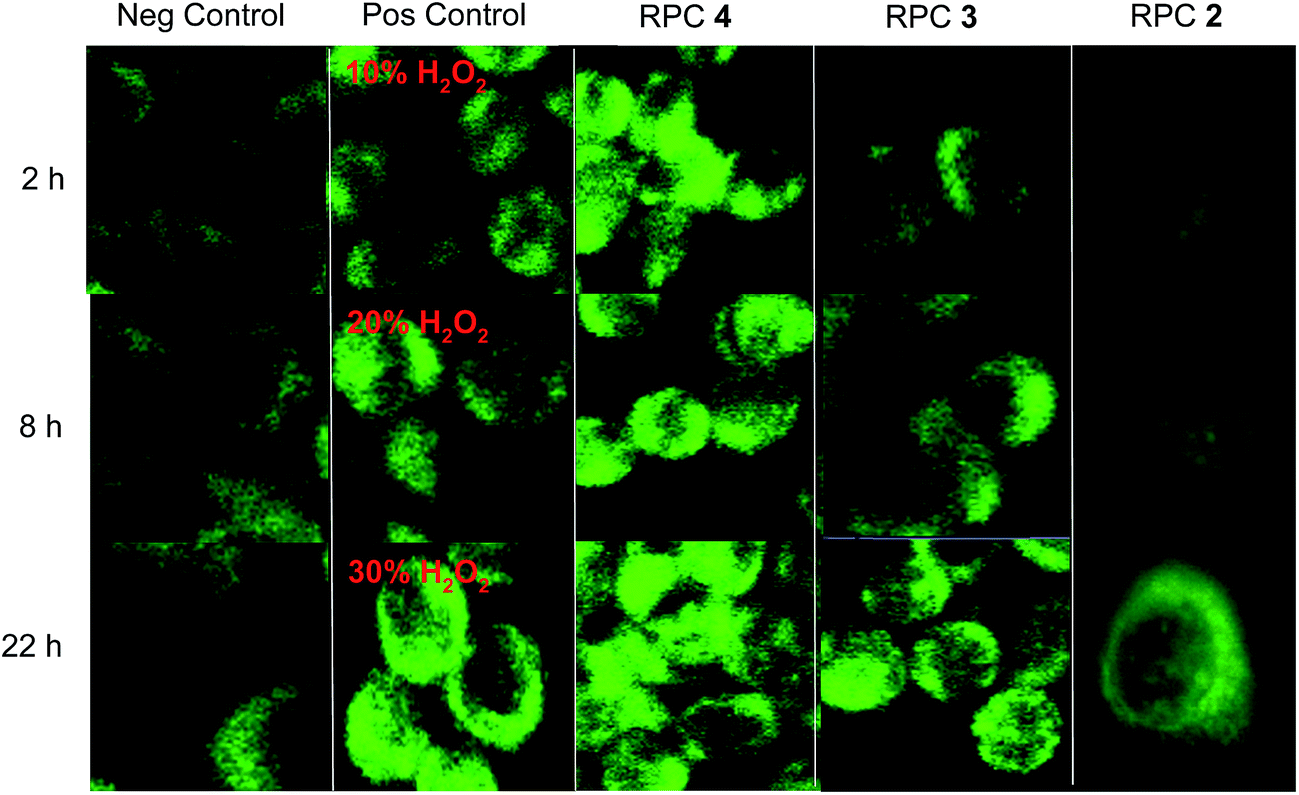 | ||
| Fig. 6 H358 cells stained with DCFH-DA to image ROS production. First column is untreated cells as a negative control. Second column is the positive control where cells were dosed with 10, 20, and 30% solutions of H2O2 for 15 min and imaged with DCFH-DA. Third, fourth, and fifth columns show H358 cells dosed with relative IC50 values of various complex as follows: 44+ (15 μM), 33+ (13 μM) and 22+ (1.7 μM) for the 3 time periods indicated. DCFH-DA was then administered for 30 min and imaged using confocal microscopy (488/519 nm). | ||
DNA cleavage activity in cultured human cancer cells
The single strand DNA cleavage observed cell-free assays and the ROS activity seen in cells, is observed to lead to DSBs in H358 cells. Oxidative DNA damage in the form of DSBs could be detected by monitoring the appearance of the phosphorylated protein, ataxia-telangiectasia mutated (pATM) and its downstream effect, γH2AX, in the nuclei of treated cells using immunofluorescent (IF) stains.127–129 DSBs induced by a number of causes including ionizing radiation and ROS are known to activate-ATM.130 As seen in Fig. 7, nuclear DSBs show up as green foci upon fixing and staining cells with ATM primary antibody where the nucleus of each cell is stained with propidium iodide (PI). Each track in the series shows the merged signals of ATM and PI. Etoposide was used as a positive control, as it is known to stabilize transient covalent complexes between topo 2 and DNA, ultimately converting them to DSBs in the S-phase cell cycle.131,132 As seen in Fig. 7, cells treated with the IC50 dose of 44+ and etoposide (1 μM) show numerous foci representing DSBs recruiting ATM at 2 h which become ever more present at longer time periods, 8 and 22 h, respectively. Cells treated with the IC50 dose of 22+ and 32+ show no or little pATM foci at 2 h, respectively, and while foci become apparent for 32+ at 8 and 22 h, they are never observed for cells treated with 22+ (up to 22 h). Again we observe distinctly different outcome for redox-active versus redox-inactive RPCs, with a noticeable lag in the activity in 32+ compared to 44+.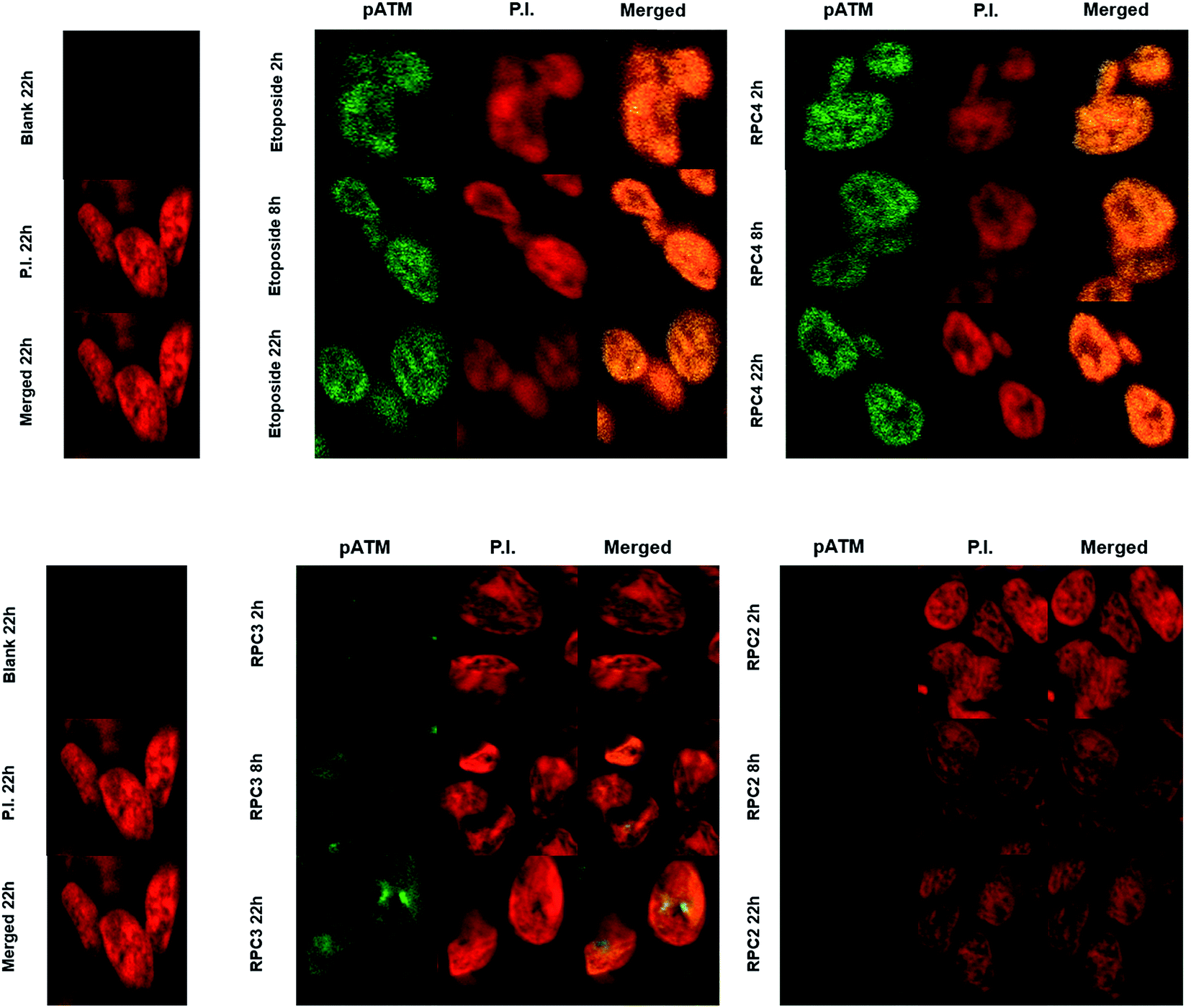 | ||
| Fig. 7 Immunofluorescence staining of pATM foci in H358 cell line. The cells were fixed stained and imaged at 2, 8, and 22 h post treatment with the IC50 values for 44+ and 32+. | ||
DSBs also cause damage at associated loci on the histone cause the histone ser-139 residues in mammalian cells to be phosphorylated which can be directly detected using the γH2AX assay.131 DSBs after activation of ATM recruits the downstream phosphorylation of γH2AX and show up as yellow-green foci upon fixing and staining H358 cells with γH2AX primary antibody. For a positive control, H358 cells were irradiated (IR) with 1.8 Gy and show numerous DSB foci within 30 minutes132–136 and is compared to the negative control of untreated H358 cells, which is dark (Fig. 8, left). As in our ATM assay, etoposide was used again as a postive control for DSB formation and in Fig. 8, cells treated with the IC50 dose (1.0 μM etoposide) show again numerous DSBs at 2 h which become substantial at longer time periods, 8 and 22 h, respectively.131,132 The next two columns in Fig. 8 show similar nuclear effects upon treating H358 cells with 44+ (15 μM) and 32+ (13 μM) at their IC50 dose. Cells treated with 44+ mimiced etoposide with numerous DSBs evident after the 2 h time period and even greater increases seen in the foci count with increasing time. In tandem with the ATM assay, RPC 32+ showed substantially fewer DSBs at the 2 and 8 h time points, than seen for etoposide or 44+. At 22 h, the appearance of numerous DSBs could be attributed to an indirect mechanism and an apoptotic cascade, however it is notable that no DSBs are seen in the nuclei of cells treated with 22+, even at 22 h. This data reveal very different mechanisms of action for 44+versus22+ and hint that the mechanism of action for 32+ may deviate from 44+. It is hard to attribute the strong 2 h DSB response seen in cells treated with 44+ to anything other than a direct response, which is mirrored by other agents directly acting on the nuclear DNA (etoposide and radiation) and these DSBs represent the primary event responsible for apoptosis.135,137
 | ||
| Fig. 8 Immunofluorescence staining of γH2AX foci in H358 cell line. The cells were fixed stained and imaged at 2, 8, and 22 h post treatment with the IC50 values for 44+ and 32+. | ||
As shown in Fig. 9, quantitation of the γH2AX foci (using the Image J software package with gives a count of the foci per 25 cells138) reveals that etoposide and 44+ show an equal response after 2 h, whereas 32+ is only slightly above the negative control. At 8 h, 44+ shows more foci than etoposide and almost 5 fold more foci than cells treated with 32+. Only after 22 h, do the foci count become near equal and the extensive number of foci (over 250 each) indicative of apoptosis. RPC 44+ clearly shows mechanistic similarities with other DNA cleavage agents and is competitive with etoposide. The DSBs for 32+, on the other hand, indicate a potential divergence in their mechanistic pathway in cells, despite near identical behavior cell-free studies. This last result is particularly intriguing as both 32+ and 44+ are essentially equitoxic as measured by IC50 values to H358 cells, and show similar tumor growth inhibition in mouse tumor models.70 Additional studies must be performed with 32+ and 44+ to determine if these divergent results are due to different transport rates and pathways, cellular localization, or simply different pro-apoptotic cascade pathways.
 | ||
| Fig. 9 Quantitative analysis of γ-H2AX foci in H358 cell line for etoposide, 44+, and 32+ using image J software. An average of 25 cells per count were used in tandem with double phase light contrast particle count. | ||
It is curious that SSBs are observed in cell-free assays, while DSBs are observed in cells. It is possible that the catalytic ROS activity of 32+ and 44+ leads to multiple SSBs resulting in DSBs, but it would be odd that this only occurs in cells. As these RPCs are known to intercalate, it could be that these RPCs induce other injuries to the cellular DNA, such as topoisomerase inhibition. In combination with the ROS generation, DSBs are effectively produced. The low cytotoxicity of other known metallointercalators with very similar structures, but lacking the redox cycling functionality (i.e., RPCs 62+, 72+, and 84+; see Table 2) suggest that intercalation in the absence of ROS production is not sufficient for a substantial cytotoxic effect. Further supporting this, fluorescent imaging of MCF7 cells treated with RPC 84+ reveals this RPC does accumulate in the cell nuclei, yet it is relatively non-cytotoxic (MCF7 IC50 138 μM).42,43 We postulate that the combination of efficient ROS production in the immediate vicinity of the DNA and inhibition of normal nuclear DNA functions by intercalation result in DSBs in cells, whereas only SSBs are seen in cell-free assays where the family of DNA associated proteins are absent.
Effects of RPCs in cancer cells within a hypoxic environment
An examination of the cytotoxicity of 32+ and 44+ in H358, HCC2998, HOP-62, Hs766t was conducted under normoxia (18% O2) and hypoxia (1.1% O2), to see if the O2 sensitivity seen in vitro in observed in cells. The bar graphs in Fig. 10 compare the IC50, as measured by MTT assay, of 32+ and 44+ when the cells are incubated under normoxic conditions (blue) and hypoxic conditions (red). A two-fold enhancement in cytotoxicity is seen for 32+ in Hs766t and HOP-62 under hypoxia compared to normoxia, however, the remaining cell lines, H358 and HCC2998, showed little difference. Surprisingly, no difference was observed in the cytotoxicity of 44+ in all four cell lines between normoxic and hypoxic conditions. Ultimately, it appears that even though DNA cleavage is enhanced in cell-free assays under hypoxic relative to normoxic conditions, the DNA damage done in cells under normoxic conditions is sufficient to trigger the same apoptotic response as seen under hypoxia. While this suggests the RPC 44+ is not selective for hypoxic cell populations, the fact that the activity of 44+ is not diminished under hypoxia is still quite attractive, as many anticancer drugs become less effective when cells are under hypoxic stress.139,140 | ||
| Fig. 10 IC50 of human malignant cell lines treated with RPCs 32+ and 44+ under normoxia (18% O2) and hypoxia (1.1% O2) represented by the blue and red bars respectively. In this case the enantiopure Δ-32+ and ΔΔ-44+ were used, which is why the IC50's reported under normoxia are lower. IC50's were determined using the MTT assay. Error bars indicate the standard deviation of IC50 as measured from three 96 well plates. Each plate contained six replicates at each concentration to determine the IC50. | ||
Conclusions
The cell-free data support a mechanistic model in which single-strand cleavage activity of DNA-bound 32+ and 44+ is observed due to redox-cycling mediated by the [GSH] and [O2]. The [GSH]/[O2] ratio dictates the steady state concentration of the three redox isomers of 32+ and 44+ in such a manner that at low [O2] and high [GSH], a pathway favorable to H2O2 production becomes increasingly favorable. The relative efficiency by which H2O2 can be activated to form hydroxy radicals over superoxide results in enhanced DNA cleavage under low O2 conditions (assuming [GSH] is held relatively constant). This redox-cycling means that the RPCs are catalytic with respect to DNA cleavage. The tatpp ligand is key to this functionality and bears some resemblance to the intercalating anthraquinone anti-cancer drugs, which also show DNA damage via redox cycling. The metal fragments impart a similar DNA binding affinity to the RPCs as the anthraquinones and the tatpp ligand imparts the redox-cycling activity. It remains to be seen how these two classes of intercalators overlap in terms of specificity, toxicity, and spectrum of use and where they diverge.The DNA cleavage activity of RPC 44+ and to a lesser extent 32+ is observed in the nuclei of H358 cells, however this time as DSBs. Within 2 h of treatment with an IC50 dose of 44+, H358 cells show elevated levels of ROS as detected by the fluorescent ROS-sensitive dye, DCFH-DA, marked phosporylation of the ATM signalling protein in the nuclei indicating DNA damage in response to ROS, and direct observation of DSBs in the nuclei using the γH2AX assay. Cells treated with 32+ also show these responses, but with a 3 to 6 h temporal delay that could indicate an indirect cleavage mechanism and which could suggest divergent reaction mechanisms for 32+ and 44+ in live cells. Cells treated with the nonredox-active RPC 22+, which is even more cytotoxic towards H358, show none of these behaviors even after 22 h treatment, suggesting the redox cycling in 32+ and 44+ is integral towards their function.
This correlation in activity between cell-free and cell studies is a first with ruthenium polypyridyl-based drugs, to our knowledge. It is interesting that the enhancement in cytotoxicity seen for RPC 32+ under hypoxia over normoxia is more pronounced than for 44+. While neither 32+ or 44+ is dramatically more cytotoxic under hypoxia if at all compared to normoxia, it is promising to note that they are not less effective under hypoxia, which is common to many O2 activating drugs.
This work also demonstrates the importance of analyzing the temporal cellular effects of treating cells with RPCs. There are numerous reports explaining how RPCs, including RPC 22+, poison mitochondria or disrupt other cellular functions, however the conclusions are based on a single time point, meaning that the observed effect could be due to apoptotic cascades induced by the RPC at any number of locations.
Notes
MacDonnell is a co-founder of Tuevol Therapeutics of Fort Worth, TX which has licensed portions of this technology from the University of Texas at Arlington.Acknowledgements
We thank Dr Rolf Brekken, Dr Brad Pierce, Dr Shreeyukta Singh, and Ms Singinee Sardar for their assistance and helpful discussions on this work. We also thank the Robert A. Welch Foundation (Y-1301) and the National Science Foundation (CHE-1301332) for financial support of this work.References
- D. Lebwohl and R. Canetta, Eur. J. Cancer, 1998, 34, 1522–1534 CrossRef CAS PubMed.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- N. Farrell, Compr. Coord. Chem. II, 2004, 9, 809–840 CAS.
- X. Meng, M. L. Leyva, M. Jenny, I. Gross, S. Benosman, B. Fricker, S. Harlepp, P. Hebraud, A. Boos, P. Wlosik, P. Bischoff, C. Sirlin, M. Pfeffer, J. P. Loeffler and C. Gaiddon, Cancer Res., 2009, 69, 5458–5466 CrossRef CAS PubMed.
- R. E. Morris, R. E. Aird, S. Murdoch Pdel, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Muller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed.
- E. Reisner, V. B. Arion, B. K. Keppler and A. J. L. Pombeiro, Inorg. Chim. Acta, 2008, 361, 1569–1583 CrossRef CAS.
- A. Bergamo, B. Gava, E. Alessio, G. Mestroni, B. Serli, M. Cocchietto, S. Zorzet and G. Sava, Internet J. Oncol., 2002, 21, 1331–1338 CAS.
- H. Depenbrock, S. Schmelcher, R. Peter, B. Keppler, G. Weirich, T. Block, J. Rastetter and A.-R. Hanauske, Eur. J. Cancer, 1997, 33, 2404–2410 CrossRef CAS PubMed.
- F. Lentz, A. Drescher, A. Lindauer, M. Henke, R. A. Hilger, C. G. Hartinger, M. E. Scheulen, C. Dittrich, B. K. Keppler and U. Jaehde, Anti-Cancer Drugs, 2009, 20, 97–103 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2015, 33, 201–214 CrossRef CAS PubMed.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- J. Fong, K. Kasimova, Y. Arenas, P. Kaspler, S. Lazic, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2015, 14, 2014–2023 CAS.
- P. Kaspler, S. Lazic, S. Forward, Y. Arenas, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2016, 15, 481–495 CAS.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- C. Moucheron, M. A. K. De and J. M. Kelly, Struct. Bonding, 1998, 92, 163–216 CrossRef CAS.
- S. Swavey and K. J. Brewer, Inorg. Chem., 2002, 41, 4044–4050 CrossRef CAS PubMed.
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 2426–2428 RSC.
- R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161–17175 CrossRef CAS PubMed.
- S. Monro, J. Scott, A. Chouai, R. Lincoln, R. Zong, R. P. Thummel and S. A. McFarland, Inorg. Chem., 2010, 49, 2889–2900 CrossRef CAS PubMed.
- G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Coord. Chem. Rev., 2015, 282–283, 127–138 CrossRef CAS.
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370–10377 CrossRef CAS PubMed.
- I. Ortmans, B. Elias, J. M. Kelly, C. Moucheron and A. Kirsch-DeMesmaeker, Dalton Trans., 2004, 4, 668–676 RSC.
- F. O'Reilly, J. Kelly and A. K.-D. Mesmaeker, Chem. Commun., 1996, 9, 1013–1014 RSC.
- E. C. Glazer, Isr. J. Chem., 2013, 53, 391–400 CrossRef CAS.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- E. Wachter and E. C. Glazer, J. Phys. Chem. A, 2014, 118, 10474–10486 CrossRef CAS PubMed.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC.
- E. Wachter, B. S. Howerton, E. C. Hall, S. Parkin and E. C. Glazer, Chem. Commun., 2014, 50, 311–313 RSC.
- A. Shulman and F. P. Dwyer, in Chelating Agents and Metal Chelates, ed. F. P. Dwyer and D. P. Mellor, 1964, ch. 9, pp. 383–435 Search PubMed.
- D. O. White, A. W. Harris, I. M. Cheyne and M. Shew, Aust. J. Exp. Biol. Med. Sci., 1969, 47, 81–89 CrossRef CAS PubMed.
- U. Schatzschneider, J. Niesel, I. Ott, R. Gust, H. Alborzinia and S. Wölfl, ChemMedChem, 2008, 3, 1104–1109 CrossRef CAS PubMed.
- F. P. Dwyer, E. Mayhew, E. M. F. Roe and A. Shulman, Br. J. Cancer, 1965, 19, 195–199 CrossRef CAS PubMed.
- J. H. Koch, W. P. Rogers, F. P. Dwyer and E. C. Gyarfas, Aust. J. Biol. Sci., 1957, 10, 342–350 CrossRef CAS.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- Y. Liu, A. Chouai, N. N. Degtyareva, D. A. Lutterman, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2005, 127, 10796–10797 CrossRef CAS PubMed.
- M. R. Gill, H. Derrat, C. G. W. Smythe, G. Battaglia and J. A. Thomas, ChemBioChem, 2011, 12, 877–880 CrossRef CAS PubMed.
- K. E. Augustyn, V. C. Pierre, J. K. Barton and T. P. Begley, in Wiley Encyclopedia of Chemical Biology, John Wiley & Sons, Inc., 2007, DOI:10.1002/9780470048672.wecb328.
- E. Baggaley, M. R. Gill, N. H. Green, D. Turton, I. V. Sazanovich, S. W. Botchway, C. Smythe, J. W. Haycock, J. A. Weinstein and J. A. Thomas, Angew. Chem., Int. Ed., 2014, 53, 3367–3371 CrossRef CAS PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- E. Meggers, Curr. Opin. Chem. Biol., 2007, 11, 287–292 CrossRef CAS PubMed.
- M. J. Pisani, D. K. Webster, K. Heimann, J. G. Collins and F. R. Keene, Metallomics, 2010, 2, 393–396 RSC.
- G. Pascu, A. Hotze, C. Sanchez-Cano, B. Kariuki and M. Hannon, Angew. Chem., Int. Ed., 2007, 46, 4374–4378 CrossRef CAS PubMed.
- C. B. Spillane, N. C. Fletcher, S. M. Rountree, H. Berg, S. Chanduloy, J. L. Morgan and F. R. Keene, J. Biol. Inorg Chem., 2007, 16, 797–807 CrossRef PubMed.
- A. H. Velders, H. Kooijman, A. L. Spek, J. G. Haasnoot, D. De Vos and J. Reedijk, Inorg. Chem., 2000, 39, 2966–2967 CrossRef CAS PubMed.
- U. McDonnell, J. M. C. A. Kerchoffs, R. P. M. Castineiras, M. R. Hicks, A. C. G. Hotze, M. J. Hannon and A. Rodger, Dalton Trans., 2008, 667–675, 10.1039/b711080d.
- O. Novakova, J. Kasparkova, O. Vrana, P. M. van Vliet, J. Reedijk and V. Brabec, Biochemistry, 1995, 34, 12369–12378 CrossRef CAS PubMed.
- C. Tan, S. Lai, S. Wu, S. Hu, L. Zhou, Y. Chen, M. Wang, Y. Zhu, W. Lian and W. Peng, J. Med. Chem., 2010, 53, 7613–7624 CrossRef CAS PubMed.
- M. R. Gill, S. N. Harun, S. Halder, R. A. Boghozian, K. Ramadan, H. Ahmad and K. A. Vallis, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- L. Zeng, Y. Chen, J. Liu, H. Huang, R. Guan, L. Ji and H. Chao, Sci. Rep., 2016, 6, 19449 CrossRef CAS PubMed.
- N. O. Brabec V, Drug Resist. Updates, 2006, 9, 111–122 CrossRef PubMed.
- A. Wragg, M. R. Gill, D. Turton, H. Adams, T. M. Roseveare, C. Smythe, X. Su and J. A. Thomas, Chem.–Eur. J., 2014, 20, 14004–14011 CrossRef CAS PubMed.
- S. P. Foxon, C. Metclafe, H. Adams, M. Webb and J. A. Thomas, Inorg. Chem., 2007, 46, 409–416 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, Bioorg. Med. Chem., 2010, 18, 3564–3569 CrossRef CAS PubMed.
- M. J. Pisani, P. D. Fromm, Y. Mulyana, R. J. Clarke, H. Körner, K. Heimann, J. G. Collins and F. R. Keene, ChemMedChem, 2011, 6, 848–858 CrossRef CAS PubMed.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2014, 134, 20376–20387 CrossRef PubMed.
- C. Tan, S. Wu, S. Lai, M. Wang, Y. Chen, L. Zhou, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, Dalton Trans., 2011, 40, 8611–8621 RSC.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, Biochemistry, 2008, 47, 11711–11716 CrossRef CAS PubMed.
- Y. Du, X. Fu, H. Li, B. Chen, Y. Guo, G. Su, H. Zhang, F. Ning, Y. Lin, W. Mei and T. Chen, ChemMedChem, 2014, 9, 714–718 CrossRef CAS PubMed.
- F. Li, E. J. Harry, A. L. Bottomley, M. D. Edstein, G. W. Birrell, C. E. Woodward, F. R. Keene and J. G. Collins, Chem. Sci., 2014, 5, 685–693 RSC.
- Y.-J. Liu, C.-H. Zeng, Z.-H. Liang, J.-H. Yao, H.-L. Huang, Z.-Z. Li and F.-H. Wu, Eur. J. Med. Chem., 2010, 45, 3087–3095 CrossRef CAS PubMed.
- T. Joshi, V. Pierroz, S. Ferrari and G. Gasser, ChemMedChem, 2014, 9, 1419–1427 CrossRef CAS PubMed.
- J. G. Vos and J. M. Kelly, Dalton Trans., 2006, 4869–4883, 10.1039/b606490f.
- T. K. Janaratne, A. Yadav, F. Ongeri and F. M. MacDonnell, Inorg. Chem., 2007, 46, 3420–3422 CrossRef CAS PubMed.
- A. Yadav, T. Janaratne, A. Krishnan, S. S. Singhal, S. Yadav, A. S. Dayoub, D. L. Hawkins, S. Awasthi and F. M. MacDonnell, Mol. Cancer Ther., 2013, 12, 643–653 CrossRef CAS PubMed.
- E. Kabingu, A. R. Oseroff, G. E. Wilding and S. O. Gollnick, Clin. Cancer Res., 2009, 15, 4460–4466 CrossRef CAS PubMed.
- F. H. Burstall, J. Chem. Soc., 1936, 173–175 RSC.
- P. A. Mabrouk and M. S. Wrighton, Inorg. Chem., 1986, 25, 526–531 CrossRef CAS.
- K. Wärnmark, O. Heyke, J. A. Thomas and J.-M. Lehn, Chem. Commun., 1996, 2603–2604 RSC.
- M.-J. Kim, R. Konduri, H. Ye, F. M. MacDonnell, F. Puntoriero, S. Serroni, S. Campagna, T. Holder, G. Kinsel and K. Rajeshwar, Inorg. Chem., 2002, 41, 2471–2476 CrossRef CAS PubMed.
- F. M. MacDonnell and S. Bodige, Inorg. Chem., 1996, 35, 5758–5759 CrossRef CAS.
- J. Bolger, A. Gourdon, E. Ishow and J.-P. Launay, Inorg. Chem., 1996, 35, 2937–2944 CrossRef CAS.
- C. Crey, P. Dumy, J. Lhomme and M. Kotera, Synth. Commun., 2003, 33, 3727–3732 CrossRef CAS.
- T. E. Goyne and D. S. Sigman, J. Am. Chem. Soc., 1987, 9, 2846–2848 CrossRef.
- K. Krumova and G. Cosa, Applications in Biosciences and Nanosciences, in Singlet Oxygen: Volume 1, The Royal Society of Chemistry, 2016, vol. 1, pp. 1–21 Search PubMed.
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215–224 RSC.
- V. H. Le, M. R. McGuire, P. Ahuja, F. M. MacDonnell and E. A. Lewis, J. Phys. Chem. A, 2015, 119, 65–71 CrossRef CAS PubMed.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–833 CrossRef CAS PubMed.
- F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- B. K. Sinha and P. M. Politi, Cancer Chemother. Biol. Response Modif., 1990, 11, 45–57 CAS.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS PubMed.
- G. T. Wondrak, Antioxid. Redox Signaling, 2009, 11, 3013–3069 CrossRef CAS PubMed.
- A. A. Oroskar, C. Lambert and M. J. Peak, Free Radical Biol. Med., 1996, 20, 751–756 CrossRef CAS PubMed.
- D. A. Bates and C. C. Winterbourn, FEBS Lett., 1982, 145, 137–142 CrossRef CAS PubMed.
- W. K. Pogozelski and T. D. Tullius, Chem. Rev., 1998, 98, 1089–1108 CrossRef CAS PubMed.
- C. V. Sonntag and SpringerLink, Free-radical-induced DNA damage and its repair: a chemical perspective, Springer, New York, Berlin, 2006 Search PubMed.
- A. R. Giandomenico, G. E. Cerniglia, J. E. Biaglow, C. W. Stevens and C. J. Koch, Free Radical Biol. Med., 1997, 23, 426–434 CrossRef CAS PubMed.
- R. Franco, M. I. Panayiotidis and J. A. Cidlowski, J. Biol. Chem., 2007, 282, 30452–30465 CrossRef CAS PubMed.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A.-F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS PubMed.
- T. K. Janaratne, A. Yadav, F. Ongeri and F. M. MacDonnell, Inorg. Chem., 2007, 46, 3420–3422 CrossRef CAS PubMed.
- C. E. Aitken, R. A. Marshall and J. D. Puglisi, Biophys. J., 2008, 94, 1826–1835 CrossRef CAS PubMed.
- R. Konduri, H. Ye, F. M. MacDonnell, S. Serroni, S. Campagna and K. Rajeshwar, Angew. Chem., Int. Ed., 2002, 41, 3185–3187 CrossRef CAS PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- T. H. James, J. M. Snell and A. Welsaberger, J. Am. Chem. Soc., 1938, 60, 2084–2093 CrossRef CAS.
- M. Akagawa, T. Shigemitsu and K. Suyama, Biosci., Biotechnol., Biochem., 2003, 67, 2632–2640 CrossRef CAS PubMed.
- G. Eberlein and T. C. Bruice, J. Am. Chem. Soc., 1982, 104, 1449–1452 CrossRef CAS.
- C. Kemal, T. W. Chan and T. C. Bruice, J. Am. Chem. Soc., 1977, 99, 7272–7286 CrossRef CAS PubMed.
- N. Kashige, T. Takeuchi, S. Matsumoto, S. Takechi, F. Miake and T. Yamaguchi, Biol. Pharm. Bull., 2005, 28, 419–423 CAS.
- S. Goldstein, D. Meyerstein and G. Czapski, Free Radical Biol. Med., 1993, 15, 435–445 CrossRef CAS PubMed.
- M. J. Burkitt and R. P. Mason, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8440–8444 CrossRef CAS.
- J. Schreiber, C. Mottley, B. K. Sinha, B. Kalyanaraman and R. P. Mason, J. Am. Chem. Soc., 1987, 109, 348–351 CrossRef CAS.
- B. Kalyanaraman, K. M. Morehouse and R. P. Mason, Arch. Biochem. Biophys., 1991, 286, 164–170 CrossRef CAS PubMed.
- P. K. Shah, K. Bhattacharjee and P. K. Shukla, RSC Adv., 2016, 6, 113620–113629 RSC.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- J. Wu, W. Chen, Y. Yin, Z. Zheng and G. Zou, BioMetals, 2014, 27, 445–458 CrossRef CAS PubMed.
- R. M. Burger, Chem. Rev., 1998, 98, 1153–1170 CrossRef CAS PubMed.
- J. Chen, M. K. Ghorai, G. Kenney and J. Stubbe, Nucleic Acids Res., 2008, 36, 3781–3790 CrossRef CAS PubMed.
- D. L. Mohler, J. R. Downs, A. L. Hurley-Predecki, J. R. Sallman, P. M. Gannett and X. Shi, J. Org. Chem., 2005, 70, 9093–9102 CrossRef CAS PubMed.
- T. J. Connolly, M. V. Baldový, N. Mohtath and J. C. Scaiano, Tetrahedron Lett., 1996, 37, 4919–4922 CrossRef CAS.
- C. F. Brayton, Cornell Vet., 1986, 76, 61–90 CAS.
- R. Konduri, T. N. R. de, K. Rajeshwar and F. M. MacDonnell, J. Am. Chem. Soc., 2004, 126, 11621–11629 CrossRef CAS PubMed.
- N. R. de Tacconi, R. O. Lezna, R. Konduri, F. Ongeri, K. Rajeshwar and F. M. MacDonnell, Chem.–Eur. J., 2005, 11, 4327–4339 CrossRef CAS PubMed.
- K. W. Wellington, RSC Adv., 2015, 5, 20309–20338 RSC.
- T. Yamaguchi, M. Eto, K. Harano, N. Kashige, K. Watanabe and S. Ito, Tetrahedron, 1999, 55, 675–686 CrossRef CAS.
- T. Yamaguchi, N. Kashige, N. Mishiro, F. Miake and K. Watanabe, Biol. Pharm. Bull., 1996, 19, 1261–1265 CAS.
- T. Yamaguchi, S. Matsumoto and K. Watanabe, Tetrahedron Lett., 1998, 39, 8311–8312 CrossRef CAS.
- T. Yamaguchi, H. Nomura, K. Matsunaga, S. Ito, J. Takata and Y. Karube, Biol. Pharm. Bull., 2003, 26, 1523–1527 CAS.
- A. Aranda, L. Sequedo, L. Tolosa, G. Quintas, E. Burello, J. V. Castell and L. Gombau, Toxicol. In Vitro, 2013, 27, 954–963 CrossRef CAS PubMed.
- T. A. Bhat, S. Kumar, A. K. Chaudhary, N. Yadav and D. Chandra, Drug Discovery Today, 2015, 20, 635–643 CrossRef CAS PubMed.
- C. Qian, J.-Q. Wang, C.-L. Song, L.-L. Wang, L.-N. Ji and H. Chao, Metallomics, 2013, 5, 844–854 RSC.
- A. Hergueta-Bravo, M. E. Jimenez-Hernandez, F. Montero, E. Oliveros and G. Orellana, J. Phys. Chem. B, 2002, 106, 4010–4017 CrossRef CAS.
- A. Maréchal and L. Zou, Cold Spring Harbor Perspect. Biol., 2013, 5, a012716 CrossRef PubMed.
- T. Tanaka, H. D. Halicka, F. Traganos, K. Seiter and Z. Darzynkiewicz, J. Med. Chem., 2007, 6, 371–376 CAS.
- W. C. Chou, L. Y. Hu, C. N. Hsiung and C. Y. Shen, Carcinogenesis, 2015, 36, 832–840 CrossRef CAS PubMed.
- A. M. Gamper, R. Rofougaran, S. C. Watkins, J. S. Greenberger, J. H. Beumer and C. J. Bakkenist, Nucleic Acids Res., 2013, 41, 10334–10344 CrossRef CAS PubMed.
- E. P. Rogakou, D. R. Pilch, A. H. Orr, V. S. Ivanova and W. M. Bonner, J. Biol. Chem., 1998, 273, 5858–5868 CrossRef CAS PubMed.
- F. Tommasino, T. Friedrich, B. Jakob, B. Meyer, M. Durante and M. Scholz, PLoS One, 2015, 10, e0129416 Search PubMed.
- S. Burma, B. P. Chen, M. Murphy, A. Kurimasa and D. J. Chen, J. Biol. Chem., 2001, 276, 42462–42467 CrossRef CAS PubMed.
- J. P. Banáth, S. H. MacPhail and P. L. Olive, Cancer Res., 2004, 64, 7144–7149 CrossRef PubMed.
- J. P. Banáth, D. Klokov, S. H. MacPhail, C. A. Banuelos and P. L. Olive, BMC Cancer, 2010, 10, 1–12 CrossRef PubMed.
- C. Redon, D. Pilch, E. Rogakou, O. Sedelnikova, K. Newrock and W. Bonner, Curr. Opin. Genet. Dev., 2002, 12, 162–169 CrossRef CAS PubMed.
- W. M. Bonner, C. E. Redon, J. S. Dickey, A. J. Nakamura, O. A. Sedelnikova, S. Solier and Y. Pommier, Nat. Rev. Cancer, 2008, 8, 957–967 CrossRef CAS PubMed.
- E. Ledesma-Fernández and P. H. Thorpe, J. Biol. Methods, 2015, 2, 62 Search PubMed.
- B. A. Teicher, Cancer Metastasis Rev., 1994, 13, 139–168 CrossRef CAS PubMed.
- B. A. Teicher, S. A. Holden, A. Al-Achi and T. S. Herman, Cancer Res., 1990, 50, 3339 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and figures for T4 ligase assay; HPLC analysis of small molecule scission products; agarose gel figures with RPCs and inhibitors sodium benzoate, sodium formate, mannitol, ethanol, DMSO, sodium pyruvate, deferoxamine, and SOD. A table containing the redox potentials and literature references of the RPCs used herein is also included. See DOI: 10.1039/c6sc04094b |
| ‡ Present addresses: University of Texas Department of Chemistry and Biochemistry, 700 Planetarium Pl., Arlington, TX 76019. |
| This journal is © The Royal Society of Chemistry 2017 |