DOI:
10.1039/C7SC00300E
(Edge Article)
Chem. Sci., 2017,
8, 3720-3725
(Phosphanyl)phosphaketenes as building blocks for novel phosphorus heterocycles†
Received
20th January 2017
, Accepted 7th March 2017
First published on 8th March 2017
Abstract
Although BH3 simply coordinates the endocyclic P of (phospholidino)phosphaketene 1Dipp, the bulkier B(C6F5)3 gives rise to a zwitterionic diphosphirenium, which is a novel type of 2π-electron aromatic system as shown by the calculated NICS values. While the reaction of 1Dipp with Na[PCO(dioxane)x] is unselective, the same reaction with the sterically bulky (phospholidino)phosphaketene 1Ar** [Ar** = 2,6-bis[di(4-tert-butylphenyl)methyl]-4-methylphenyl selectively affords a sodium bridged dimer containing a hitherto unknown λ3,λ5,λ3-triphosphete core. The latter formally results from “P−” addition to a 1,3-P/C-dipole. Similarly, adamantyl isonitrile adds to 1Dipp giving a 4-membered phosphacycle. In contrast to 1, the phosphaketene derived from the electrophilic diazaphospholidine-4,5-dione is unstable and reacts with a second molecule of Na[PCO(dioxane)x] to afford a 1,3,4-oxadiphospholonide derivative.
Introduction
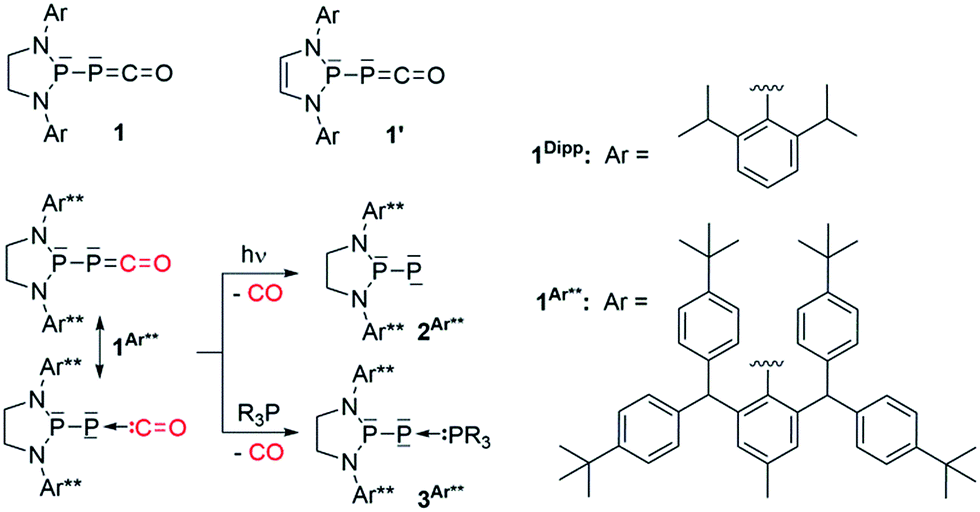
Compared to the well-known isocyanates [R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO], the chemistry of their heavier homologues, namely phosphaketenes [R–PCO], has been largely unexplored. This is presumably the result of limited synthetic access and poor stability of their alkyl and aryl substituted derivatives.1 Indeed, pioneering work by Appel et al. showed that although the very bulky [C6H3(tBu)3]–PCO can be isolated at room temperature, tBu-PCO dimerizes above −60 °C.2 However, the recent discovery of efficient preparation3 of phosphaethynolate salts (PCO−M+)4 has allowed access to group 14- (Si,5 Sn,6 Ge,7 Pb5) and transition metal- (Re, Au, Co, W)8 substituted phosphaketenes. In addition, the reaction of chlorodiaza-phospholidines and -phospholines9 with Na[PCO(dioxane)x] has allowed for the isolation of (phosphino)phosphaketenes 110 and 1′,11 respectively (Scheme 1). Although the phosphaketene moiety of 1′ reacts with the unsaturated backbone to give various rearrangement products,11 (phospholidino)-phosphaketene 1Dipp is thermally very stable (heating a toluene solution of 1Dipp overnight at 80 °C does not lead to decomposition or any rearrangement products), which allows for studying the reactivity of the [P]–PCO moiety. We have already reported that elimination of CO occurred under irradiation of 1Ar**, affording the corresponding room temperature stable phosphinidene 2Ar**,10a while addition of phosphines to 1Ar** and 1Dipp leads to adducts 3.12 In both of these reactions, the phosphaketene group acts as a phosphinidene-carbonyl adduct. This behavior is reminiscent of the chemistry of transition metal carbonyl complexes, and it is noteworthy that before our work the chemistry of main group carbonyl compounds13 was essentially limited to boranes,14 polyboranes15 and carbenes.16 Herein we report that the P–PCO scaffold can also react without loss of CO to give access to a variety of hitherto unknown phosphorus heterocycles.
CO], the chemistry of their heavier homologues, namely phosphaketenes [R–PCO], has been largely unexplored. This is presumably the result of limited synthetic access and poor stability of their alkyl and aryl substituted derivatives.1 Indeed, pioneering work by Appel et al. showed that although the very bulky [C6H3(tBu)3]–PCO can be isolated at room temperature, tBu-PCO dimerizes above −60 °C.2 However, the recent discovery of efficient preparation3 of phosphaethynolate salts (PCO−M+)4 has allowed access to group 14- (Si,5 Sn,6 Ge,7 Pb5) and transition metal- (Re, Au, Co, W)8 substituted phosphaketenes. In addition, the reaction of chlorodiaza-phospholidines and -phospholines9 with Na[PCO(dioxane)x] has allowed for the isolation of (phosphino)phosphaketenes 110 and 1′,11 respectively (Scheme 1). Although the phosphaketene moiety of 1′ reacts with the unsaturated backbone to give various rearrangement products,11 (phospholidino)-phosphaketene 1Dipp is thermally very stable (heating a toluene solution of 1Dipp overnight at 80 °C does not lead to decomposition or any rearrangement products), which allows for studying the reactivity of the [P]–PCO moiety. We have already reported that elimination of CO occurred under irradiation of 1Ar**, affording the corresponding room temperature stable phosphinidene 2Ar**,10a while addition of phosphines to 1Ar** and 1Dipp leads to adducts 3.12 In both of these reactions, the phosphaketene group acts as a phosphinidene-carbonyl adduct. This behavior is reminiscent of the chemistry of transition metal carbonyl complexes, and it is noteworthy that before our work the chemistry of main group carbonyl compounds13 was essentially limited to boranes,14 polyboranes15 and carbenes.16 Herein we report that the P–PCO scaffold can also react without loss of CO to give access to a variety of hitherto unknown phosphorus heterocycles.
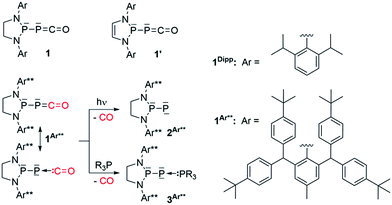 |
| | Scheme 1 Recently reported (phosphino)phosphaketenes 1 (ref. 10) and 1′,11 elimination and substitution of CO leading to the stable phosphinidene 2Ar** and adducts 3Ar**, respectively. | |
Results and discussion
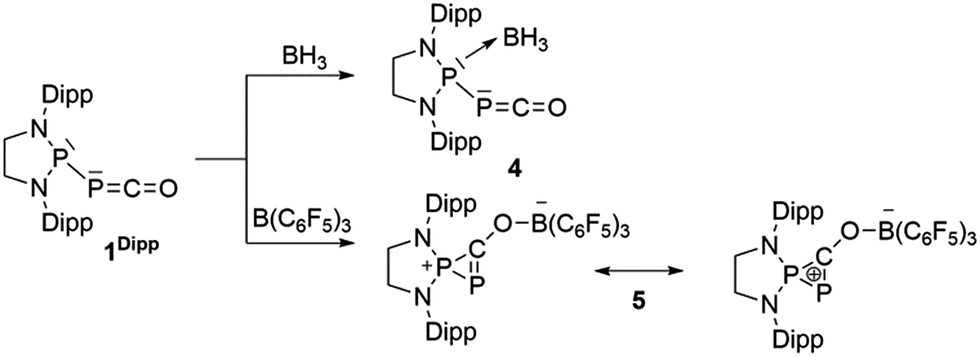
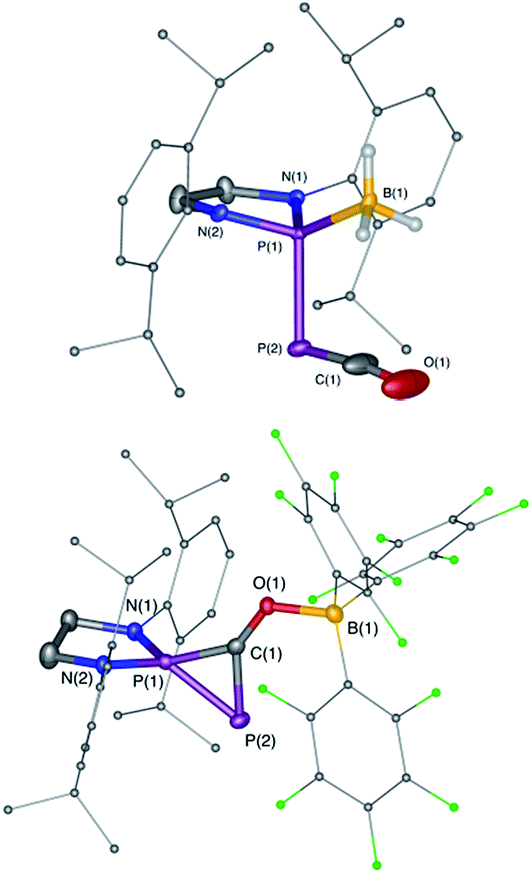
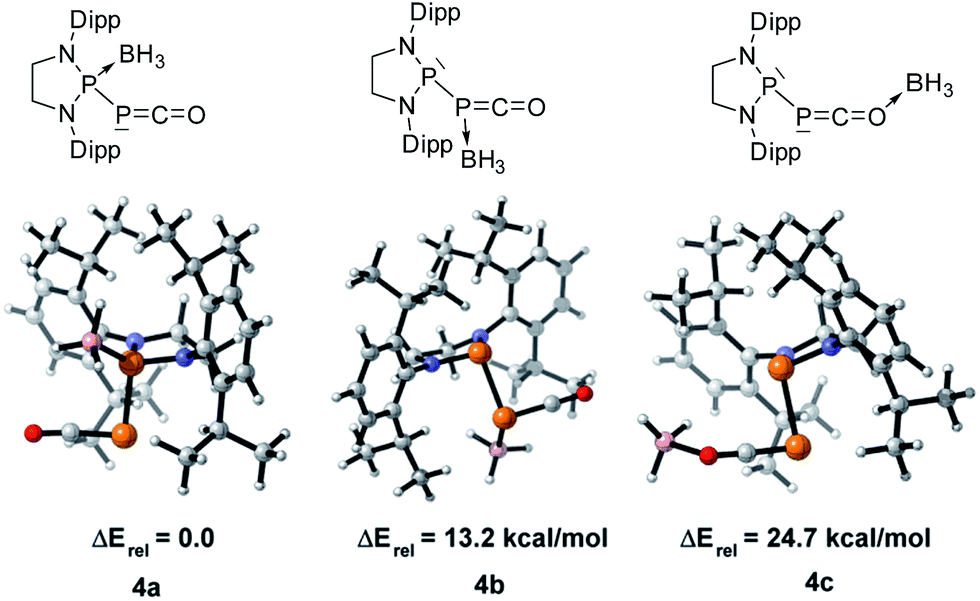
We started our investigation by studying the electrophilic activation of the [P]–PCO moiety of 1Dipp, with the aim of triggering the loss of carbon monoxide. We chose two different boron-derived Lewis acids. Upon addition of excess BH3, simple coordination to the endocyclic P center occurs giving 4, as shown by the 31P NMR spectrum [−226 ppm (d), +131 ppm (br. d), JPP = 295 Hz] and by a single crystal X-ray diffraction study (Scheme 2; Fig. 1, top). To understand the regioselectivity of the reaction, three BH3 adduct isomers were optimized at the B3LYP-D3BJ/def2-TZVP level of theory (Fig. 2). The results show that the observed product 4 is more thermodynamically stable than 4b and 4c by +13.2 and +24.7 kcal mol−1 (gas-phase electronic energies), respectively. Moreover, since the absolute coefficient of the HOMO of 1Dipp at the endocyclic P (0.42) is much larger than those at the phosphorus of PCO (0.32) and at O (0.11), 4 is also the kinetic product of the reaction.
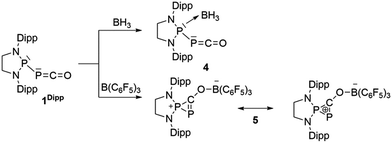 |
| | Scheme 2 Reactivity of 1Dipp with boranes. | |
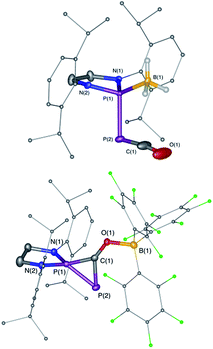 |
| | Fig. 1 Solid-state structures of 4 (top) and 5 (bottom). Hydrogen atoms are omitted for clarity. Ellipsoids shown at 50% probability. Selected bond parameters for 4 in [Å] and [°]: P1–P2 2.2657(16), P2–C1 1.533(6), C1–O1 1.165(8), P1–B1 1.906(6), P1–P2–C1 96.8(2), P2–C1–O1 171.9(6), B1–P1–P2 113.0(2); and 5: P1–P2 2.0804(14), P2–C1 1.748(4), P1–C1 1.727(4), C1–O1 1.289(4), O1–B1 1.560(5), P2–P1–C1 53.67(13), P1–C1–P2 73.55(15), P1–C1–O1 143.7(3), C1–O1–B1 125.0(3). | |
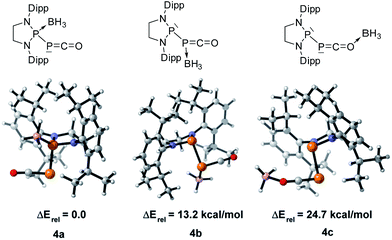 |
| | Fig. 2 Optimized structures of three BH3 adduct isomers and their relative electronic energies at the B3LYP/def2-TZVP level of theory. | |
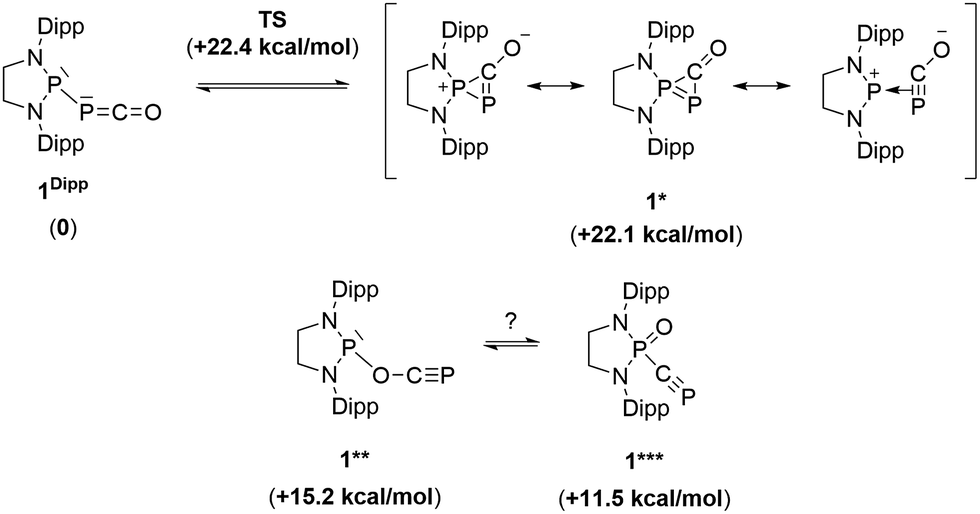
Due to the steric environment around the endocyclic P atom, we wondered whether a larger borane would react at a different site (Scheme 2). Mixing 1Dipp and B(C6F5)3 resulted in a new product as observed by 31P NMR spectroscopy with two sharp doublets at δ = +206 and −11 ppm (JPP = 215 Hz). An X-ray diffraction study revealed the formation of the unusual zwitterionic diphosphirenium 5 (Fig. 1, bottom).17 The PP bond distance (2.0804(14) Å) becomes significantly shorter than in 1Dipp (2.3782(8) Å)10 and is in the outer range for PP double bonds (1.985–2.050 Å).18 Concomitantly, the CO bond elongates from 1.170(3) Å in 1Dipp to 1.289(4) Å in 5. It is important to note that the computed nucleus independent chemical shift (NICS)19 values for the central three-membered ring are negative [NICS(0) = −17.33 and NICS(1) = −11.71 ppm], which suggests that the three-membered ring of 5 is a 2π-electron aromatic system. Mechanistically, the interaction of the borane with the oxygen atom induced a ring closure between the carbon ketene atom and the endocyclic P center. Alternatively, a reviewer suggested that the borane abstracts the PCO− moiety to form a close ion contact-pair [P]+[PCO-BR3]− followed by coordination of the phosphaalkyne to the electrophilic phosphorus center.20 However, DFT calculations indicate that the heterolytic cleavage of the P–P bond is energetically very costly. Moreover, a transition state in agreement with a concerted Lewis acid activation of 1 has been located using the small BF3 Lewis acid as a model (Fig. S1†). Interestingly, 5 can be regarded as 1*, the cyclic isomer of 1 trapped by a Lewis acid. DFT calculations predict an energy barrier of 22.4 kcal mol−1 for the endergonic interconversion of 1 into its cyclic isomer 1* (ΔE = 22.1 kcal mol−1) (Scheme 3). Note that the [P]–OCP (1**) and [PO]–CP (1***) isomers are predicted to be 15.2 and 11.5 kcal mol−1, respectively, higher in energy than 1.21
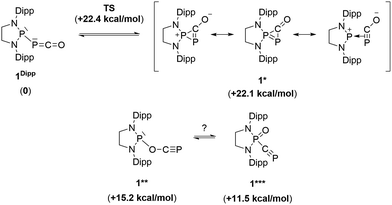 |
| | Scheme 3 Possible isomers of the P–PCO moiety and their relative electronic energies (in brackets) compared to 1Dipp (B3LYP-D3BJ/cc-pVDZ level of theory). | |
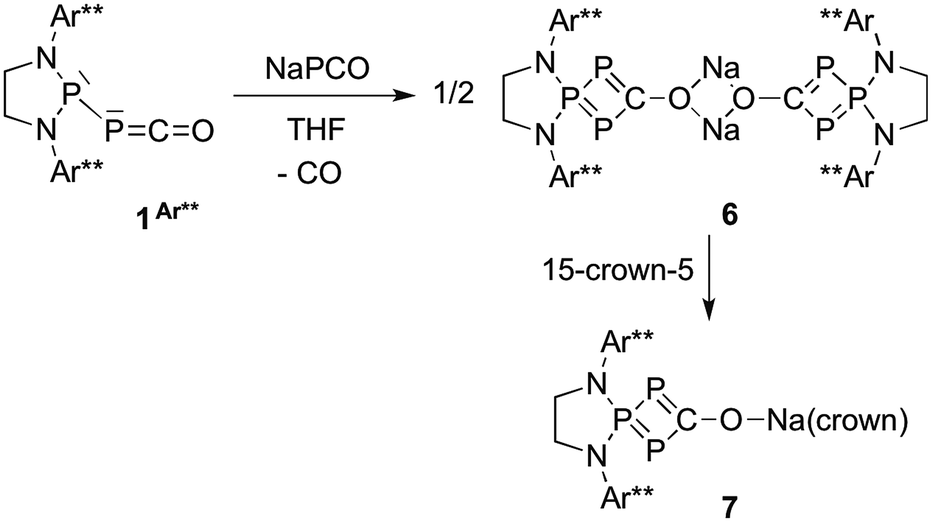
While the reaction of 1Dipp with Na[PCO(dioxane)x] is unselective, giving rise to several compounds, we observed that the same reaction with the sterically bulky (phosphino)phosphaketene 1Ar**, featuring 2,6-bis[di(4-tert-butylphenyl)methyl]-4-methylphenyl substituents,22 was highly selective. Independent of the excess Na[PCO(dioxane)x] used (or one equivalent), the 31P NMR spectrum showed the formation of a single product [+126.1 (d), +69.6 ppm (t), JPP = 302 Hz]. An X-ray diffraction study revealed that it was the sodium bridged dimer 6 containing the hitherto unknown λ3,λ5,λ3-triphosphete core (Scheme 4, Fig. 3). Upon addition of 15-crown-5 to 6 a slight shift of the 31P NMR signals [+119.7 (d) and +78.8 ppm (t), JPP = 310 Hz] was observed and the corresponding monomer 7 could be characterized by X-ray diffraction (Fig. 4).
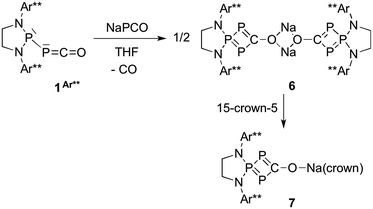 |
| | Scheme 4 Synthesis of the 1,2,3-triphosphete 7via a formal [1 + 3]-cycloaddition of P− to the PPC unit of 1Ar**. | |
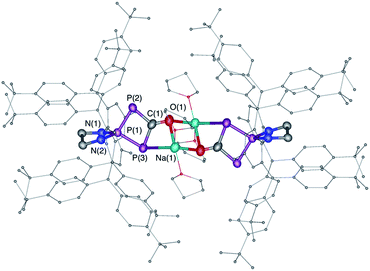 |
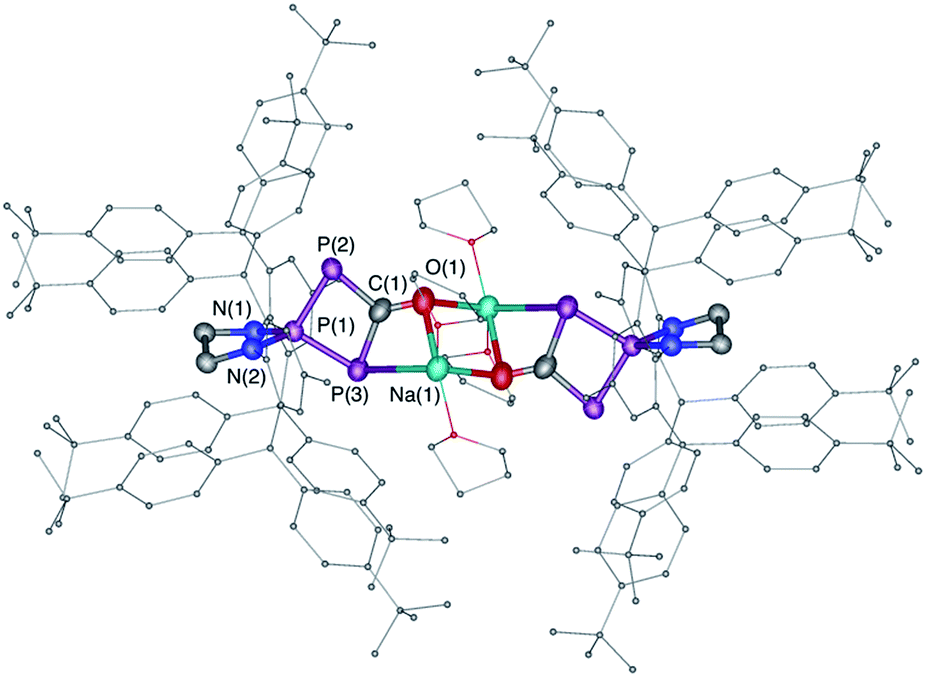
| | Fig. 3 Solid-state structure of 6. Hydrogen atoms are omitted for clarity. Ellipsoids shown at 50% probability. Selected bond parameters in [Å] and [°]: P1–P2 2.1213(15), P1–P3 2.1371(14), P2–C1 1.820(5), C1–P3 1.854(5), C1–O1 1.257(5), O1–Na1 2.305(4), P3–P1–P2 91.44(5), P1–P2–C1 78.76(15). | |
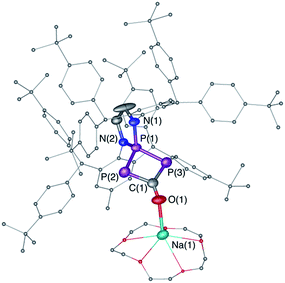 |
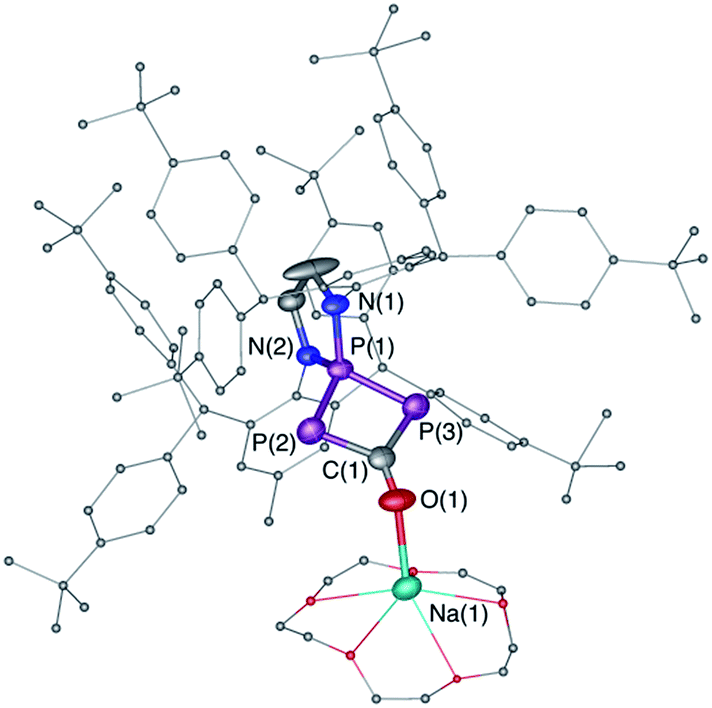
| | Fig. 4 Solid-state structure of 7. Hydrogen atoms are omitted for clarity. Structural data are not presented due to the low quality of the X-ray data. | |
The formation of the triphosphete scaffold formally results from a [1 + 3]-cycloaddition of “P−”23 (from NaPCO) to the PPC unit. Indeed, when the 13C labeled phosphaketene 1Ar** was reacted with non-13C-labeled Na[PCO(dioxane)x], we observed an intense broad resonance24 at δ13C = 250.2 ppm demonstrating that the PPCO moiety remains, at least to a large extent, in the final product. Mechanistically, DFT calculations indicate that the reaction involves an initial attack onto the carbon atom of the PCO moiety followed by cyclization with simultaneous loss of CO (ΔG‡ = 16.1 kcal mol−1) (see Fig. S2 in ESI†). Aside from the formation of a novel type of phosphorus heterocycle, these results are interesting because they give important information on the synthesis of (phosphino)phosphaketenes 1. Indeed, to prepare the latter, it is crucial to use only one equivalent of Na[PCO(dioxane)x] and toluene as the solvent in which Na[PCO(dioxane)x] is only poorly soluble. Otherwise, instead of 1, heterocycles of type 6 are formed as the major product.
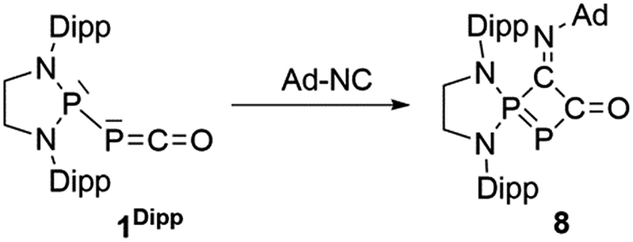
Serendipitously, we also prepared another novel type of phosphorus heterocycle formally resulting from a [1 + 3]-cycloaddition. As isonitriles and carbon monoxide are isoelectronic, we were interested in the thermal substitution of the CO in phosphaketene 1Dipp by an isonitrile, using our recently reported ligand exchange strategy.12 Surprisingly, the isonitrile does not add at the phosphorus center of PCO to displace CO, as previously observed with phosphines, but attacks at the carbon.25 This is followed by a cyclization involving the endocyclic P and the resulting heterocycle 8 was isolated in 85% yield (δ31P 148.0 and 88.3 ppm, J = 370 Hz) (Scheme 5, Fig. 5).
 |
| | Scheme 5 Synthesis of heterocycle 8via a formal [1 + 3]-cycloaddition of adamantyl isonitrile to the PPC unit of 1Ar**. | |
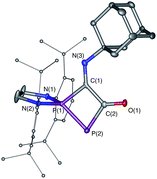 |
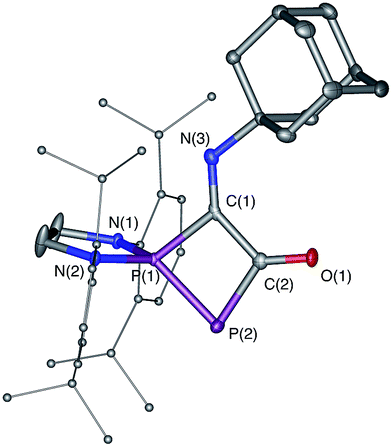
| | Fig. 5 Solid-state structure of 8. Hydrogen atoms are omitted for clarity. Ellipsoids shown at 50% probability. Selected bond lengths [Å] and angles [°]: P1–P2 2.1028(11), P1–C1 1.837(3), C1–C2 1.535(4), C2–P2 1.832(3), C2–O1 1.225(4), C1–N3 1.265(4), C2–P2–P1 77.96(11), P2–P1–C1 84.17(10). | |
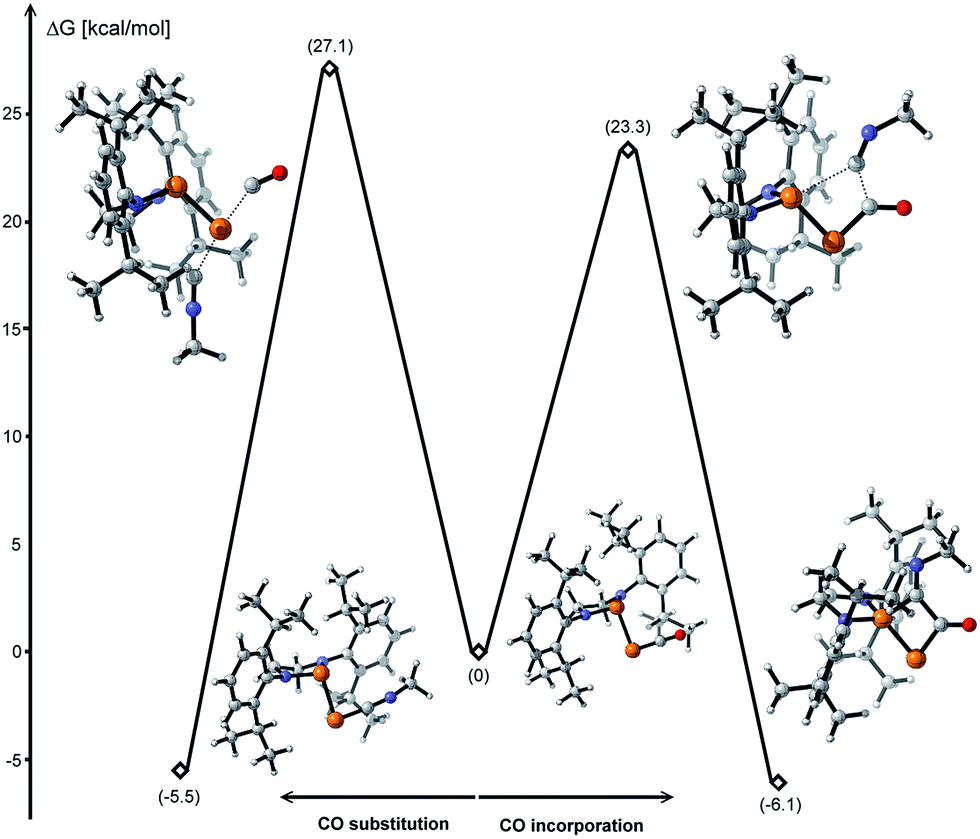
The formal insertion of an isonitrile giving 8 can be rationalized by a mechanism similar to that postulated for the insertion of P- leading to 6. According to DFT calculations, this process is exergonic by 6.1 kcal mol−1 with an energy barrier of 23.3 kcal mol−1 (Fig. 6, right). Note that direct substitution of CO by the isonitrile is also exergonic by 5.5 kcal mol−1 but with a higher activation energy barrier (27.1 kcal mol−1) (Fig. 6, left).
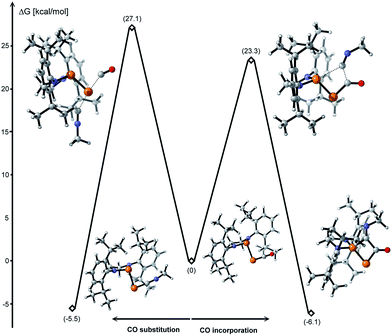 |
| | Fig. 6 Energy profiles for the CO substitution and incorporation of an isonitrile to 1 calculated at the B3LYP-D3BJ/cc-pVDZ level of theory. | |
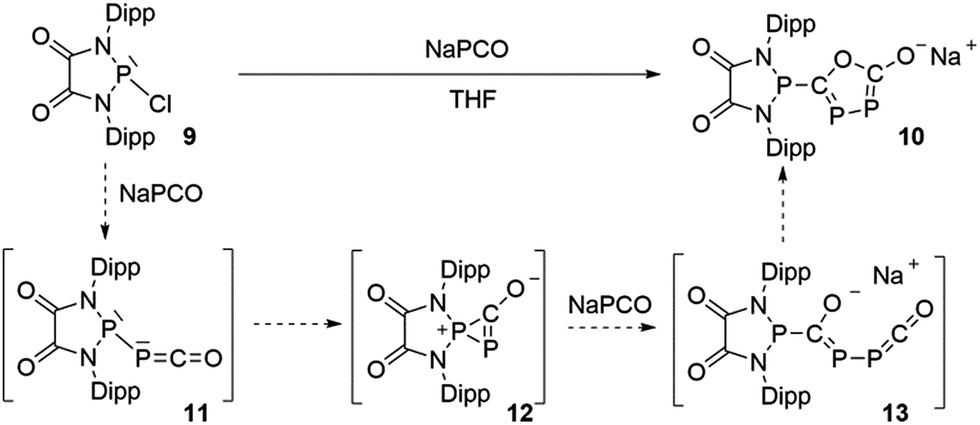
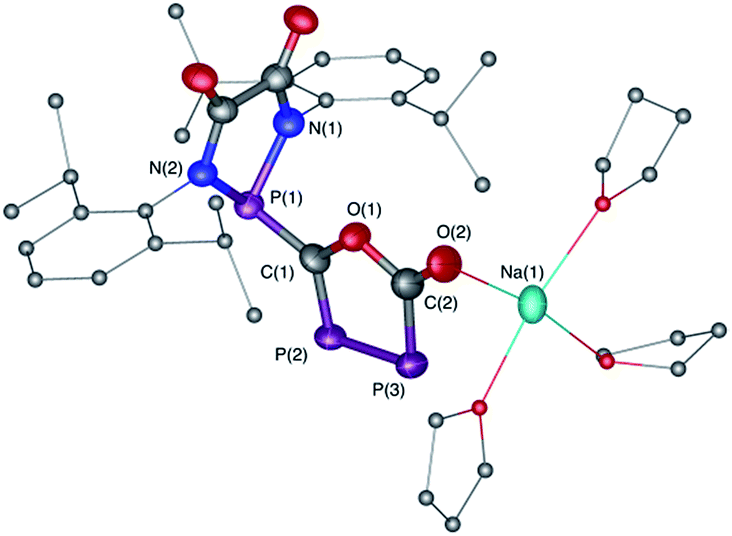
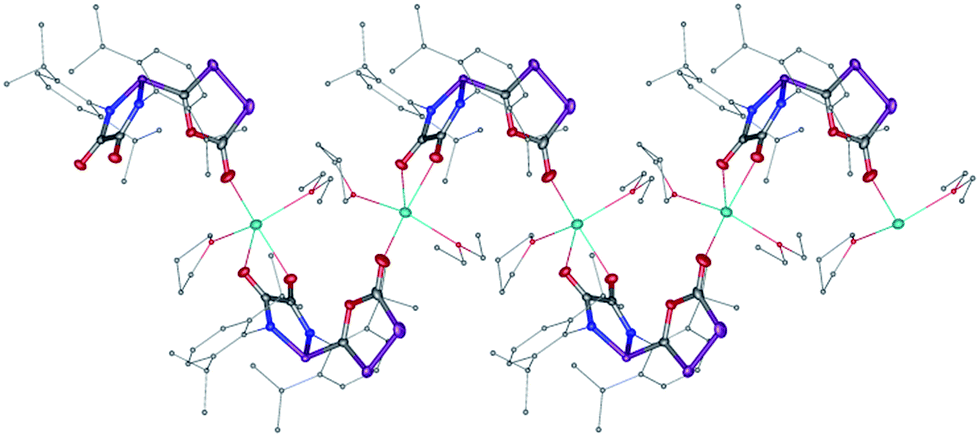
The difficulty in synthesizing (phosphino)phosphaketenes is illustrated by our attempt to prepare 11 derived from the electrophilic diazaphospholidine-4,5-dione (Scheme 6). A single product was formed upon mixing 9 with NaPCO, but the 31P NMR spectrum revealed the presence of three different phosphorus nuclei [31P NMR δ = +323 (dd, J = 466 Hz, 282 Hz); +48 (d, J = 282 Hz); +45 (d, J = 466 Hz) ppm]. An X-ray diffraction study revealed the 1,3,4-oxadiphospholonide core 10 (Fig. 7), a type of heterocycle previously only observed by Grützmacher et al. in the reaction of NaPCO with tetraphenyl-cyclopentadienone.26 Interestingly, in the solid-state this compound features a linear polymeric network structure in which the sodium cation is bridging between the diketone moiety and the phosphorus heterocycle (Fig. 8).
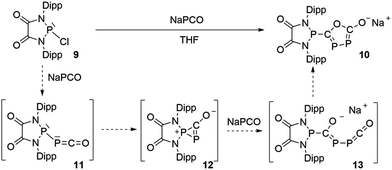 |
| | Scheme 6 Attempted synthesis of (phopshino)phosphaketene 11 and the formation of heterocycle 10. | |
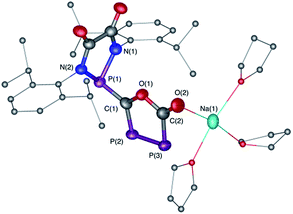 |
| | Fig. 7 Solid-state structure of 10 crystalized from THF. Hydrogen atoms and THF molecules coordinated are omitted for clarity. Ellipsoids shown at 50% probability. Selected bond parameters for 10 in [Å] and [°]: P1–C1 1.759(5), C1–P2 1.696(5), P2–P3 2.096(3), P3–C2 1.795(6), C2–O1 1.405(6), C2–O2 1.231(7), O1–C1 1.389(6), O2–Na1 2.329(4), P1–C1–P2 118.3(3), C1–P2–P3 94.9(2), P2–P3–C2 93.5(2). | |
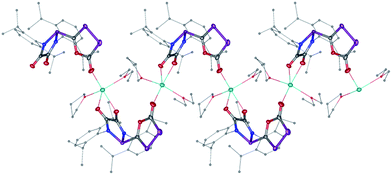 |
| | Fig. 8 Stolid-state structure of 10 crystalized from THF/Et2O/CH3CN showing the polymeric chain nature. Ellipsoids shown at 50% probability. | |
Mechanistically, it seems reasonable to postulate the initially formed (phosphino)phosphaketene 11 spontaneously rearranges into the spirocyclic zwitterionic derivative 12 which resembles the borane adduct 5. Then, a second equivalent of NaPCO induces a ring opening giving 13, which undergoes a ring closure leading to the observed product 10.
Conclusions
This work has shown that (phosphino)phosphaketenes are powerful building blocks in heterocyclic chemistry. In contrast to our recent reported CO substitution approach,12 this work demonstrates the feasibility of nucleophiles to add to carbon on the phosphaketene moiety. The endocyclic P center can either activate the phosphaketene by forming highly reactive diphosphirenium species or engage in ring closing reactions. Importantly the stability and chemical behaviour of these novel heterocycles is strongly dependent on the nature of the phosphino substituents.
Acknowledgements
Thanks are due to the NSF (CHE-1359809) for financial support. MMH is thankful to the Alexander von Humboldt foundation for a Feodor-Lynen scholarship and LL to the China Scholarship Council for a graduate fellowship. We are grateful to the Keck Foundation for funding the KeckII computer center. Dr Milan Gembicky and Dr Curtis Moore are greatly acknowledged for their help with X-ray diffraction.
Notes and references
- For reviews on phosphaketenes, see:
(a) J. Escudié, H. Ranaivonjatovo and L. Rigon, Chem. Rev., 2000, 100, 3639–3696 CrossRef;
(b) R. Appel and F. Knoll, Adv. Inorg. Chem., 1989, 33, 259–361 CrossRef CAS.
-
(a) R. Appel, In Multiple Bonds and Low Coordination in Phosphorus Chemistry, ed. M. Regitz and O. J. Scherer, Thieme, Stuttgart, Germany, 1990, pp. 188–190 Search PubMed;
(b) R. Appel, B. Laubach and M. Siraay, Tetrahedron Lett., 1984, 25, 4447–4448 CrossRef CAS;
(c) R. Appel and W. Paulen, Tetrahedron Lett., 1983, 24, 2639–2642 CrossRef CAS;
(d) R. Appel and W. Paulen, Angew. Chem., 1983, 95, 807–808 CrossRef;
(e) V. Plack, J. R. Görlich, A. Fischer and R. Schmutzler, Z. Anorg. Allg. Chem., 1999, 625, 1979–1984 CrossRef CAS;
(f) A. H. Cowley, B. Pellerin, J. L. Atwood and S. G. Bott, J. Am. Chem. Soc., 1990, 112, 6734–6735 CrossRef CAS;
(g) M.-A. David, D. S. Glueck, G. P. A. Yap and A. L. Rheingold, Organometallics, 1995, 14, 4040–4042 CrossRef CAS;
(h) M.-A. David, D. K. Wicht and D. S. Glueck, Organometallics, 1997, 16, 4768–4770 CrossRef CAS;
(i) R. Appel, P. Fölling, B. Josten, M. Siray, V. Winkhaus and F. Knoch, Angew. Chem., 1984, 96, 620–621 CrossRef CAS;
(j) R. Appel, C. Porz and F. Knoch, Chem. Ber., 1986, 119, 2748–2755 CrossRef CAS.
-
(a) Z.-J. Quan and X.-C. Wang, Org. Chem. Front., 2014, 1, 1128–1131 RSC;
(b) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed;
(c) A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed;
(d) D. Heift, Z. Benkö, H. Grützmacher, A. R. Jupp and J. M. Goicoechea, Chem. Sci., 2015, 6, 4017–4024 RSC;
(e) D. Heift, Z. Benkö and H. Grützmacher, Dalton Trans., 2014, 43, 831–840 RSC.
- For the first preparation of the phosphaethynolate sodium salt: G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS.
- D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 5920–5928 RSC.
- D. Heift, Z. Benkő and H. Grützmacher, Chem.–Eur. J., 2014, 20, 11326–11330 CrossRef CAS PubMed.
-
(a) S. Yao, Y. Xiong, T. Szilvasi, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 4781–4785 CrossRef CAS PubMed;
(b) Y. Wu, L. Liu, J. Su, J. Zhu, Z. Ji and Y. Zhao, Organometallics, 2016, 35, 1593–1596 CrossRef CAS;
(c) N. D. Rio, A. Baceiredo, N. Saffon-Merceron, D. Hashizume, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 4753–4758 CrossRef PubMed.
- For transition-metal PCO complexes, see:
(a) L. Liu, D. A. Ruiz, F. Dahchech, G. Bertrand, R. Suter, A. M. Tondreau and H. Grützmacher, Chem. Sci., 2016, 7, 2335–2341 RSC;
(b) S. Alidori, D. Heift, G. Santiso-Quinones, Z. Benkő, H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem.–Eur. J., 2012, 18, 14805–14811 CrossRef CAS PubMed;
(c) A. R. Jupp, M. B. Geeson, J. E. McGrady and J. M. Goicoechea, Eur. J. Inorg. Chem., 2016, 2016, 639–648 CrossRef CAS PubMed;
(d) C. Camp, N. Settineri, J. Lefèvre, A. R. Jupp, J. M. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379–6384 RSC.
- D. Gudat, Dalton Trans., 2016, 45, 5896–5907 RSC.
-
(a) L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CrossRef CAS;
(b) M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 8356–8359 CrossRef CAS PubMed.
- Z. Li, X. Chen, M. Bergeler, M. Reiher, C.-Y. Su and H. Grützmacher, Dalton Trans., 2015, 44, 6431–6438 RSC.
- M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS PubMed.
- For a comparison of transition metal reactivity with main group compounds see:
(a) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed;
(b) D. Martin, M. Soleihavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
-
(a) M. Finze, E. Bernhardt, A. Terheiden, M. Berkei, H. Willner, D. Christen, H. Oberhammer and F. Aubke, J. Am. Chem. Soc., 2002, 124, 15385–15398 CrossRef CAS PubMed;
(b) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed.
- J. D. Glore, J. W. Rathke and R. Schaeffer, Inorg. Chem., 1973, 12, 2175–2178 CrossRef CAS.
-
(a) V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488–3491 CrossRef CAS PubMed;
(b) C. Goedecke, M. Leibold, U. Siemeling and G. Frenking, J. Am. Chem. Soc., 2011, 133, 3557–3569 CrossRef CAS PubMed;
(c) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS PubMed.
-
(a) F. Castan, A. Baceiredo, J. Fischer, A. De Cian, G. Commenges and G. Bertrand, J. Am. Chem. Soc., 1991, 113, 8160–8161 CrossRef CAS; for a review on 3-membered phosphorus cycles, see:
(b) F. Mathey, Chem. Rev., 1990, 90, 997–1025 CrossRef CAS.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- B. Breit, U. Bergsträsser, G. Maas and M. Regitz, Angew. Chem., Int. Ed., 1992, 31, 1055–1058 CrossRef.
- The [P]–PCO to [P]–OCP energy difference is in good agreement with the recently calculated value, see ref. 11.
- S. G. Weber, C. Loos, F. Rominger and B. F. Straub, ARKIVOC, 2012, 2, 226–242 Search PubMed.
- For P- transfer from PCO see for example:
(a) A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC;
(b) T. P. Robinson, M. J. Cowley, D. Scheschkewitz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 683–686 CrossRef CAS PubMed.
- The 13C NMR signal is broad as a result of the coupling with several different phosphorus nuclei (see ESI†).
- For the addition of nucleophiles to the carbon atom of phosphaketenes, see:
(a) Z. Li, X. Chen, Z. Benkő, L. Liu, D. A. Ruiz, J. L. Peltier, G. Bertrand, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 6018–6022 CrossRef CAS PubMed;
(b) R. Appel, P. Foelling, B. Josten, W. Schuhn, H. V. Wenzel and F. Knoch, Z. Anorg. Allg. Chem., 1988, 556, 7–22 CrossRef CAS;
(c) R. Appel, P. Foelling, W. Schuhn and F. Knoch, Tetrahedron Lett., 1986, 27, 1661–1664 CrossRef CAS;
(d) R. Appel, C. Porz and F. Knoch, Chem. Ber., 1986, 119, 2748–2755 CrossRef CAS;
(e) R. Appel, P. Foelling, B. Josten, M. Siray, V. Winkhaus and F. Knoch, Angew. Chem., Int. Ed. Engl., 1984, 96, 619–621 CrossRef.
- X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkö, Z. Li, G. Becker, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data, crystallographic data and Cartesian coordinates of DFT optimized structures. CCDC 1432815(4), 1432814 (5), 1528506 (6), 1432817 (8), 1528508 (10) and 1528507 (10′). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00300e |
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Liu Leo Liu,
Rodolphe Jazzar
,
Liu Leo Liu,
Rodolphe Jazzar