
Gemini quaternary ammonium salt waterborne biodegradable polyurethanes with antibacterial and biocompatible properties†
Yi
Zhang
ab,
Wei
He
a,
Jiehua
Li
*a,
Kunjie
Wang
c,
Jianshu
Li
a,
Hong
Tan
*a and
Qiang
Fu
a
aCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China. E-mail: hongtan@scu.edu.cn; jiehua_li@scu.edu.cn
bHigh and New Technology Research Center of Henan Academy of Sciences, Zhengzhou 450002, China
cDepartment of Urology, West China Hospital, Huaxi Clinical College, Sichuan University, Chengdu 610065, China
First published on 15th August 2016
Abstract
In this study, a series of waterborne biodegradable polyurethanes with antibacterial and biocompatible properties were developed. To obtain these polyurethanes, lysine-derivative gemini quaternary ammonium salt (GQAS) chain extenders with different hydrophobic alkyl chain lengths (named EGn, where n = 8, 12, 16; the hydrophobic alkyl chain length of GQAS) were designed and synthesized. Then, waterborne biodegradable polyurethanes (PCLPUn) were prepared using isophorone diisocyanate (IPDI), poly(ε-caprolactone) (PCL), poly(ethylene glycol) (PEG), L-lysine and EGn. The antibacterial activities of these EGn and PCLPUn emulsions were evaluated by a minimal inhibitory concentration (MIC) method, and the antibacterial and antifouling functionalities of the PCLPUn film surfaces were confirmed by a contact-active antibacterial and culture-based method using both Gram-positive and Gram-negative bacteria. The in vitro degradation and cytotoxicity of these obtained polyurethanes were also systematically investigated. These results indicated that the PCLPUn films possessed good antibacterial and antifouling abilities, biodegradability, and good biocompatibility. Notably, PCLPU12 shows the best antibacterial activity and cytocompatibility. Such antibacterial materials could be degraded to non-toxic components, and potentially be widely used in medical and environmental applications, especially as biodegradable coatings of surgical equipment and medical implants.
Introduction
Infection related to biomaterial implants, such as urinary catheters, intravascular catheters,1–4 biomedical devices,5 and orthopaedic implants,6 remains one of the most severe and devastating complications in a clinical environment. Despite the establishment of many advanced sterilization and aseptic techniques to minimize the possibilities of biomaterial associated infection during the surgery, unfortunately, these are still unable to eradicate this completely. As infection occurs, implant removal and substitution, with accompanying medical risks and complications, is often inevitable. Therefore, integrating implants with good anti-infection activity during the service period is of great importance. The positively charged antibacterial agents quaternary ammonium salts (QAS), with broad antibacterial spectrum, good environmental stability and biological activity,7 which do not easily form resistance,8–11 has already been applied in many commercial products, such as deodorants, mouth and hair rinse products, contact lens solutions, and so on. Moreover, extensive research concerning QAS antibacterial materials have been done and is demonstrated constantly.12–14 Of particular note is the fact that QAS antibacterial surfaces are apt to killed microorganisms due to the positive charge of QAS, which could block the functional groups and trigger immune responses and inflammatory reactions.15 As a result, these antibacterial materials as implants cannot fulfill both requirements of biocompatibility and non-fouling ability. It is worth noting that these surfaces constructed from biodegradable polymers could be gradually decomposed and degraded under enzymatic attack, which could release the attached living organisms.16–19 Thus, biodegradable antibacterial materials with good biocompatibility that could maintain an anti-infection effect in the serving time, and then degrade to non-toxic small molecules seem to be attractive materials to the fight infection of biomaterials.20In addition, the biological activity of QAS compounds depends on the nature of the organic groups attached to the nitrogen positive ion (N+), the number of N+ present, and the counter ion.21,22 Gemini quaternary ammonium salts (GQAS) possessing two hydrophobic hydrocarbon chains and two hydrophilic quaternary ammonium groups have much stronger surface activity and permanent excellent antibacterial activity compared to single-chain QAS.23,24 It has been shown that an increase of alkyl chain length of QAS is followed by an increase in the antimicrobial activity of the compound.15,25–29 The hydrophobic alkyl chain length of QAS not only affects antibacterial activity, but also has an effect on the material biocompatibility.29 Therefore, development of biocompatible antibacterial materials requires the investigation of the effect of the hydrophobic alkyl chain length of QAS on the antibacterial activity and biocompatibility.
In this study, to develop biocompatible antibacterial and antifouling GQAS waterborne polyurethanes, lysine-derivative chain extenders containing GQAS side chains consisting of two hydrophilic head groups and two hydrophobic alkyl tails (denoted as EGn, n = 8, 12, 16; the length of GQAS hydrophobic alkyl tails) were designed and synthesized. Then, a series of biocompatible degradable antibacterial waterborne polyurethanes (named as PCLPUn, n = 8, 12, 16, the length of GQAS hydrophobic alkyl tails) were prepared using isophorone diisocyanate (IPDI), poly(ethylene glycol) (PEG), poly(ε-caprolactone) diols (PCL), L-lysine and EGn as primary materials. The antibacterial and antifouling abilities of these obtained polyurethanes were determined by the minimal inhibitory concentration (MIC), contact-active antibacterial and a culture-based antibacterial and antifouling assay. In vitro extensive degradation and cytotoxicity analysis studies on the obtained PCLPUn samples were also performed to study the effects of different hydrophobic alkyl chains of GQAS on the degradation and biocompatibility of PCLPUn.
Experimental section
Materials
Unless otherwise noted, all chemical reagents and reagent grade solvents were obtained from commercial suppliers and used without additional purification. 1,3-Propane diamine, 1-bromooctane, 1-bromododecane and 1-bromohexadecane were purchased from Aladdin Reagent. Isophorone diisocyanate (IPDI) (BASF) was redistilled under vacuum before use. Poly(ethylene glycol) (PEG, molecular weight 1450, Dow Chemical) and poly(ε-caprolactone) diols (PCL, molecular weight 2000, Dow Chemical) were dehydrated at 80–90 °C under vacuum for 2 h before use. L-Lysine and N,N,N′,N′-tetramethyl-L-lysine ethyl ester (LE) was used as received. (Boc)2 lysine was synthesized using a conventional manner of preparing peptides in our laboratory. Lipase from Pseudomonas cepacia (lipase PS, ≥30 U mg−1) was purchased from Sigma.Characterization
Proton nuclear magnetic resonance (1H NMR, 400 MHz) spectra were recorded on a Bruker AV II-400 MHz spectrometer in DMSO-d6. Gel permeation chromatography (GPC) was performed by a Waters-1515 using N,N-dimethylformamide (DMF)/LiBr as an eluent and polymethyl methacrylate (PMMA) as a reference. The sample concentration was 2–3 mg mL−1, and the flow rate was 1.000 mL min−1 at 40 °C. Scanning electron microscopy (SEM) pictures were taken using a SEM instrument (Inspect F, FEI Company) at an accelerated voltage of 15 kV.Synthesis of antibacterial waterborne polyurethanes
To obtain a novel series of biodegradable waterborne PCLPUn with long-term contact-active antibacterial and antifouling properties, the lysine-derivative GQAS monomer containing two primary amine groups with different hydrophobic alkyl chain lengths (EGn, n = 8, 12 and 16; the length of GQAS hydrophobic alkyl tails) were first designed and synthesized. The synthesis details (synthesis route: Scheme S1, ESI†) and 1H NMR information (Fig. S1, ESI†) are provided in the ESI.† Subsequently, biodegradable antibacterial and antifouling PCLPUn based on IPDI, PEG, PCL and the chain extender lysine and EGn were synthesized using a two-step polymerization. The feed ratios are shown in Table 1, and were decided by considering both the antibacterial and biocompatibility of the polyurethanes from our previous work.30 Firstly, IPDI and 0.1% stannous octoate were added to the stirred and dried PEG and PCL at 70–75 °C under a dry nitrogen atmosphere. After reaction for 1 h at 70–75 °C, the reaction mixture was cooled down to room temperature, and the chain extender EGn was added in the mixture with continuous stirring for 20–30 min. Finally, the prepolymer was poured into L-lysine aqueous solution to emulsify with high-speed stirring (600 rpm) and ultrasound (200 W), simultaneously, dilute sodium hydroxide solution was added dropwise into the aqueous emulsion to neutralize the carboxyl groups of L-lysine under room temperature for 2 h. Polyurethane without GQAS (PCLPU0) as a control was also prepared with a similar process as described above.| Samples | Molar ratio of IPDI/PEG/PCL/EGn/lysine | Chain extender EGn | EGn (wt%) | M n (g mol−1) | M w/Mn | MIC (µg mL−1) | |
|---|---|---|---|---|---|---|---|
| S. aureus | E. coli | ||||||
| PCLPU0 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.33:0.67:0.5:0.5 :0.33:0.67:0.5:0.5 |
0 | 0 | 62419 |
1.41 | >1000 | >1000 |
| PCLPU8 | 2:0.33:0.67:0.5:0.5 |
EG8 | 14.40 | 91912 |
2.06 | 100 | 400 |
| PCLPU12 | 2:0.33:0.67:0.5:0.5 |
EG12 | 15.83 | 55265 |
1.53 | 45.7 | 91.4 |
| PCLPU16 | 2:0.33:0.67:0.5:0.5 |
EG16 | 17.41 | 101085 |
1.52 | 48.1 | 192.4 |
Antibacterial surfaces coated with PCLPUn films were prepared through casting the PCLPUn emulsions on surfaces of siliconized culture dishes which were then dried at room temperature for 2 d, then put into an oven at 60 °C for 2 d, followed by treatment of 60 °C under vacuum for 2 d. The films were cut into sheets of 1 × 1 cm in size and approximately 0.5 mm thickness for physicochemical characterization. PCLPU0 was used as a control. Certain polyurethane films (1 × 1 cm, 0.5 mm thickness) were immersed in water in a horizontal laboratory shaker (110 rpm, 37 °C) for 60 s, and then dried at 60 °C under vacuum for 2 d before testing.
Antibacterial studies
Degradation test
PCLPUn and PCLPU0 films were prepared through casting the polyurethane emulsions onto the surfaces of siliconized culture dishes and drying at room temperature for 2 d. These were then put into an oven at 60 °C for 2 d, followed by treatment of 60 °C under vacuum for 2 d. The films were cut in sheets of 1 × 1 cm in size and approximately 0.5 mm thickness (without washing before the test) for the degradation test.The degradation of PCLPUn in vitro was evaluated as weight loss of these polyurethane films (10 × 10 mm in size, with approximately 0.5 mm thickness) in phosphate buffer solution alone (PBS, pH = 7.4) and enzyme medium (PBS with lipase PS, 0.3 mg mL−1), then the vials were incubated with shaking at 37 °C.33–35 PCLPU0 was used as a control. The samples were taken out at a predetermined time, washed with distilled water three times, and dried in a vacuum oven at 25 °C to a constant weight. The residual weight was calculated as:
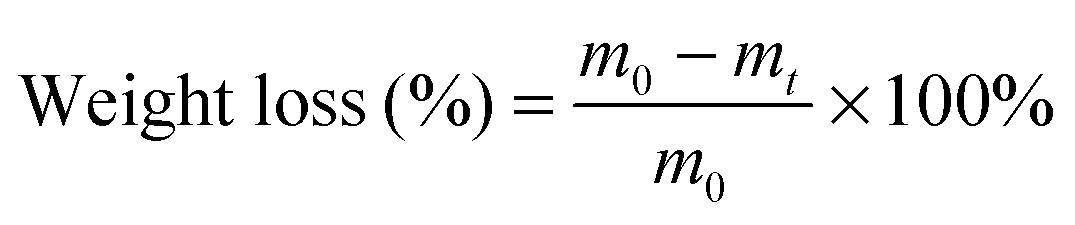
Biocompatibility of PCLPUn
All obtained films (PCLPUn, PCLPU0 films and latex rubber film) were immersed in water in a horizontal laboratory shaker (110 rpm, 37 °C) for 60 s, and then dried at 60 °C under vacuum for 2 d before the biocompatibility test.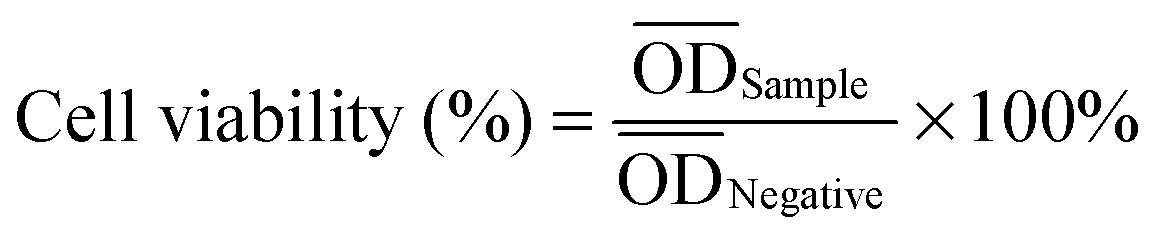 000 times with culture media. Sterile PBS was similarly diluted with media as a control. The solutions were added to the cells cultured in the 96-well plates (100 µL per well), which were then incubated at 37 °C, 95% relative humidity, and 5% CO2 for 24 h. The cell viability was evaluated by employing a MTT assay as described previously. In addition, the cytocompatibility of EGn was also assessed. Solutions of gemini in culture media were prepared with concentrations ranging from 1 to 1 × 10−4 mg mL−1, and the cytotoxicity was tested similarly as the degradation solutions.
000 times with culture media. Sterile PBS was similarly diluted with media as a control. The solutions were added to the cells cultured in the 96-well plates (100 µL per well), which were then incubated at 37 °C, 95% relative humidity, and 5% CO2 for 24 h. The cell viability was evaluated by employing a MTT assay as described previously. In addition, the cytocompatibility of EGn was also assessed. Solutions of gemini in culture media were prepared with concentrations ranging from 1 to 1 × 10−4 mg mL−1, and the cytotoxicity was tested similarly as the degradation solutions.
Statistical data analysis
The results for the antibacterial test, protein and adsorption cytotoxicity experiments are expressed as mean values ± standard deviations (SD, n = 3 for each analysis). Statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS, version 17.0) software. Statistical comparisons were made by the Student's t-test or one-way analysis of variance (ANOVA). In all cases, p < 0.05 was considered as statistically significant.Results and discussion
Synthesis and characterization of waterborne PCLPUn
To obtain new biocompatible materials with good antibacterial and antifouling properties, the lysine-derivative GQAS monomers containing two primary amine groups and different lengths of hydrophobic alkyl chains as chain extenders (EGn) were first designed and synthesized. Their synthesis details, structures, and 1H NMR information are provided in the ESI† (Scheme S1 and Fig. S1). Subsequently, a series of waterborne biodegradable polyurethanes containing various GQASs have been prepared (Fig. 1). PCLPU0 without a GQAS chain extender was also prepared for comparison. The characteristic peaks at 3.99 (–COOCH2–), 2.27 (–CH2COO–), 1.55 (–CH2CH2CH2–), and 1.29 (–CH2CH2CH2–) ppm are assigned to methylene protons of the PCL blocks in the 1H NMR spectra (Fig. 2). The peaks at 3.49 ppm are assigned to methylene protons (–CH2CH2O) of the PEG units. The peak at 3.81 ppm is ascribed to the terminal methylene groups (–NHCOO–CH2–) in PEG and PCL. The peaks at 3.00–3.19 ppm, originated from methyl and methylene groups of the N positive ions of the GQAS chain extenders, and can only be found in the PCLPUn samples. The 1H NMR spectra show that all gemini chain extenders (EG8, EG12, and EG16) have been copolymerized into chains of these polyurethanes, as compared to that of PCLPU0 (Fig. 2). The number-average molecular weight (Mn) of these resulting PCLPUn and PCLPU0 determined by GPC range from 55265 to 101085 presenting monodispersed and narrow molecular weight distributions (Table 1). More detailed information on the bulk structures of these polyurethanes (DSC and FTIR results) is presented in the ESI† (Fig. S2 and S3). These results demonstrate that these waterborne antibacterial biodegradable polyurethanes have been successfully synthesized.
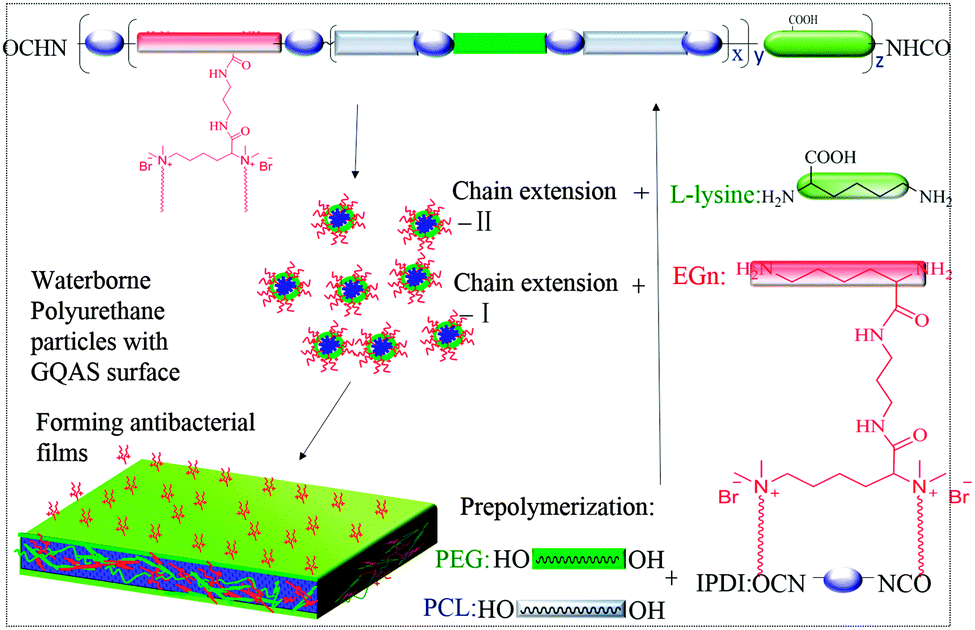 | ||
| Fig. 1 Schematic structure of gemini quaternary ammonium salt waterborne polyurethane with antibacterial surfaces. | ||
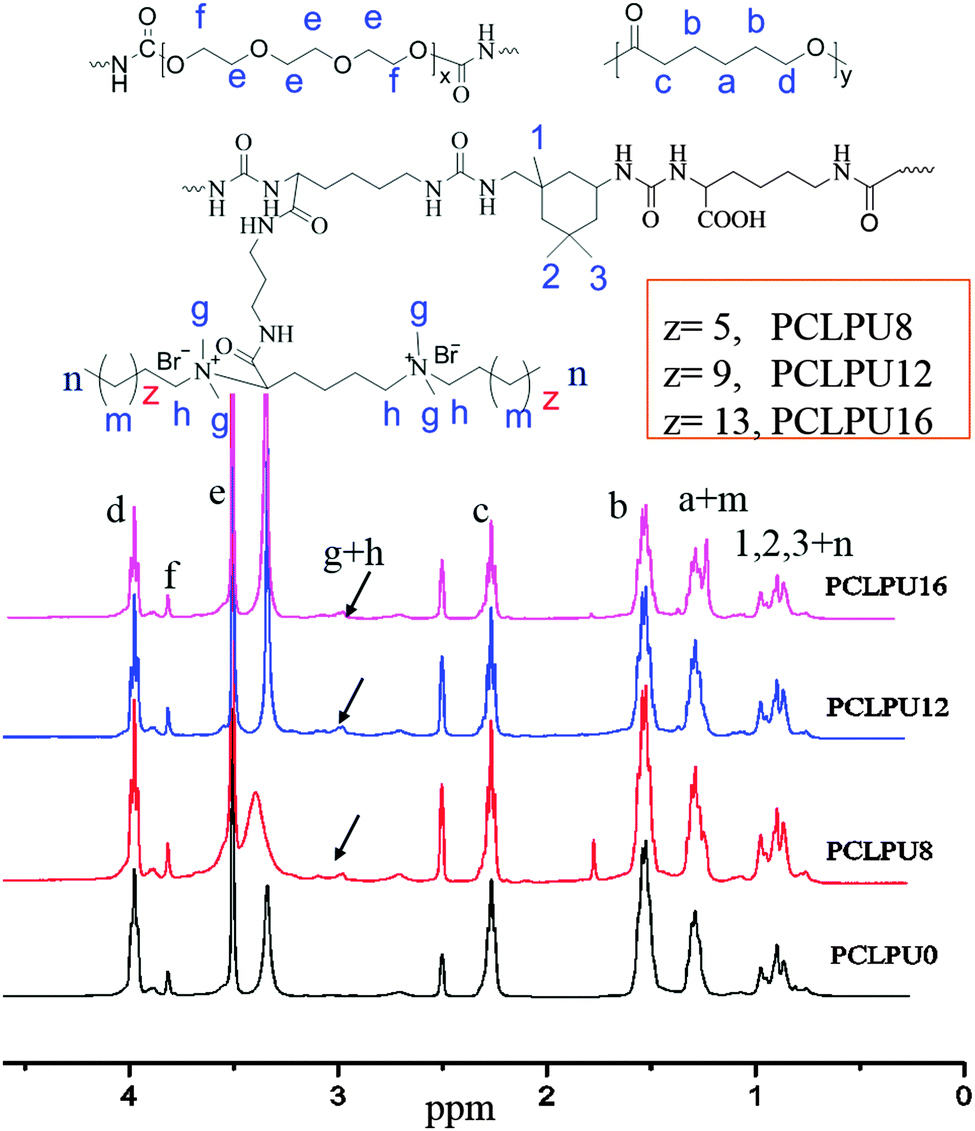 | ||
| Fig. 2 The structures and 1H NMR spectra of PCLPUn samples recorded in DMSO-d6. | ||
Antibacterial and antifouling activities of obtained waterborne PCLPUn films
To verify the antibacterial properties of these biodegradable waterborne PCLPUn, we first measured the MICs of these polyurethane emulsions against suspensions of ubiquitous infectious bacteria, including Staphylococcus aureus (S. aureus; Gram-positive), Escherichia coli (E. coli; Gram-negative), using EGn as controls. The MICs of EGn were in the 4–16 µg mL−1 range for S. aureus, and 8–64 µg mL−1 for E. coli, and EG12 showed more efficient antibacterial activity than EG8 and EG16 (Table S1, ESI†), since the length of hydrophobic alkyl chain had a strong impact on the antibacterial activity.25,36,37 Biodegradable waterborne polyurethanes containing GQAS were in the 45.7–100 µg mL−1 range for S. aureus, and 91.4–400 µg mL−1 for E. coli (Table 1), indicating that these polyurethanes have good antibacterial activities consistent with our previous work.30 In particular, PCLPU12 shows the best antibacterial efficiency of these polyurethanes owing to the chain extender EG12, as described above. Similarly, these GQAS polyurethanes had better antibacterial activity to Gram-positive bacteria than to Gram-negative bacteria,32 since Gram-negative bacteria have very sophisticated outer cell walls to effectively keep out antibacterial agents.22The antifouling and antibacterial potential of these films was also evaluated by protein adsorption and a shake-flask method,32,38 and the results are illustrated in Fig. 3 and Fig. S5 (ESI†). Nonspecific protein adsorption is a key factor in material surface fouling. Because this surface has the ability to inhibit nonspecific protein adsorption, it will to some degree, resist bacterial adhesion under condition with no antibacterial moieties.39,40 The adsorption of protein BSA on PCLPUn and TPEU films surfaces is shown in Fig. S5 (ESI†). The amount of protein adsorption on PCLPU0 surfaces is about 0.17 µg cm−2, while the adsorbed protein on the commercial TPEU surfaces is higher, 1.31 µg cm−2, owing to the rearrangement of PEG and the –COOH repulsion of PCLPU0 reducing the adsorption of proteins from solution after contact with water.41,42 However, the introduction of GQAS in polyurethanes had a remarkable increase in the protein adsorption on these surfaces compared to that on the surfaces of PCLPU0, because the transition from a negatively charged surface to positively charged surface could enhance electrostatic interaction between BSA and the surface (BSA, negatively charged at pH 7).43 In addition, the amount protein adsorption of PCLPUn samples was increased with the length increase of these GQAS hydrophobic alkyl chains, since the longer hydrophobic alkyl chains are more likely to enhance the protein–surface hydrophobic interactions.44 Interestingly, the increased protein adsorption on the surfaces of gemini quaternary ammonium salt polyurethanes would not much affect their antifouling and antibacterial activities. The culture-based antibacterial and antifouling results are shown in Fig. 3A. The density of live bacteria attached on the control surfaces (without PEG) was 2.90 × 106 CFU cm−2E. coli and 4.0 × 105 CFU cm−2S. aureus, of which E. coli and S. aureus were about 6.6 and 2 times higher than those on PCLPU0 surfaces (4.4 × 105 CFU cm−2 live E. coli and 2.1 × 105 CFU cm−2 live S. aureus), respectively, indicating that PEG and carboxy groups could endow antifouling properties for material surfaces,45,46 distinctly, these surfaces should possess antibacterial activities. Thus, there were no live E. coli or S. aureus cells detected on all surfaces of the PCLPUn containing different GQASs. A glass slide spreading method was also employed to investigate the contact-active antibacterial activity of these obtained PCLPUn films,47 and using PCLPU0 as a negative control. All PCLPUn films containing GQAS show excellent contact-active antibacterial activity meaning that no bacteria colonies were observed on their surface (Fig. 3B). To further test which GQAS chain extender introduced can form a better antibacterial activity for polyurethanes, a series of PCLPUn films with varying GQAS content (5, 6, 7 times dilution) were prepared by changing the weight ratios of PCLPUn and PCLPU0. After 6 times dilution, PCLUP12 still has a good contact-active antibacterial activity. The results show that PCLPU12 has the best contact-active antibacterial activity (Fig. 3C), and the antibacterial activity of PCLPU16 is relatively the weakest among these obtained PCLPUn films. The contact-active antibacterial mechanism of the QAS polymers is that the positively charged quaternary ammonium salts on the polyurethane surface first interact with the negatively charged phospholipid head groups of the bacterial cellular membrane, causing a general perturbation of the lipid bilayer. The long hydrophobic alkyl chains of QAS then pierce the membranes of these surface-attached bacteria, forming holes that cause cytoplasm leakage, lysis, and death.48 Therefore, the antibacterial activity is heavily impacted by the hydrophobic alkyl chains and cellular membrane thickness of bacteria,37,49,50 the optimal alkyl chain of QAS could achieve the best antibacterial activity.
 | ||
| Fig. 3 (A) Antibacterial and antifouling activity of PCLPUn films: live bacteria attached on surfaces with and without GQAS and PEG. (B) Contact-active antibacterial and antifouling activity of PCLPUn films. (C) Killing efficiency and contact-active antibacterial activity of PCLPUn films with various GQAS content, which were obtained via changing the weight ratios of PCLPUn and PCLPU0. | ||
Degradation properties of PCLPUn
From what has been discussed before, a polymer surface with biodegradable property could be gradually decomposed and degraded and release the attached living organisms.16–19 Thus investigation of the degradation properties of these antibacterial PCLPUn films is of great importance. The hydrolytic and enzymatic degradation characteristics of obtained PCLPUn have been preliminarily explored in vitro. The degradation rates of the obtained PCLPUn films were determined by measuring their weight losses during degradation (Fig. 4). For both hydrolytic and enzymatic degradation, weight loss ratios of all polyurethanes increased sharply in the first day, and then slowed down afterward, possibly due to the good solubility and relatively small molecular weight components leaching out from the polyurethane matrix at the initial phase of degradation.51 The degradation rates of these polyurethanes observed in lipase PS were higher than those in PBS, indicating that PCLPUn are facilely degraded in enzymatic solution by the acceleration of lipase PS compared to PCL-based polymers biodegradation.33,52,53 It is valuable to note that the degradation rates of PCLPUn had an increase in lipase PS than those in PBS. Also, the enzymatic degradation rate of PCLPU12 is the highest of all the samples. The probable reason is that the more hydrophilic surfaces of these polyurethanes are more liable to have accelerated degradation by lipase PS (Fig. S4, ESI†). The good hydrophilicity is due to the higher migration ratio of PEG and gemini quaternary ammonium on thePCLPU12 surfaces,51 which will be investigated in our further work.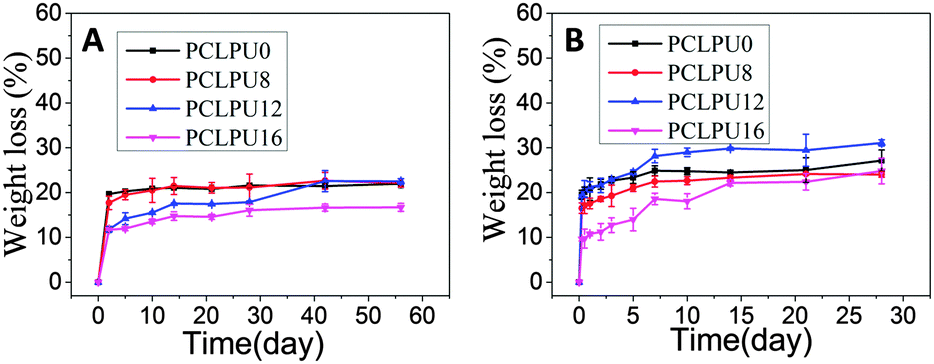 | ||
| Fig. 4 In vitro hydrolytic and enzymatic degradation profiles of PCLPUn and PCLPU0 films. (A) Hydrolytic degradation in PBS; (B) enzymatic degradation in lipase PS. Error bars represent means ± standard deviation for n = 3. | ||
To further investigate the degradation mechanism under different conditions, SEM was used to observe the surface morphology changes of all PCLPUn samples. Fig. 5 shows the SEM images of non-degraded and degraded PCLPUn films surfaces in PBS and lipase PS media. After 28 day of hydrolytic degradation, these samples show rough and porous surfaces, and even cracks to channel-like structures (Fig. 5a1–d1), which are attributed to the leaching of water-soluble oligomers or monomers formed after hydrolytic degradation from amorphous areas of these film surfaces into the surrounding media.51,54 However, enzymatic degradation shows a superficial erosion over the whole surface and all samples exhibit rough surfaces without any pores or cracks in the structure under lipase PS conditions (Fig. 5a2–d2). This occurs especially for bacterial enzymes,34 because these enzymes adsorb onto the surface before initiating polyurethane hydrolysis.55 These results are in good agreement with the commonly recognized fact of superficial erosion in the case of enzymatic degradation.56 More importantly, this degradation pattern can give PCLPUn films antifouling properties in the bacterial growth environment by releasing the attached bacteria with the whole superficial erosion degradation, and further studies are currently being carried out in our group to investigate the antifouling mechanism.
 | ||
| Fig. 5 Superficial SEM micrographs of PCLPUn films: (A–D) non-degraded samples; (a1–d1): samples degraded in PBS medium for 28 days; (a2–d2) samples degraded for 28 days in lipase PS. Bar = 100 µm. The enlarge images bar = 10 µm. | ||
Biocompatibility of PCLPUn films
The study of the biocompatibility of the PCLPUn films was carried out by evaluating the cytotoxicity of their extracts and degradation liquor by MTT assay, which was frequently applied to screen the polymer cytotoxicity for its reliability and sensitivity. L929 mouse fibroblasts are used in cytotoxicity assays. The cytotoxicity of the GQAS chain extenders were also performed through MTT assay (Fig. S6, ESI†) and a GQAS chain extender with a longer hydrophobic alkyl chain has a higher toxicity.The extraction procedures were carried out to simulate clinical conditions to evaluate the release of leachable toxic components from these PCLPUn matrices without damaging their physicochemical or mechanical properties. The effect of the extract concentrations on the proliferation of L929 mouse fibroblasts is assessed, as shown in Fig. 6. The extracts of the PCLPUn samples show an increasing cytotoxicity against fibroblasts with increase in GQAS hydrophobic alkyl chain length. However, more than 90% cell viability could be retained after 5 times dilution with a 24 or 72 h incubation for PCLPU8 and PCLPU12, suggesting that GQAS polyurethanes containing shorter hydrophobic alkyl chain length of GQAS are able to have good biocompatibility.
 | ||
| Fig. 6 Cell viability measured by MTT assay after 24 h (A) and 72 h (B) of incubation with serial dilutions of samples extracts. Error bars represent means ± standard deviation for n = 3. Statistical significance: p < 0.05. | ||
To further evaluate the biocompatibility of these PCLPUn samples, an accelerated degradation in a strong basic solution was carried out to completely decompose the polymers and yield the maximum release of degradation products. All these polyurethane degradation solutions were performed using the MTT assay and these results showed no apparent inhibition effect for the L929 fibroblasts when the degradation solutions are diluted to 0.25 mg mL−1 (Fig. 7 and Fig. S7, ESI†). The lowered cell viability at a high concentration is probably caused by the inherent toxicity of most polycations51 and large amounts of sodium chloride in the degradation solutions. The blank control sample (NaOH + HCl) without dilution shows high cytotoxicity and can confirm this phenomenon. Therefore, in view of clinical usage and the low degradation ratio of the samples, these polyurethanes and their degradation products can potentially meet the safety requirements of biomedical applications. In addition, the effect of GQAS content on antibacterial and biocompatibility was further studied in our ongoing work.
 | ||
| Fig. 7 Cell viability measured by MTT assay after 24 h (A) and 72 h (B) of incubation with different concentrations of PCLPUn degradation products. Error bars represent means ± standard deviation for n = 3. Statistical significance: p < 0.05. | ||
Conclusions
In summary, a series of antibacterial and antifouling waterborne biodegradable polyurethanes containing GQAS have been successfully synthesized using PCL, PEG, lysine and IPDI by a facile polymerization strategy. The incorporation of different hydrophobic alkyl chain lengths of GQAS has significant impacts on the properties of these waterborne polyurethanes, including the antibacterial activities, antifouling, biodegradability, and biocompatibility. The obtained PCLPUn films could be degraded via a superficial erosion mechanism in lipase PS, thus this degradation pattern is favorable for these surfaces to possess a good antifouling ability in a bacterial growth environment to release the attached bacteria accompanied by the layer degradation of PCLPUn films. Hence, these waterborne polyurethanes, especially PCLPU12 with the best antibacterial activity and the relatively good biocompatibility, could potentially be applied as coatings on implant surfaces and tissue engineering scaffolds to prevent biomaterial related infections.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51173118, 51273124, and 51273126), and the National Science Fund for Distinguished Young Scholars of China (51425305).Notes and references
- R. Djeribi, W. Bouchloukh, T. Jouenne and B. Menaa, Am. J. Infect. Control, 2012, 40, 854–859 CrossRef PubMed.
- X. Ding, C. Yang, T. P. Lim, L. Y. Hsu, A. C. Engler, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2012, 33, 6593–6603 CrossRef CAS PubMed.
- N. M. O'Brien-Simpson, Polym. Chem., 2015, 6, 198–212 RSC.
- T. D. Michl, K. E. Locock, N. E. Stevens, J. D. Hayball, K. Vasilev, A. Postma, Y. Qu, A. Traven, M. Haeussler and L. Meagher, Polym. Chem., 2014, 5, 5813–5822 RSC.
- F. Paladini, M. Pollini, A. Sannino and L. Ambrosio, Biomacromolecules, 2015, 16, 1873–1885 CrossRef CAS PubMed.
- F. Pishbin, V. Mouriño, S. Flor, S. Kreppel, V. Salih, M. P. Ryan and A. R. Boccaccini, ACS Appl. Mater. Interfaces, 2014, 6, 8796–8806 CAS.
- M. L. Knetsch and L. H. Koole, Polymers, 2011, 3, 340–366 CrossRef CAS.
- P. Li, Y. F. Poon, W. Li, H.-Y. Zhu, S. H. Yeap, Y. Cao, X. Qi, C. Zhou, M. Lamrani and R. W. Beuerman, Nat. Mater., 2010, 10, 149–156 CrossRef PubMed.
- H. Tan, Z. Peng, Q. Li, X. Xu, S. Guo and T. Tang, Biomaterials, 2012, 33, 365–377 CrossRef CAS PubMed.
- S. Buffet-Bataillon, P. Tattevin, M. Bonnaure-Mallet and A. Jolivet-Gougeon, Int. J. Antimicrob. Agents, 2012, 39, 381–389 CrossRef CAS PubMed.
- C. Zhu, Q. Yang, L. Liu, F. Lv, S. Li, G. Yang and S. Wang, Adv. Mater., 2011, 23, 4805–4810 CrossRef CAS PubMed.
- Y. Zhang, M. Ding, L. Zhou, H. Tan, J. Li, H. Xiao, J. Li and J. Snow, Polym. Chem., 2012, 3, 907 RSC.
- L. A. T. W. Asri, M. Crismaru, S. Roest, Y. Chen, O. Ivashenko, P. Rudolf, J. C. Tiller, H. C. van der Mei, T. J. A. Loontjens and H. J. Busscher, Adv. Funct. Mater., 2014, 24, 346–355 CrossRef CAS.
- M. Patel, R. Patel, W. S. Chi, J. H. Kim and J.-S. Sung, Chin. J. Polym. Sci., 2015, 33, 265–274 CrossRef CAS.
- G. Cheng, H. Xue, Z. Zhang, S. Chen and S. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8831–8834 CrossRef CAS PubMed.
- L. Xie, F. Hong, C. He, C. Ma, J. Liu, G. Zhang and C. Wu, Polymer, 2011, 52, 3738–3744 CrossRef CAS.
- C. Ma, H. Yang, X. Zhou, B. Wu and G. Zhang, Colloids Surf., B, 2012, 100, 31–35 CrossRef CAS PubMed.
- C. Ma, L. Xu, W. Xu and G. Zhang, J. Mater. Chem. B, 2013, 1, 3099 RSC.
- F. Hong, L. Xie, C. He, J. Liu, G. Zhang and C. Wu, J. Mater. Chem. B, 2013, 1, 2048 RSC.
- S. Q. Liu, C. Yang, Y. Huang, X. Ding, Y. Li, W. M. Fan, J. L. Hedrick and Y.-Y. Yang, Adv. Mater., 2012, 24, 6484–6489 CrossRef CAS PubMed.
- G. Li, J. Shen and Y. Zhu, J. Appl. Polym. Sci., 2000, 78, 668–675 CrossRef CAS.
- C. Z. Chen, N. C. Beck-Tan, P. Dhurjati, T. K. van Dyk, R. A. LaRossa and S. L. Cooper, Biomacromolecules, 2000, 1, 473–480 CrossRef CAS.
- H. Kourai, T. Yabuhara, A. Shirai, T. Maeda and H. Nagamune, Eur. J. Med. Chem., 2006, 41, 437–444 CrossRef CAS PubMed.
- H. Tan and H. Xiao, Tetrahedron Lett., 2008, 49, 1759–1761 CrossRef CAS.
- T. Abel, J. I. Cohen, R. Engel, M. Filshtinskaya, A. Melkonian and K. Melkonian, Carbohydr. Res., 2002, 337, 2495–2499 CrossRef CAS PubMed.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, ACS Macro Lett., 2015, 4, 961–964 CrossRef CAS.
- T. C. Tseng, L. Tao, F. Y. Hsieh, Y. Wei, I. M. Chiu and S. H. Hsu, Adv. Mater., 2015, 27, 3518–3524 CrossRef CAS PubMed.
- M. Garcıa, I. Ribosa, T. Guindulain, J. Sanchez-Leal and J. Vives-Rego, Environ. Pollut., 2001, 111, 169–175 CrossRef.
- L. Pérez, M. T. Garcia, I. Ribosa, M. P. Vinardell, A. Manresa and M. R. Infante, Environ. Toxicol. Chem., 2002, 21, 1279–1285 CrossRef.
- Y. Zhang, Y. Li, J. Li, Y. Gao, H. Tan, K. Wang, J. Li and Q. Fu, Sci. Bull., 2015, 60, 1114–1121 CrossRef CAS.
- Y.-J. Jeon, P.-J. Park and S.-K. Kim, Carbohydr. Polym., 2001, 44, 71–76 CrossRef CAS.
- H. Bakhshi, H. Yeganeh, S. Mehdipour-Ataei, M. A. Shokrgozar, A. Yari and S. N. Saeedi-Eslami, Mater. Sci. Eng., C, 2013, 33, 153–164 CrossRef CAS.
- J. R. Porter, A. Henson and K. C. Popat, Biomaterials, 2009, 30, 780–788 CrossRef CAS PubMed.
- J. Zeng, X. Chen, Q. Liang, X. Xu and X. Jing, Macromol. Biosci., 2004, 4, 1118–1125 CrossRef CAS PubMed.
- X. Gu, J. Wu and P. T. Mather, Biomacromolecules, 2011, 12, 3066–3077 CrossRef CAS PubMed.
- J. C. Tiller, C.-J. Liao, K. Lewis and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5981–5985 CrossRef CAS PubMed.
- J. C. Tiller, S. B. Lee, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2002, 79, 465–471 CrossRef CAS PubMed.
- G. Gao, D. Lange, K. Hilpert, J. Kindrachuk, Y. Zou, J. T. J. Cheng, M. Kazemzadeh-Narbat, K. Yu, R. Wang, S. K. Straus, D. E. Brooks, B. H. Chew, R. E. W. Hancock and J. N. Kizhakkedathu, Biomaterials, 2011, 32, 3899–3909 CrossRef CAS PubMed.
- G. Cheng, Z. Zhang, S. Chen, J. D. Bryers and S. Jiang, Biomaterials, 2007, 28, 4192–4199 CrossRef CAS PubMed.
- E. E. MacKintosh, J. D. Patel, R. E. Marchant and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2006, 78, 836–842 CrossRef PubMed.
- N. Faucheux, R. Schweiss, K. Lutzow, C. Werner and T. Groth, Biomaterials, 2004, 25, 2721–2730 CrossRef CAS PubMed.
- M. Ding, Z. Qian, J. Wang, J. Li, H. Tan, Q. Gu and Q. Fu, Polym. Chem., 2011, 2, 885 RSC.
- K. Rezwan, L. P. Meier and L. J. Gauckler, Biomaterials, 2005, 26, 4351–4357 CrossRef CAS PubMed.
- P.-S. Liu, Q. Chen, S.-S. Wu, J. Shen and S.-C. Lin, J. Membr. Sci., 2010, 350, 387–394 CrossRef CAS.
- K. Hilpert, M. R. Elliott, R. Volkmer-Engert, P. Henklein, O. Donini, Q. Zhou, D. F. H. Winkler and R. E. W. Hancock, Chem. Biol., 2006, 13, 1101–1107 CrossRef CAS PubMed.
- C. Wu, Chin. J. Polym. Sci., 2014, 32, 1575–1580 CrossRef CAS.
- C. J. Waschinski, J. Zimmermann, U. Salz, R. Hutzler, G. Sadowski and J. C. Tiller, Adv. Mater., 2008, 20, 104–108 CrossRef CAS.
- L. Qian, Y. Guan, B. He and H. Xiao, Polymer, 2008, 49, 2471–2475 CrossRef CAS.
- H. Murata, R. R. Koepsel, K. Matyjaszewski and A. J. Russell, Biomaterials, 2007, 28, 4870–4879 CrossRef CAS PubMed.
- J. C. Tiller, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5981–5985 CrossRef CAS PubMed.
- M. Ding, J. Li, X. Fu, J. Zhou, H. Tan, Q. Gu and Q. Fu, Biomacromolecules, 2009, 10, 2857–2865 CrossRef CAS PubMed.
- N. Jiang, S. Jiang, Y. Hou, S. Yan, G. Zhang and Z. Gan, Polymer, 2010, 51, 2426–2434 CrossRef CAS.
- H. L. Zeng, C. Gao and D. Y. Yan, Adv. Funct. Mater., 2006, 16, 812–818 CrossRef CAS.
- L. Zhou, D. Liang, X. He, J. Li, H. Tan, J. Li, Q. Fu and Q. Gu, Biomaterials, 2012, 33, 2734–2745 CrossRef CAS PubMed.
- L. A. Bosworth and S. Downes, Polym. Degrad. Stab., 2010, 95, 2269–2276 CrossRef CAS.
- I. Castilla-Cortázar, J. Más-Estellés, J. M. Meseguer-Dueñas, J. L. Escobar Ivirico, B. Marí and A. Vidaurre, Polym. Degrad. Stab., 2012, 97, 1241–1248 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00039h |
| This journal is © the Partner Organisations 2017 |