
Supramolecular hybrids of carbon dots with doxorubicin: synthesis, stability and cellular trafficking
Tingting
Sun
ab,
Min
Zheng
*c,
Zhigang
Xie
*a and
Xiabin
Jing
a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun, Jilin 130022, P. R. China. E-mail: xiez@ciac.ac.cn; Fax: +86 431 85262775; Tel: +86 431 85262775
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cChemistry and Life Science School, Advanced Institute of Materials Science, Changchun University of Technology, 2055 Yanan Street, Changchun, Jilin 130012, P. R. China. E-mail: zhengmin@ccut.edu.cn
First published on 15th August 2016
Abstract
Supramolecular hybrids of carbon dots (CDs) and doxorubicin (Dox) were successfully prepared via π–π stacking and electrostatic interactions. The hybrids were characterized by the changes in size, morphology and zeta potential, and further validated by the absorption and photoluminescence spectra. A binding constant of 63 L g−1 between CDs and Dox was calculated from the Stern–Volmer plot. The hybrids between CDs and Dox (CDs–Dox) showed pH-dependent drug loading and release behaviors in aqueous solution. The poor stability of CDs–Dox in fetal bovine serum (FBS) solution led to the rapid separation of Dox and CDs, facilitating the release of Dox and its entrance into cellular nuclei as revealed by confocal laser scanning microscopy (CLSM). After being coated with polydopamine (CDs–Dox@PDA), the stability of the hybrids in cell culture media was enhanced, as supported by the slow release of Dox in living cells. This work highlights the potential of using CDs to synthesize functional nanoparticles through supramolecular interactions.
Introduction
Carbon nanomaterials are a class of low-dimensional materials, which include fullerenes (zero-dimensional, 0D), carbon nanotubes (1D), graphene (2D), and recently carbon dots (0D/2D).1–7 Nanodiamonds, which consist of mostly sp3 carbon atoms, have also received much research interest.8 More recently, a new type of 1D nanostructure comprised of crystalline sp3 carbon only, named carbon nanothreads, has been reported.9 Owing to the quantum confinement effect, these carbon nanomaterials have a wide range of applications in electronics, photonics, sensors, renewable energy, and biomedicine.10–14 Among different subtypes of graphene-based nanomaterials, graphene oxide (GO), with large specific surface area (2630 m2 g−1) and abundant functional groups (hydroxyl, epoxide, and carboxylic groups), has found great potential in drug delivery.15–17 The unique structural features endow GO with physiological stability and solubility, excellent biocompatibility, and capability of loading drugs or genes via physisorption approaches.18 It was demonstrated that graphene materials can be loaded with aromatic ring-containing anticancer drugs with high efficiency through supramolecular interactions.15,17–22 For example Dai et al. for the first time employed polyethylene glycol (PEG) modified nanoscale GO as a drug carrier to load doxorubicin (Dox)16 and a camptothecin (CPT) analogue SN3817 by simple non-covalent adsorption, including π–π stacking and hydrophobic interactions.Supramolecular stacking of Dox on carbon-based nanomaterials for biomedical applications has been studied since the pioneering work by Liu et al., who reported the supramolecular hybrids of Dox and carbon nanotubes for cancer therapy.23 PEGylated carbon nanodots have been used for real-time monitoring of drug delivery and two-photon imaging.24 Besides, graphene quantum dots (GQDs) with fascinating electronic properties have been used as multifunctional drug nanocarriers to load Dox for cancer therapy.25
Recently, carbon dots (CDs) have attracted a great deal of attention owing to their unique properties, such as high solubility in water, nontoxicity, excellent biocompatibility, and good cell permeability.26–28 They exhibit extensive potential applications in energy conversion, ion detection, and bioimaging fields.5,29–31 In order to be used as drug carriers, CDs are usually chemically modified through the surface functional groups, such as hydroxyl, amino and carboxyl groups. For example, we have reported a theranostic agent integrating oxaliplatin by using luminescent CDs.26 Dox and the targeting peptide have been conjugated on CDs for nucleus targeted delivery and cancer therapy.32 Alternatively, it would be useful to develop supramolecular methods to load drug molecules on CDs. Lately, Guldi et al. have demonstrated that carbon nanodots could form supramolecular electron donor–acceptor hybrids with electron-accepting perylenediimides (PDI) via electrostatic and π–π stacking interactions.2 However, the interplay of CDs and Dox was not studied in detail, and the stability of hybrids under physiological conditions is unknown yet. So it is important to understand the supramolecular interactions between CDs and Dox for developing a CD-based drug delivery system. In this work, Dox was selected as a model drug and its interactions with CDs were investigated via size, morphology, zeta potential and spectral analysis. The stability of the hybrids between CDs and Dox (CDs–Dox) under different conditions was monitored by photoluminescence (PL) spectroscopy. The cellular uptake and cytotoxicities were also evaluated in detail.
Experimental
Materials
Dox was purchased from Zhejiang Hisun Pharmaceutical Co. Ltd (Taizhou city, China). 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Shanghai Yuanye Co. Ltd (Shanghai, China). MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetra-zolium bromide) was purchased from Amresco (USA). Ultrapure water was prepared from a Milli-Q system (Millipore, USA). All other chemicals were of analytical grade or above.Methods
Ultraviolet-visible (UV-vis) absorption spectra were obtained by using a Shimadzu UV-2450 PC UV-vis spectrophotometer. Fluorescence intensity tests were performed on a PerkinElmer LS-55 spectrofluorophotometer. The size and size distribution of the nanoparticles were determined by dynamic light scattering (DLS) using a Malvern Zeta-sizer Nano. The measurement was carried out at 25 °C and the scattering angle was fixed at 90°. The morphology of the nanoparticles was measured by transmission electron microscopy (TEM) performed on a JEOL JEM-1011 electron microscope operating at an acceleration voltage of 100 kV. To prepare the specimen for TEM, a drop of solution was deposited onto a copper grid with a carbon coating. The specimen was air-dried and measured at room temperature.Preparation of CDs
CDs were prepared according to the reported method.33D-Glucose (2.5 mmol) and L-glutamic acid (2.5 mmol) were loaded into a beaker and then 1 M sodium hydrate (3.0 mL) was added. The transparent solution was heated to 125 °C and kept for 30 min, then heated to 200 °C and maintained for 20 min. The final reaction products were completely solubilized, and then placed in a dialysis bag (cutoff Mw: 3.5 kDa) and dialyzed against water to remove small molecules for 1 d. The resulting product was freeze-dried to obtain a brown solid with a yield of 29.8%.Dox loading and release behaviors of CDs–Dox
Dox loading and release behaviors of CDs–Dox were investigated according to the literature.34 Dox loading onto CDs was achieved by simple mixing of CDs with Dox solution in water or phosphate buffered saline (PBS) at pH 5.0 and 7.4. After shaking overnight, excess Dox molecules were removed by centrifugation and washed thoroughly with water or PBS until the supernatant became free of reddish color (corresponding to free Dox). The resulting CDs–Dox hybrid was redispersed in water or PBS for subsequent tests of Dox release and cytotoxicities in vitro. The loading content of Dox was determined by the absorbance at 480 nm for Dox, according to the standard calibration curve of free Dox. The drug loading efficiency (DLE) was calculated according to the following equation:| DLE% = (weight of Dox in CDs–Dox/weight of Dox added) × 100% |
To measure the drug release, CDs–Dox hybrids were dispersed in 3 mL of PBS solutions (pH 5.0 or 7.4) at an initial concentration of 1.07 mg mL−1, sealed in a dialysis bag (Mw cutoff: 3.5 kDa) and incubated in PBS (17 mL) at 37 °C under oscillation at 90 rpm. At selected time intervals, 1 mL of buffer solution outside the dialysis bag was removed for UV-vis analysis and replaced with 1 mL of fresh buffer solution. The released amount of Dox was determined from the absorbance at 480 nm with the help of a standard calibration curve of Dox in the same buffer. Then the accumulative weight and relative percentage of the released Dox were calculated as a function of incubation time.
Preparation of CDs–Dox with polydopamine (PDA) coatings (CDs–Dox@PDA)
CDs–Dox@PDA was prepared according to the reported method.35,36 About 0.3 mg of CDs–Dox were incubated in 2 mL of 0.5 mg mL−1 dopamine hydrochloride solution in Tris buffer (10 mM, pH 8.5). After being stirred under air at room temperature for 3 h, the obtained CDs–Dox@PDA coatings was purified by centrifugation and redispersed in water or PBS for further experiments.Cell lines and cell culture
Human cervical carcinoma (HeLa) cells were grown in Dulbecco's modified Eagle's medium (DMEM, GIBCO) supplemented with 10% heat-inactivated fetal bovine serum (FBS, GIBCO), and the culture medium was replaced once every day.MTT assays
Cells harvested in a logarithmic growth phase were seeded in 96-well plates at a density of 2 × 103 cells per well and incubated in DMEM for 24 h. The medium was then replaced by CDs, free Dox, CDs–Dox or CDs–Dox@PDA at various concentrations. The incubation was continued for 48 h. Then, 20 µL of MTT solution in PBS at the concentration of 5 mg mL−1 was added and the plates were incubated for another 4 h at 37 °C, followed by the removal of the culture media containing MTT and addition of 150 µL of dimethyl sulfoxide (DMSO) to each well to dissolve the formazan crystals formed. Finally, the plates were shaken for 5 min, and the absorbance of the formazan product was measured at 490 nm using a microplate reader.Confocal laser scanning microscopy (CLSM)
Cellular uptake by HeLa cells was examined using CLSM. HeLa cells were seeded in 6-well culture plates (a clean cover slip was put in each well) at a density of 2 × 105 cells per well and allowed to adhere for 24 h. The medium was then replaced by Dox, CDs–Dox or CDs–Dox@PDA solutions at a final Dox concentration of 15 µg mL−1 for each. After incubation for different time periods at 37 °C, the supernatant was carefully removed and the cells were washed three times with PBS. Subsequently, the cells were fixed with 1 mL of 4% paraformaldehyde in each well for 10 min at room temperature and washed twice with PBS again. DAPI was used to stain the nuclei. Samples were examined by CLSM using a Zeiss LSM 700 (Zurich, Switzerland).Measurements of the fluorescence intensity changes of CDs–Dox or CDs–Dox@PDA
CDs–Dox or CDs–Dox@PDA were dispersed in DMEM supplemented with 10% heat-inactivated FBS or in PBS solutions (pH 7.4) with different volume ratios (0, 5%, 10% and 50%) of FBS, and then the fluorescence intensity was measured at different time periods.Results and discussion
Preparation and characterization of CDs and CDs–Dox
CDs were prepared according to the reported method33 with a diameter of about 2 nm. CDs showed two broad absorption peaks at 277 and 338 nm and the maximum emission was at 547 nm under 480 nm excitation (Fig. 1a). For preparation of CDs–Dox, Dox was dissolved in deionized water, mixed with CD aqueous solution and shaked overnight at room temperature, and excess Dox molecules were removed by centrifugation.34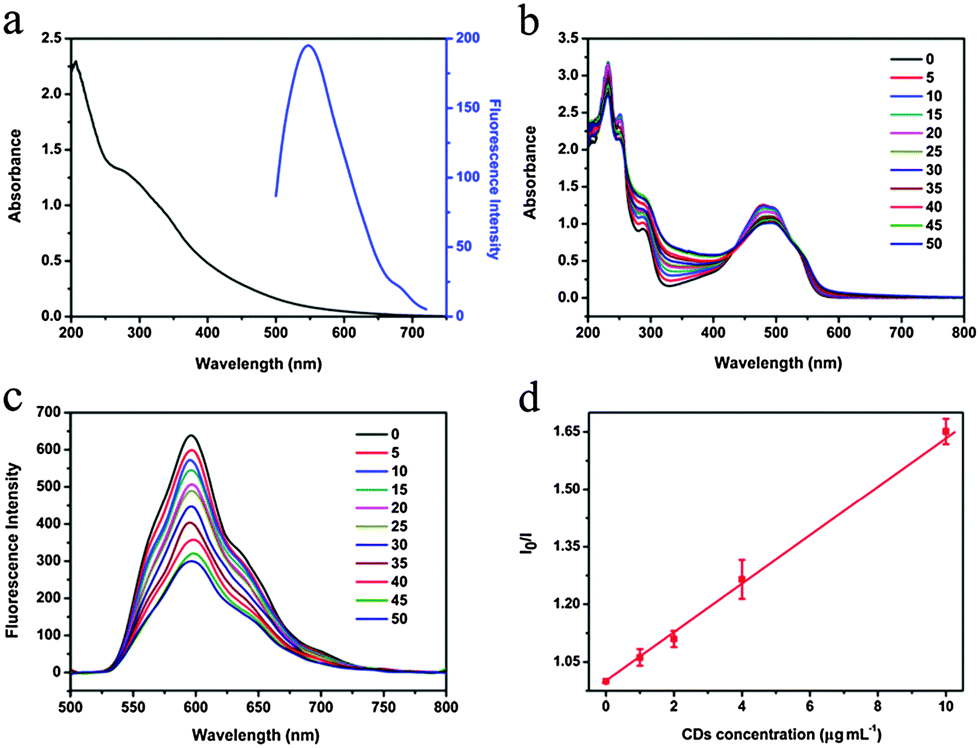 | ||
| Fig. 1 (a) Absorption and PL (excited at 480 nm) spectra of CDs. (b) Absorption and (c) PL spectra of Dox (66.7 µg mL−1) excited at 480 nm during the course of a titration with CDs (0–50 µg mL−1) in deionized water at room temperature. (d) Relationship of I0/I versus the concentration of CDs used to determine the binding constant, where I0 is the fluorescence intensity of Dox without CDs, and I is that of Dox after titration with different concentrations of CDs. | ||
Optical absorption and PL spectra were used to analyze the interactions between CDs and Dox. Steady-state absorption titration assays provided particular information about the non-covalent assembly of CDs with Dox. The concentration of Dox was kept constant at 66.7 µg mL−1, CDs with various concentrations (0–50 µg mL−1) were added and the absorption changes were recorded in Fig. 1b. An obvious decrease of the Dox-centered absorption was observed with increasing concentrations of CDs, which was a typical indicator for the non-covalent complexation of CDs and Dox.2,37 Moreover, a shift of the absorption maxima from 480 to 494 nm, together with the appearance of two isosbestic points at 432 and 530 nm suggested significant electronic communication between CDs and Dox.38 It was found that titration of CDs into the aqueous solution of Dox led to the fluorescence quenching of Dox. Fig. 1c shows the PL spectra of Dox excited at 480 nm with various concentrations of CDs. With increasing concentrations of CDs, the fluorescence intensity of Dox gradually decreased. The Stern–Volmer plot of I0/I (I0 is the fluorescence intensity of Dox without CDs, and I is the fluorescence intensity of Dox after titration with different concentrations of CDs) versus the concentration of CDs as shown in Fig. 1d indicates a binding constant of 63 L g−1.
The morphology and size distribution of CDs–Dox at a weight ratio of CDs and Dox of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 were characterized by TEM and DLS, as shown in Fig. 2. The TEM image (Fig. 2a) showed that small aggregates with sizes around 100 nm were formed, and the DLS plot (Fig. 2b) presented a similar result. Besides, the zeta potential of CDs was −27.9 mV, which changed to −14.1 mV after mixing with Dox at a weight ratio of 3:4. The changes in size, morphology and zeta potential indicated that hybrids between CDs and Dox were successfully prepared.
:4 were characterized by TEM and DLS, as shown in Fig. 2. The TEM image (Fig. 2a) showed that small aggregates with sizes around 100 nm were formed, and the DLS plot (Fig. 2b) presented a similar result. Besides, the zeta potential of CDs was −27.9 mV, which changed to −14.1 mV after mixing with Dox at a weight ratio of 3:4. The changes in size, morphology and zeta potential indicated that hybrids between CDs and Dox were successfully prepared.
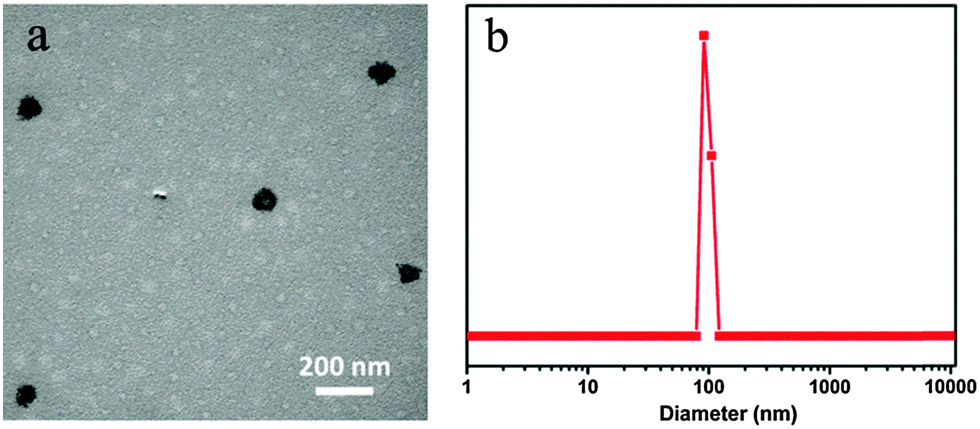 | ||
| Fig. 2 TEM image (a) and size distribution (b) of CDs–Dox at a weight ratio of CDs and Dox of 3:4. | ||
Dox loading and release behaviors of CDs–Dox
A quantitative study of the loading behaviors of Dox on CDs in PBS at pH 5.0 and 7.4 was performed. The loaded Dox was determined via a standard curve. As shown in Fig. 3a, the amount of Dox loaded on the CDs gradually increased with increasing initial concentrations of Dox, and the drug loading ratio (the weight ratio of Dox to CDs) reached as high as 260 wt% at pH 7.4. It was found that the amount of Dox loaded on CDs was pH-dependent (Fig. 3a). The loading amount of Dox at pH 7.4 was dramatically higher than that at pH 5.0 when the concentrations of Dox were higher than 300 µg mL−1. This trend was attributed to the protonation of Dox at lower pH, and the low pH subsequently reduced the interactions between Dox and CDs. Similar types of pH-dependent loading of nanoscale drug carriers have been reported previously.19,39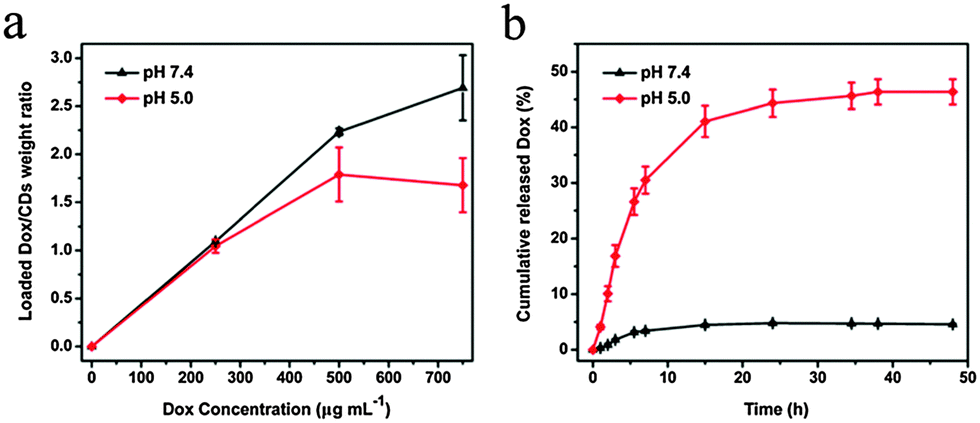 | ||
| Fig. 3 (a) Quantification of various concentrations of Dox (0–750 µg mL−1) loaded on CDs at different pH values (pH 5.0 and 7.4) at a fixed concentration of CDs (200 µg mL−1). (b) Dox release profiles of CDs–Dox in PBS (pH 5.0 and 7.4, 20 mM). | ||
A sustained-release character is indispensable for an efficient drug delivery system.34 The drug release behaviors of CDs–Dox were evaluated in PBS solutions at different pH values (pH 5.0 and 7.4). Fig. 3b shows the release profiles of the Dox at pH 5.0 and 7.4 at 37 °C. The release rate of Dox at pH 5.0 was significantly higher than that at pH 7.4. Only about 5% of Dox was released from CDs–Dox within 48 h at pH 7.4. While under acidic conditions of pH 5.0, Dox was quickly released in the first 7 hours, and about 46% of the Dox loaded was released in 48 h, indicating the sustained drug-release behavior of CDs–Dox. This is because of the protonation of Dox at a lower pH value, which increases the hydrophilicity of Dox and leads to rapid release of the loaded Dox. Such a release behavior of Dox is important, because the tumor extracellular environment is more acidic (pH 6.0–6.5) than blood (pH ∼ 7.4), and the pH values of endosomes and lysosomes are even lower (pH 5.0–5.5),40–43 which is beneficial for cancer therapy.34
Cytotoxicity study
The biocompatibility of nanomaterials is of great importance for biomedical applications,44,45 so we first tested the cytotoxicity of CDs toward HeLa cells by an MTT assay. As shown in Fig. 4a, no obvious cytotoxicity was observed even at a concentration of 1 mg mL−1 for CDs after incubation for 48 h, indicating excellent biocompatibility of CDs. The cytotoxicities of CDs–Dox and free Dox against HeLa cells determined by MTT assays after 48 h culture are illustrated in Fig. 4b. It can be seen that cytotoxicities are all concentration dependent. The inhibition efficacy of CDs–Dox is slightly lower than that of free Dox at lower concentrations (0.005–0.5 µg mL−1). However, it exhibits similar activities to free Dox at a concentration of 5 µg mL−1. This result demonstrates that CDs–Dox still possesses the ability to kill cancer cells.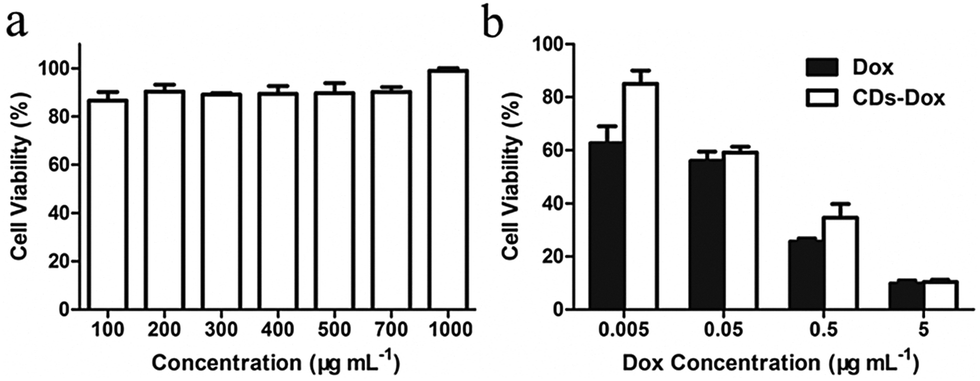 | ||
| Fig. 4 In vitro (a) biocompatibility of CDs against HeLa cells and (b) viabilities of HeLa cells incubated with free Dox and CDs–Dox for 48 h at 37 °C. All the results were repeated three times and presented as mean ± SD. | ||
Cellular uptake and stability of CDs–Dox
To obtain insight into the intracellular fate of CDs–Dox, we analyzed their cellular uptake by virtue of the inherent fluorescence of Dox using CLSM. The cellular nuclei were selectively stained with DAPI. CLSM images of HeLa cells incubated with CDs–Dox for 10 min, 30 min, 1 h and 1.5 h are shown in Fig. 5. Poor red fluorescence was observed in the cells after incubation for 10 min. And the intensity of red fluorescence increased obviously along with the incubation time. Nearly all the fluorescence located in the cellular nuclei, which might be ascribed to the rapid dissociation of CDs–Dox in the living cells.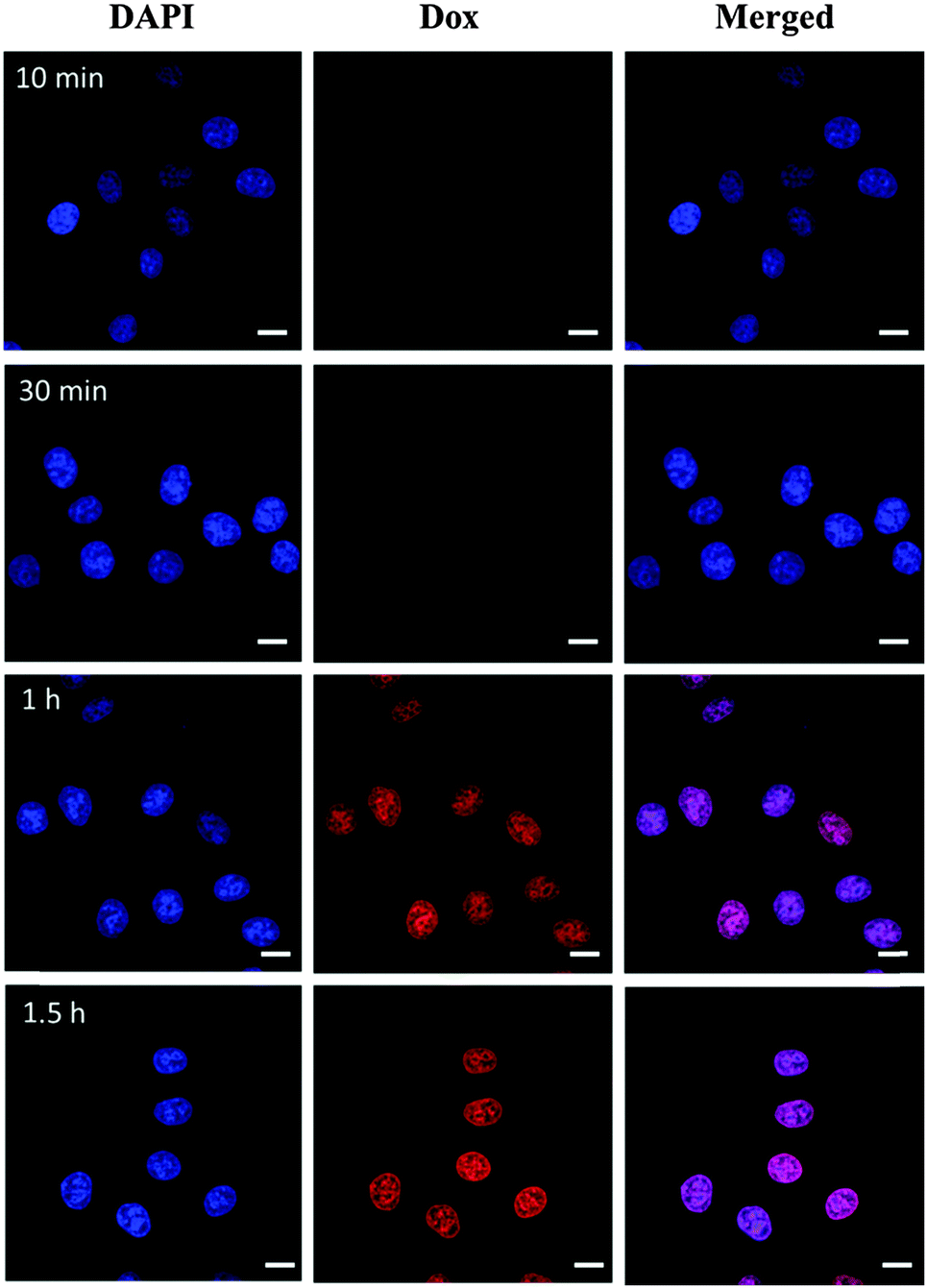 | ||
| Fig. 5 CLSM images of HeLa cells incubated with CD–Dox for different time periods at 37 °C at a Dox concentration of 15 µg mL−1. Cells are viewed in the blue channel for DAPI and in the red channel for Dox. Scale bars represent 20 µm in all images. | ||
We wondered whether CDs–Dox was stable in culture media, or part of the Dox molecules were released before CDs–Dox was internalized by the cells. We tested the fluorescence intensity changes of CDs–Dox hybrids in PBS solutions (pH 7.4) with different volume ratios (0, 5%, 10% and 50%) of FBS, as shown in Fig. 6. The fluorescence intensity of CDs–Dox in PBS solutions (pH 7.4) with FBS (5%, 10% and 50%) increased with time in the beginning, and then remained constant. With the increase of the content of FBS, more rapid changes in fluorescence intensity were observed. While nearly no fluorescence intensity change of CDs–Dox in PBS solutions in the absence of FBS could be detected. A quantitative comparison of fluorescence intensity changes in the form of I/I0 (I0 is the initial fluorescence intensity of CDs–Dox immediately dispersed in different solutions, and I is that of CDs–Dox after different time periods) versus time is shown in Fig. 6e. These results confirmed that CDs–Dox was not very stable and part of the Dox molecules could be released in the presence of serum.
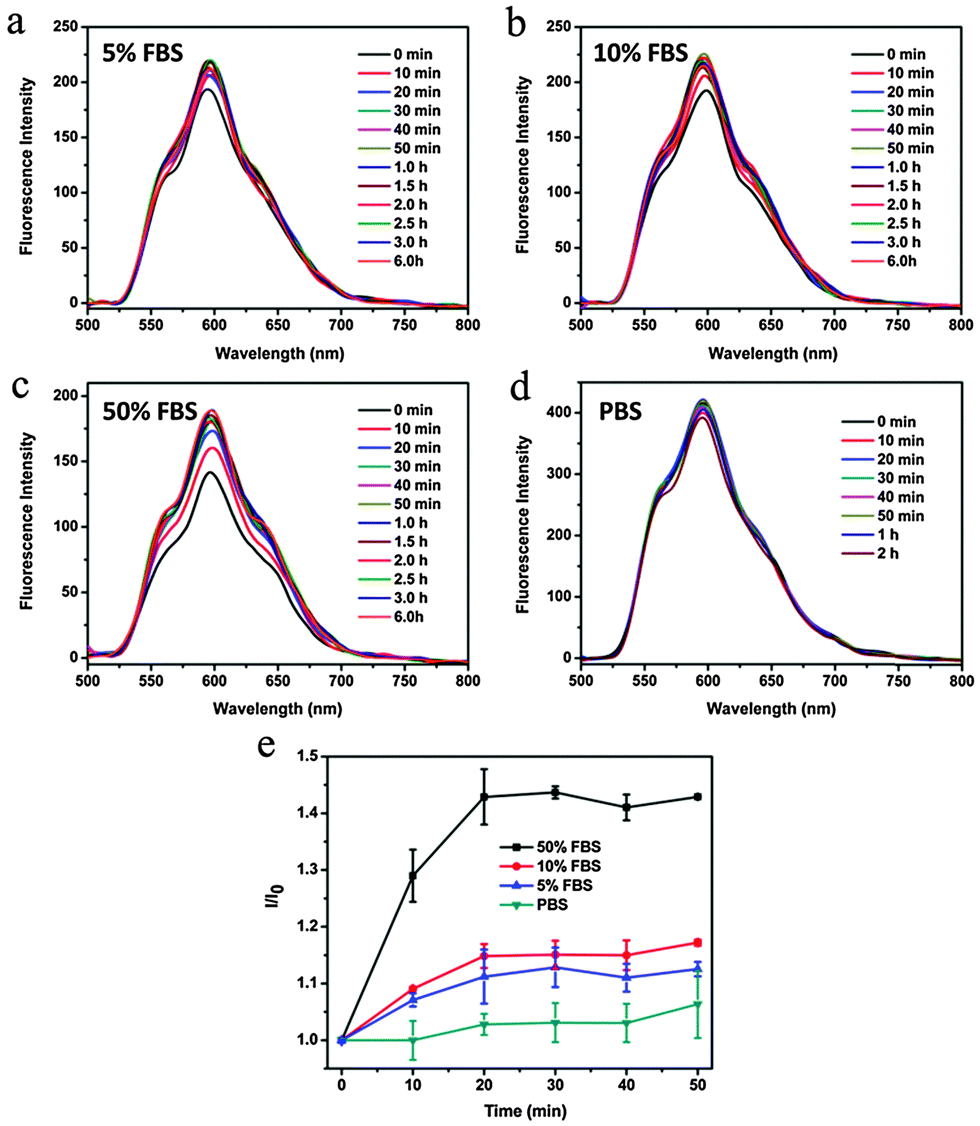 | ||
| Fig. 6 The fluorescence intensity changes of CDs–Dox in PBS (pH 7.4) solutions with volume ratios of FBS at 5% (a), 10% (b), 50% (c) and 0 (d). (e) Quantitative comparison of I/I0versus time under the above conditions in the first 50 minutes, where I0 is the initial fluorescence intensity of CDs–Dox immediately dispersed in different solutions, and I is that of CD–Dox after different time periods. | ||
The low stability of nanomedicine is undesired when they are injected into the blood before reaching the disease sites. To improve the stability of CDs–Dox, we employed a simple and versatile surface modification method based on dopamine polymerization.35 Research studies have confirmed that PDA can serve as a multifunctional nanocarrier for cancer therapy46 and PDA coatings possess good biocompatibility and promise for tissue engineering.36 Dopamine can form a stable, robust anchor on the surface of nanoparticles without regard to surface functional groups.47 Under weak alkaline conditions (∼pH 8–8.5) dopamine catechol is oxidized to quinone, which reacts with other catechols and/or quinones to form polymerized dopamine.35,48,49 CDs–Dox@PDA was prepared according to the reported method35,36 (Scheme 1a), and characterized by using TEM and DLS (Fig. 7a). Next, we evaluated the stability of CDs–Dox and CDs–Dox@PDA in DMEM supplemented with 10% heat-inactivated FBS, as shown in Fig. 7b. CDs–Dox@PDA exhibited improved stability in DMEM compared to CDs–Dox. The cellular uptake of CDs–Dox@PDA was then evaluated by using CLSM (Scheme 1b) after HeLa cells were incubated with CDs–Dox@PDA for 10 min, 30 min, 1 h and 1.5 h. As can be seen from Fig. 8, nearly no fluorescence of Dox could be detected at 10 min, and only weak red fluorescence could be observed in the cytoplasm at 30 min. While after 1.5 h incubation, bright red fluorescence could be seen both in the cellular nuclei and cytoplasm of HeLa cells clearly. For the control experiment, almost all the red fluorescence appeared in the cell nuclei for HeLa cells treated with free Dox for 30 min. These results proved that the PDA coatings could slow down the release of Dox and increase the stability of CDs–Dox in the culture media.
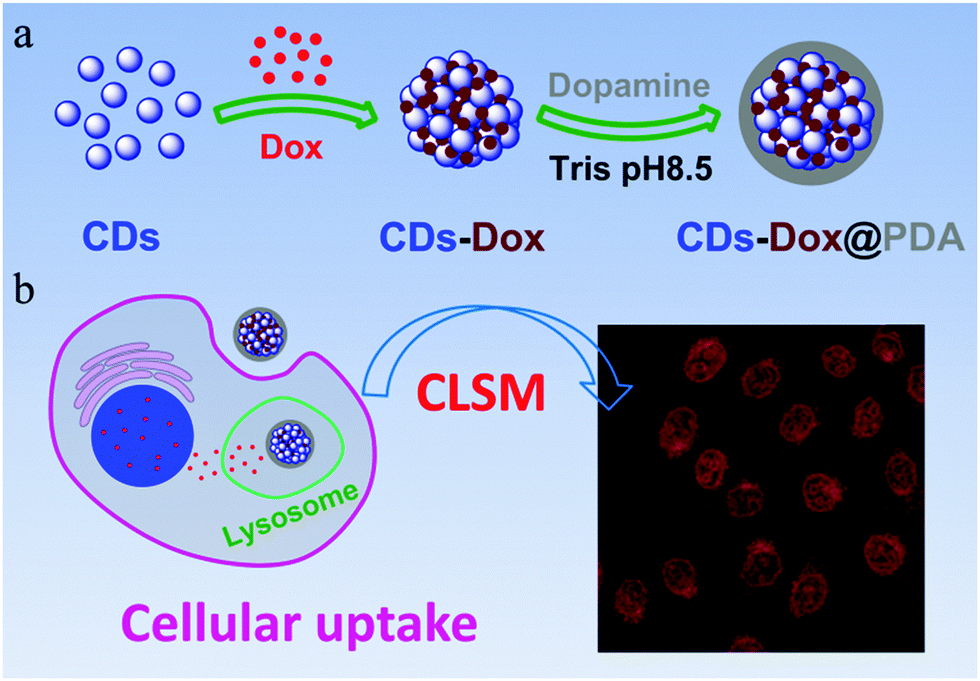 | ||
| Scheme 1 A schematic illustration of the preparation (a) and cellular uptake (b) of CDs–Dox@PDA. | ||
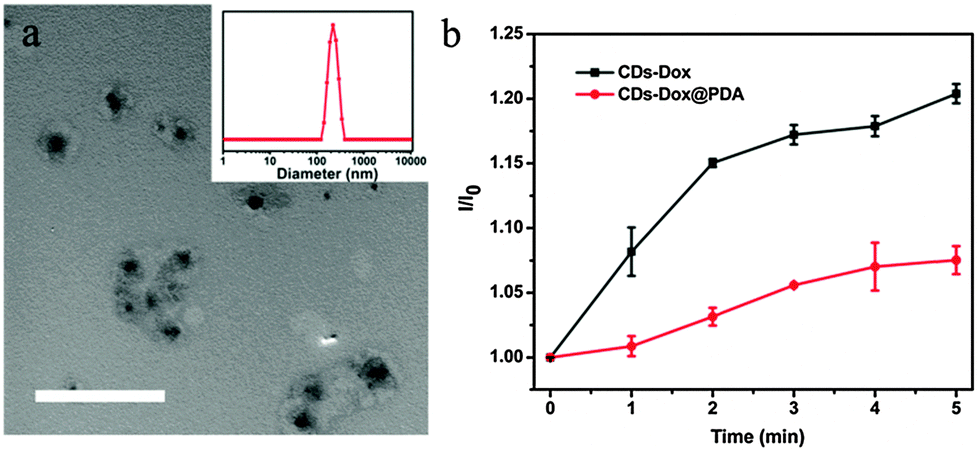 | ||
| Fig. 7 (a) TEM image of CDs–Dox@PDA. Scale bar represents 500 nm. Inset: size distribution of CDs–Dox@PDA. (b) Quantitative comparison of I/I0versus time for CDs–Dox and CDs–Dox@PDA in DMEM, where I0 is the initial fluorescence intensity of CDs–Dox or CDs–Dox@PDA immediately dispersed in DMEM, and I is that after different time periods. | ||
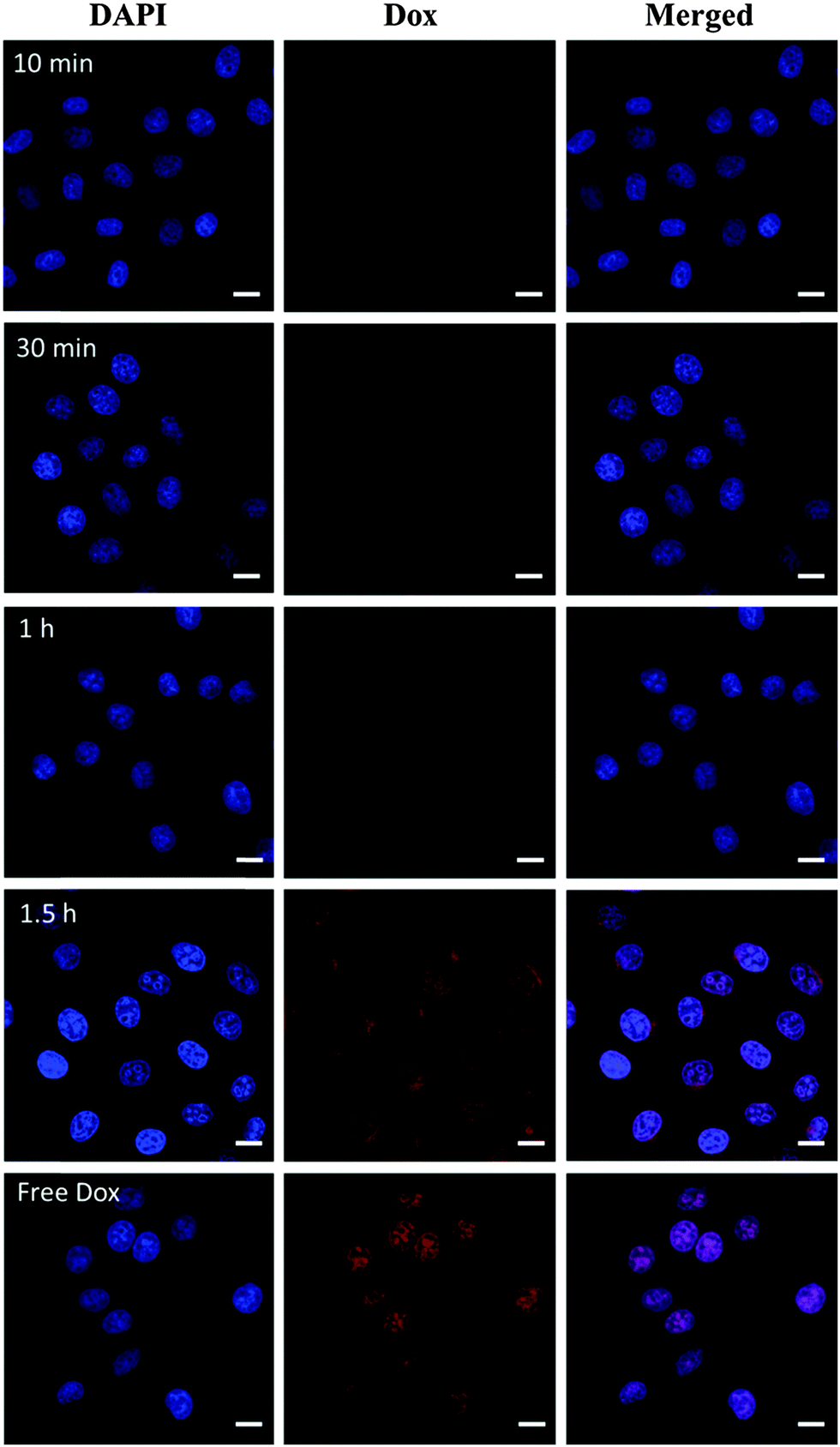 | ||
| Fig. 8 CLSM images of HeLa cells incubated with CDs–Dox@PDA for different time periods and free Dox for 30 min at 37 °C at a Dox concentration of 15 µg mL−1. Cells are viewed in the blue channel for DAPI and in the red channel for Dox. Scale bars represent 20 µm in all images. | ||
Subsequently, the anti-proliferative properties of CDs–Dox@PDA against HeLa cells were also assessed by MTT assays. Fig. 9 shows the cell viabilities of HeLa cells incubated with CDs–Dox@PDA or CDs–Dox at various concentrations for 48 h at 37 °C. CDs–Dox@PDA exhibited slightly lower cytotoxicity compared with CDs–Dox, especially at lower concentrations, which might be attributed to the slower release of Dox from CDs–Dox@PDA.
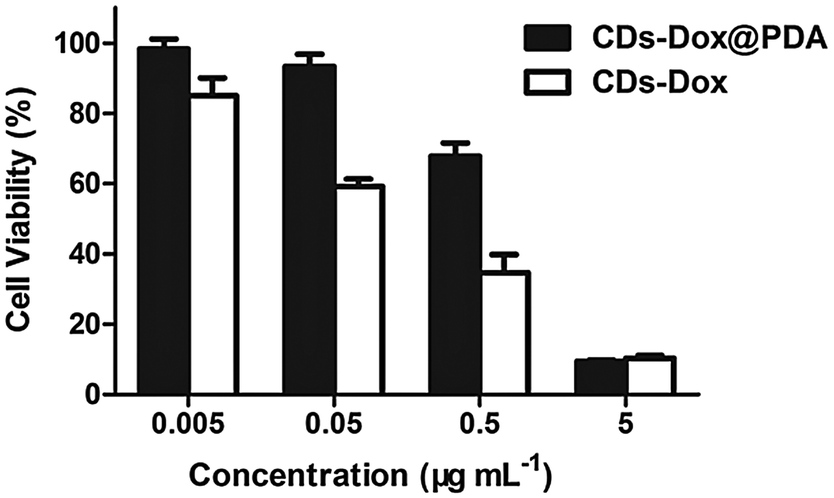 | ||
| Fig. 9 Viabilities of HeLa cells incubated with CDs–Dox@PDA and CDs–Dox for 48 h at 37 °C. All the results were repeated three times and presented as mean ± SD. | ||
Conclusions
In conclusion, we have successfully prepared the CDs–Dox supramolecular hybrids via π–π stacking and electrostatic interactions. CDs–Dox showed pH-dependent loading and release behaviors. The further modification of CDs–Dox with PDA coatings made the hybrid more stable in cell culture media, and this hybrid could be used for efficient drug delivery. Our proof of concept herein shows that the method, adsorption of drug molecules onto CDs and then coating with PDA, represents an attractive approach to be optimized and developed in the future.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Project No. 51522307 and 91227118).Notes and references
- K. Shinke, K. Ando, T. Koyama, T. Takai, S. Nakaji and T. Ogino, Colloids Surf., B, 2010, 77, 18–21 CrossRef CAS PubMed.
- V. Strauss, J. T. Margraf, K. Dirian, Z. Syrgiannis, M. Prato, C. Wessendorf, A. Hirsch, T. Clark and D. M. Guldi, Angew. Chem., Int. Ed., 2015, 54, 8292–8297 CrossRef CAS PubMed.
- X. T. Zheng, A. Ananthanarayanan, K. Q. Luo and P. Chen, Small, 2015, 11, 1620–1636 CrossRef CAS PubMed.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- P. Miao, K. Han, Y. Tang, B. Wang, T. Lin and W. Cheng, Nanoscale, 2015, 7, 1586–1595 RSC.
- X. Wang, X. Liu and H. Han, Colloids Surf., B, 2013, 103, 136–142 CrossRef CAS PubMed.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11–23 CrossRef CAS PubMed.
- T. C. Fitzgibbons, M. Guthrie, E.-s. Xu, V. H. Crespi, S. K. Davidowski, G. D. Cody, N. Alem and J. V. Badding, Nat. Mater., 2015, 14, 43–47 CrossRef CAS PubMed.
- H. Wang and H. Dai, Chem. Soc. Rev., 2013, 42, 3088–3113 RSC.
- M. Zheng, Z. Xie, D. Qu, D. Li, P. Du, X. Jing and Z. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 13242–13247 CAS.
- S. A. Kumar, S.-F. Wang, Y.-T. Chang, H.-C. Lu and C.-T. Yeh, Colloids Surf., B, 2011, 82, 526–531 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, S. M. Tabakman, K. Yang and H. Dai, Mater. Today, 2011, 14, 316–323 CrossRef CAS.
- Z.-Q. Xu, J.-Y. Lan, J.-C. Jin, T. Gao, L.-L. Pan, F.-L. Jiang and Y. Liu, Colloids Surf., B, 2015, 130, 207–214 CrossRef CAS PubMed.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- H. Shen, L. Zhang, M. Liu and Z. Zhang, Theranostics, 2012, 2, 283–294 CrossRef CAS PubMed.
- S. Some, A.-R. Gwon, E. Hwang, G.-h. Bahn, Y. Yoon, Y. Kim, S.-H. Kim, S. Bak, J. Yang and D.-G. Jo, Sci. Rep., 2014, 4, 6314 CrossRef CAS PubMed.
- N. G. Sahoo, H. Bao, Y. Pan, M. Pal, M. Kakran, H. K. F. Cheng, L. Li and L. P. Tan, Chem. Commun., 2011, 47, 5235–5237 RSC.
- L. Feng and Z. Liu, Nanomedicine, 2011, 6, 317–324 CrossRef CAS PubMed.
- H. Bao, Y. Pan and L. Li, Nano LIFE, 2012, 2, 1230001 CrossRef.
- Z. Liu, A. C. Fan, K. Rakhra, S. Sherlock, A. Goodwin, X. Chen, Q. Yang, D. W. Felsher and H. Dai, Angew. Chem., Int. Ed., 2009, 48, 7668–7672 CrossRef CAS PubMed.
- J. Tang, B. Kong, H. Wu, M. Xu, Y. Wang, Y. Wang, D. Zhao and G. Zheng, Adv. Mater., 2013, 25, 6569–6574 CrossRef CAS PubMed.
- C.-L. Huang, C.-C. Huang, F.-D. Mai, C.-L. Yen, S.-H. Tzing, H.-T. Hsieh, Y.-C. Ling and J.-Y. Chang, J. Mater. Chem. B, 2015, 3, 651–664 RSC.
- M. Zheng, S. Liu, J. Li, D. Qu, H. Zhao, X. Guan, X. Hu, Z. Xie, X. Jing and Z. Sun, Adv. Mater., 2014, 26, 3554–3560 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- M. Tan, X. Li, H. Wu, B. Wang and J. Wu, Colloids Surf., B, 2015, 136, 141–149 CrossRef CAS PubMed.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- L. Li, G. Wu, G. Yang, J. Peng, J. Zhao and J.-J. Zhu, Nanoscale, 2013, 5, 4015–4039 RSC.
- X. Gong, Q. Zhang, Y. Gao, S. Shuang, M. M. F. Choi and C. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 11288–11297 Search PubMed.
- L. Yang, Z. Wang, J. Wang, W. Jiang, X. Jiang, Z. Bai, Y. He, J. Jiang, D. Wang and L. Yang, Nanoscale, 2016, 8, 6801–6809 RSC.
- M. Zheng, S. Ruan, S. Liu, T. Sun, D. Qu, H. Zhao, Z. Xie, H. Gao, X. Jing and Z. Sun, ACS Nano, 2015, 9, 11455–11461 CrossRef CAS PubMed.
- J. Bai, Y. Liu and X. Jiang, Biomaterials, 2014, 35, 5805–5813 CrossRef CAS PubMed.
- J. Park, T. F. Brust, H. J. Lee, S. C. Lee, V. J. Watts and Y. Yeo, ACS Nano, 2014, 8, 3347–3356 CrossRef CAS PubMed.
- X. Liu, J. Cao, H. Li, J. Li, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 9384–9395 CrossRef CAS PubMed.
- C. Ehli, C. Oelsner, D. M. Guldi, A. Mateo-Alonso, M. Prato, C. Schmidt, C. Backes, F. Hauke and A. Hirsch, Nat. Chem., 2009, 1, 243–249 CrossRef CAS PubMed.
- M. Supur and S. Fukuzumi, J. Phys. Chem. C, 2012, 116, 23274–23282 CAS.
- F. Peng, Y. Su, X. Wei, Y. Lu, Y. Zhou, Y. Zhong, S. T. Lee and Y. He, Angew. Chem., Int. Ed., 2013, 52, 1457–1461 CrossRef CAS PubMed.
- X. Hu, X. Guan, J. Li, Q. Pei, M. Liu, Z. Xie and X. Jing, Chem. Commun., 2014, 50, 9188–9191 RSC.
- M. Meyer, A. Philipp, R. Oskuee, C. Schmidt and E. Wagner, J. Am. Chem. Soc., 2008, 130, 3272–3273 CrossRef CAS PubMed.
- J.-Z. Du, T.-M. Sun, W.-J. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 3621–3626 CrossRef CAS PubMed.
- Y. Lee, T. Ishii, H. Cabral, H. J. Kim, J.-H. Seo, N. Nishiyama, H. Oshima, K. Osada and K. Kataoka, Angew. Chem., Int. Ed., 2009, 48, 5309–5312 CrossRef CAS PubMed.
- Z. Li, M. Zheng, X. Guan, Z. Xie, Y. Huang and X. Jing, Nanoscale, 2014, 6, 5662–5665 RSC.
- T. Sun, X. Guan, M. Zheng, X. Jing and Z. Xie, ACS Med. Chem. Lett., 2015, 6, 430–433 CrossRef CAS PubMed.
- X. Zhong, K. Yang, Z. Dong, X. Yi, Y. Wang, C. Ge, Y. Zhao and Z. Liu, Adv. Funct. Mater., 2015, 25, 7327–7336 CrossRef CAS.
- J. Li, C.-Y. Hong, S.-X. Wu, H. Liang, L.-P. Wang, G. Huang, X. Chen, H.-H. Yang, D. Shangguan and W. Tan, J. Am. Chem. Soc., 2015, 137, 11210–11213 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2017 |