
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemote C–H insertion of vinyl cations leading to cyclopentenones†
Sarah E.
Cleary
,
Magenta J.
Hensinger
and
Matthias
Brewer
 *
*
Department of Chemistry, University of Vermont, Burlington, Vermont 05405, USA. E-mail: matthias.brewer@uvm.edu
First published on 3rd August 2017
Abstract
We report a Lewis acid catalyzed reaction sequence involving a 1,2-shift and subsequent C–H insertion that gives monocyclic and fused bicyclic cyclopentenone products. This reaction sequence, which is initiated by treating β-hydroxy-α-diazo ketones with a Lewis acid, proceeds through vinyl cation intermediates that insert at non-activated gamma C–H bonds. This reaction represents an alternative strategy to exploit the diazo functional group in an intramolecular C–H insertion, and can provide products not accessible by transition metal catalyzed C–H insertions. This remote C–H activation process provides good yields of bicyclic cyclopentenone products that contain 7- and 8-membered rings, and monocyclic prostaglandin analogs.
Introduction
The insertion of a hypovalent atom into an unactivated C–H bond is an attractive strategy for bond formation as it can lead to more efficient and more concise synthetic routes.1–5 Importantly, unlike most bond forming reactions that require the presence of two reactive functional groups, insertion reactions take advantage of a typically inert C–H bond and a single additional functional group. Minimizing the number of functional groups present in a system decreases the need for protecting groups and oxidation state changes during a synthetic sequence, which makes a synthetic route greener and more efficient from a time and cost perspective.6–8 This type of reactivity is exemplified by the intramolecular insertion of metal carbenes, derived from diazo precursors, into gamma C–H bonds as reported by Wenkert9,10 and Taber11 (Fig. 1a). Advances in efficiency and stereoselectivity have since made rhodium carbene insertions valuable synthetic transformations especially for the formation of γ-lactones and γ-lactams.1–3 The strategic advantages that C–H insertion reactions offer synthetic chemists make identifying additional methods to effect these types of ring forming reactions a worthwhile pursuit, particularly if these new methods can provide structures that existing methods cannot. Herein we report an alternative strategy to exploit the diazo functional group in a C–H insertion reaction that can provide products not accessible by the transition metal catalyzed procedures. Specifically, β-hydroxy-α-diazo ketones react with Lewis acids by a rearrangement and subsequent C–H insertion to give cyclopentenone products in good yield (Fig. 1b). | ||
| Fig. 1 C–H insertions leading to 5-membered carbocycles and examples of biologically active cyclopentanes. | ||
Cyclopentenones are versatile synthetic intermediates that are commonly used to make more-highly functionalized cyclopentanes, which are one of the most common ring systems found in biologically active organic structures (Fig. 1c).12 Cyclopentenones are often prepared by the Nazarov cyclization13–15 or the Pauson–Khand reaction,16–20 but reactivity and selectivity issues and the need for stoichiometric quantities of reagents can limit the generality of these reactions.
Results and discussion
Our discovery of the insertion sequence shown in Fig. 1b stemmed from our work on the Lewis acid mediated ring fragmentation of γ-silyloxy-β-hydroxy-α-diazo esters (Fig. 2).21–24 In that reaction, a vinyl cation,25,26 formed from the diazo alcohol by the process described by Pellicciari27 and Padwa,28 underwent a Grob-type fragmentation that was facilitated by the γ-silyloxy group. We hypothesized that a β-hydroxy-α-diazo ketone that lacks a fragmentation-promoting group at the γ position might instead react by a C–H insertion pathway. This hypothesis was based on Schegolev's initial reports29–31 that vinyl cations, generated by the acylation of alkynes, can participate in C–H insertion reactions.32,33 We began our studies by preparing diazo ketones 1 and 3 (Scheme 1) by adding lithiated 1-diazo-3,3-dimethyl-2-butanone or lithiated 1-diazo-3-methyl-2-butanone to cyclohexanone. Treating these materials with SnCl4 at −20 °C gave the hexahydro-1-azulenones 2 and 4 in 83% and 70% yield respectively, thus confirming that β-hydroxy-α-diazo ketones could serve as C–H insertion precursors. Importantly, cyclopentenone 2, which has a quaternary carbon adjacent to the carbonyl, could not be made by a rhodium catalyzed C–H insertion. | ||
| Fig. 2 Lewis acid mediated ring fragmentation of a γ-silyloxy-β-hydroxy-α-diazo ester. | ||
 | ||
| Scheme 1 Lewis acid mediated conversion of β-hydroxy-α-diazo ketones to bicyclic cyclopentenones. | ||
We propose that this transformation occurs by the sequence shown in Scheme 2.28 Lewis acid mediated elimination of the tertiary hydroxide would give vinyl diazonium ion 5, which would lose nitrogen to give vinyl cation 6. Carbonyl groups typically stabilize cations on adjacent carbons through resonance.34 However, in this system the p-orbitals of the carbonyl would be orthogonal to the cation's vacant p-orbital, thus preventing resonance stabilization. Instead, the cation would be destabilized by the inductively withdrawing carbonyl, which would facilitate a 1,2-shift across the alkene25 to give the ring expanded cyclic vinyl cation 7. Remote C–H insertion of the vinyl cation into a non-activated pendent methyl C–H bond would provide tertiary cation 8, and subsequent loss of a proton would give the cyclopentenone product (4). In line with this sequence, vinyl chloride 9 (Scheme 2), which could be formed by trapping vinyl cation 6 with chloride, was also isolated from the reaction mixture.
 | ||
| Scheme 2 Proposed sequence for bicyclic cyclopentenone and vinyl chloride formation. | ||
To optimize hexahydro-1-azulenone formation we evaluated other Lewis acid promoters. While BF3·Et2O, In(OTf)3, and FeCl3 were all less effective at promoting the reaction, tris(pentafluorophenyl)borane (BCF) gave 2 and 4 in 88% and 82% yield respectively (Table 1, entries 1 and 2). We next examined how temperature affected the conversion of 3 to 4 with both SnCl4 and BCF as Lewis acids. There was negligible change in yield when the reaction was run at temperatures between −35 °C and room temperature, but for both Lewis acids the yield decreased to 71% when the reaction was run at 40 °C (entry 3). It is noteworthy that reducing the quantity of BCF to 20 mol% provided the desired product (4) in only a slightly diminished yield of 67% (entry 4), while running the reaction with 30 mol% BCF in the presence of 1 equiv. of MgSO4 returned the product in 80% yield (entry 5). We also prepared two analogs of 1 that have substituents at the 4 position of the ring (10 and 12; entries 6 and 7). Both of these substrates reacted productively, albeit in slightly diminished yields as compared to the unsubstituted analog.

To assess the scope of the reaction with respect to the diazo ketone portion of the molecule, we prepared diazo ketones 14, 16, and 18 (shown in Schemes 3 and 4) and treated them with both SnCl4 and BCF. The ethyl ketone derivative (14) gave cyclopentenone 15 in 62% yield on treatment with SnCl4, whereas BCF returned the product in 70% yield. The trend that insertion products 2, 4, and 15 are isolated in decreasing yields (88%, 82% and 70% respectively) might be a reflection of the changing number of possible sites of insertion for these different substrates, or could indicate that the Thorpe–Ingold effect is playing a role in promoting the ring closure. The n-propyl ketone derivative (16) was prepared to determine if insertion could occur at a methylene rather than methyl position. In this case, the SnCl4 mediated reaction returned the desired insertion product in only 32% yield, whereas the BCF mediated reaction gave the desired product in a more useful 55% yield. Further investigation will be required to determine why insertion at a methylene position is less productive than insertion at a methyl position. Finally, we prepared isobutyl ketone derivative 18 (Scheme 4) to see if insertion would occur at a tertiary carbon to give an all carbon quaternary center. When reacted with BCF, this material returned diene 19 in 77% yield, whereas treatment this material with SnCl4 returned diene 19 and chloride trapped products as an inseparable mixture. To form 19, it appears that hydride transfer occurred from the tertiary center to the vinyl cation, but ring closure did not ensue, which may be a ramification of the stability of the tertiary carbocation formed upon hydride transfer. A similar hydride transfer – elimination sequence was not apparent for n-propyl substrate 16, which indicates that the degree of concertedness of the insertion step likely depends on the nature of the point of insertion.
 | ||
| Scheme 3 Variation at the site of insertion. | ||
 | ||
| Scheme 4 Hydride transfer from a tertiary carbon. | ||
With these results in hand, we prepared a series of diazo ketones by adding lithiated 1-diazo-3-methyl-2-butanone to various cyclic ketones and have reacted each of these diazo ketones with SnCl4 and BCF (Table 2). When treated with SnCl4, the cyclobutanone and cyclopentanone derivatives (20 and 23) gave their respective insertion products in only trace quantities and 21% yield respectively (entries 1 and 2). In these smaller ring systems, rearrangement of the initially formed linear vinyl cation to the cyclic vinyl cation intermediate would be less favorable because of increased ring strain in the rearranged product, and the initially formed linear vinyl cations were instead captured by chloride to give vinyl chlorides 22 and 25 as the major products of the reactions. Switching the Lewis acid to BCF did not result in the productive formation of cyclopentenone 21 (entry 4), but the yield of cyclopentenone 24 increased to 60% (entry 5). The increased yield in this case can be attributed to the fact that BCF does not contain a moiety that could readily capture the vinyl cation intermediate, thus allowing the rearrangement and insertion to occur. The cycloheptyl derivative (26) reacted with SnCl4 to give the desired 5,8-fused bicycle 27 in 42% yield along with a significant quantity (24% yield) of vinyl chloride 28 (entry 3). It is not clear to us why capture by chloride is an important pathway in this larger ring system. In the presence of BCF, diazo ketone 26 returned cyclopentenone 27 in 66% yield (entry 6).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | n | Lewis acid | Product | Yield | Product | Yield |
| 1 | 20 | 1 | SnCl4 | 21 | <5% | 22 | 68% |
| 2 | 23 | 2 | SnCl4 | 24 | 21% | 25 | 30% |
| 3 | 26 | 4 | SnCl4 | 27 | 42% | 28 | 24% |
| 4 | 20 | 1 | (C6F5)3B | 21 | 0% | ||
| 5 | 23 | 2 | (C6F5)3B | 24 | 60% | ||
| 6 | 26 | 4 | (C6F5)3B | 27 | 66% | ||

In view of the important role that monocyclic cyclopentenone derivatives play as synthetic intermediates, and the fact that prostaglandins and their analogs (e.g.Fig. 1c35) are important biologically active compounds derived from these scaffolds, we were interested to determine if this reaction could provide monocyclic cyclopentenone products. To this end, we prepared diazo ketones 29 and 31 (Scheme 5) from 4-heptanone. Treating these substrates with BCF returned the desired 2,3-disubstituted cyclopentenones 30 and 32 respectively, indicating the potential of this methodology to provide new access to prostaglandin analogs.
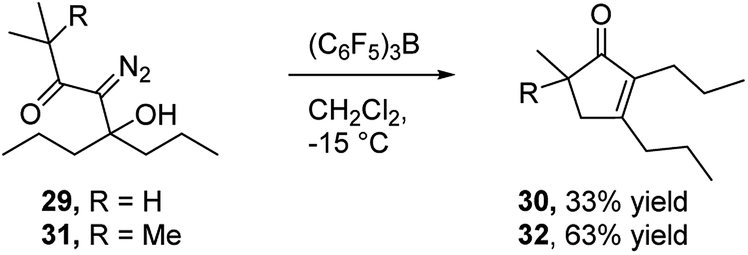 | ||
| Scheme 5 Preparation of monocyclic cyclopentenone derivative. | ||
Conclusions
In summary, we have shown that β-hydroxy-α-diazo ketones, which are simple to prepare, can serve as convenient vinyl cation precursors for C–H insertion reactions that lead to monocyclic or bicyclic cyclopentenone products. This reaction sequence appears to be fairly general, and gives good to excellent yields of ring fused 5,6-, 5,7-, and 5,8-bicyclic and 2,3-disubstituted monocyclic products. While insertion occurred productively at methyl and methylene positions, a methyne position acted as a hydride donor without subsequent C–C bond formation. In this later case, a concerted insertion process is clearly precluded, but further studies are needed to probe the mechanism of the insertion step at methyl and methylene positions. Overall, this reaction sequence represents a new approach to making common and important cyclopentenone scaffolds, including systems that cannot be made by transition metal catalyzed C–H insertion reactions. In addition to studying this reaction further, we are currently investigating alternative uses for vinyl cation intermediates formed from β-hydroxy-α-diazo carbonyl compounds.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the NIH (National Institute of General Medical Sciences Award Number R01GM092870). The content is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS or NIH. Mass spectrometry data was acquired by Bruce O'Rourke on instruments purchased through instrumentation grants provided by the National Institutes of Health (NIH) (S10 OD018126).References
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed.
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 177, 9247–9301 CrossRef PubMed.
- T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931–942 RSC.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed.
- E. Wenkert, L. L. Davis, B. L. Mylari, M. F. Solomon, R. R. Da Silva, S. Shulman, R. J. Warnet, P. Ceccherelli, M. Curini and R. Pellicciari, J. Org. Chem., 1982, 47, 3242–3247 CrossRef CAS.
- P. Ceccherelli, M. Curini, M. C. Marcotullio, O. Rosati and E. Wenkert, J. Org. Chem., 1990, 55, 311–315 CrossRef CAS.
- D. F. Taber and E. H. Petty, J. Org. Chem., 1982, 47, 4808–4809 CrossRef CAS.
- S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744–5893 CrossRef CAS PubMed.
- A. J. Frontier and C. Collison, Tetrahedron, 2005, 61, 7577–7606 CrossRef CAS.
- M. A. Tius, Acc. Chem. Res., 2003, 36, 284–290 CrossRef CAS PubMed.
- S. E. Denmark and T. K. Jones, J. Am. Chem. Soc., 1982, 104, 2642–2645 CrossRef CAS.
- I. U. Khand, G. R. Knox, P. L. Pauson, W. E. Watts and M. I. Foreman, J. Chem. Soc., Perkin Trans. 1, 1973, 977–981 RSC.
- K. M. Brummond and J. L. Kent, Tetrahedron, 2000, 56, 3263–3283 CrossRef CAS.
- J. Blanco-Urgoiti, L. Anorbe, L. Perez-Serrano, G. Dominguez and J. Perez-Castells, Chem. Soc. Rev., 2004, 33, 32–42 RSC.
- N. E. Schore, in Organic Reactions, John Wiley & Sons, Inc., 2004, DOI:10.1002/0471264180.or040.01.
- V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2009, 109, 6809–6857 CrossRef CAS PubMed.
- C. Draghici and M. Brewer, J. Am. Chem. Soc., 2008, 130, 3766–3767 CrossRef CAS PubMed.
- A. Bayir, C. Draghici and M. Brewer, J. Org. Chem., 2010, 75, 296–302 CrossRef CAS PubMed.
- N. P. Tsvetkov, A. Bayir, S. Schneider and M. Brewer, Org. Lett., 2012, 14, 264–267 CrossRef CAS PubMed.
- A. Bayir and M. Brewer, J. Org. Chem., 2014, 79, 6037–6046 CrossRef CAS PubMed.
- P. J. Stang, Z. Rappoport, M. Hanack and L. B. Subramanian, Vinyl Cations, Academic Press, New York, 1979 Search PubMed.
- Z. Rappoport and P. J. Stang, Dicoordinated carbocations, Wiley, Chichester, New York, 1997 Search PubMed.
- R. Pellicciari, B. Natalini, B. M. Sadeghpour, G. C. Rosato and A. Ursini, Chem. Commun., 1993, 1798–1800 RSC.
- R. Pellicciari, B. Natalini, B. M. Sadeghpour, M. Marinozzi, J. P. Snyder, B. L. Williamson, J. T. Kuethe and A. Padwa, J. Am. Chem. Soc., 1996, 118, 1–12 CrossRef CAS.
- A. A. Schegolev, W. A. Smit, G. V. Roitburd and V. F. Kucherov, Tetrahedron Lett., 1974, 15, 3373–3376 CrossRef.
- A. A. Schegolev, W. A. Smit, V. F. Kucherov and R. Caple, J. Am. Chem. Soc., 1975, 97, 6604–6606 CrossRef.
- M. I. Kanishchev, A. A. Shegolev, W. A. Smit, R. Caple and M. J. Kelner, J. Am. Chem. Soc., 1979, 101, 5660–5671 CrossRef CAS.
- Metzger, et al., have shown that vinyl cations generated by the addition of carbocations to alkynes undergo intramolecular insertion reactions to give cyclopentane products in the presence of a reductant. See ref. 33.
- U. Biermann, R. Koch and J. O. Metzger, Angew. Chem., Int. Ed. Engl., 2006, 45, 3076–3079 CrossRef CAS PubMed.
- D. A. Dixon, R. A. Eades, R. Frey, P. G. Gassman, M. L. Hendewerk, M. N. Paddon-Row and K. N. Houk, J. Am. Chem. Soc., 1984, 106, 3885–3891 CrossRef CAS.
- D. Yariv, WO2006113571, 2006.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02768k |
| This journal is © The Royal Society of Chemistry 2017 |