
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed asymmetric allylic alkylations via C–H activation of N-allyl imines with glycinates†
Barry M.
Trost
 * and
Xiaoxun
Li
* and
Xiaoxun
Li
Department of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: bmtrost@stanford.edu
First published on 15th August 2017
Abstract
Herein is reported the first example of palladium-catalyzed asymmetric allylic alkylation (AAA) reactions involving 2-aza-π-allyl palladium intermediates. The 2-aza-π-allyl complex was generated via a novel mode of activation of N-allyl imines. Pd-catalyzed C(sp3)–H activation of N-allyl imines and subsequent nucleophilic attack by glycinates delivered vicinal diamino derivatives as the sole regioisomers with high levels of diastereo- and enantio-control in the presence of the chiral, bidentate (S,S)-Cy-DIOP ligand. This procedure is highly atom economical and could also be performed by a simple one-pot operation starting from aldehydes, allyl amines and glycinates under mild conditions. The products of this transformation could be converted into various useful derivatives, where the allyl substitution serves as a unique tool for differentiating the two amino moieties in the products.
Introduction
Metal-catalyzed asymmetric allylic alkylation (AAA) reactions fall among one of the most active areas in the realm of organic synthesis due to their broad utility in the construction of a diverse array of enantioenriched products from achiral starting materials.1 A variety of transition metals, such as palladium,2 copper,3 iridium,4 and molybdenum,5 have been applied in these transformations. At present, a number of modes exist for the formation of the π-allyl intermediates. The most common has been the use of an allylic leaving group,2–5 which undergoes ionization in the presence of a Pd(0) complex. A more atom economical strategy6 uses protonation of a 1,3- or 1,2- (i.e. an allene) diene to form the π-allyl intermediate.7 An interesting attractive method uses C–H activation at the allylic position of a simple olefin. Indeed, our initial work in 1973 on π-allyl palladium chemistry established the feasibility of this process but required a stoichiometric amount of palladium.8 Recently, catalytic processes involving an allylic hydrogen atom as a “leaving group” under a direct C(sp3)–H activation method has emerged as an active area.9,10 Irrespective of the catalyst, however, one common feature of these AAA reactions is the involvement of all-carbon π-allyl complexes (Fig. 1, path a). To the best of our knowledge, there is only one asymmetric study of the aza analogue of the all-carbon π-allyl complex, and that approach involved the generation of an aza-π-allyl complex by ionization of an acetoxy leaving group (path b).11 Recently, our group disclosed a general and efficient way to access the 2-aza-π-allyl palladium complex, which was employed in the preparation of either a 1-aza-1,3-diene or a 2-aza-1,3-diene as the sole regioisomer based on the choice of nucleophile (path b),12 although in a racemic manner. The importance of nitrogen-containing compounds,13 especially chiral vicinal diamino derivatives,14 raises the question of an asymmetric transformation using a 2-aza-π-allyl complex. Enantioselective direct Mannich reactions15 of glycinate Schiff bases with activated imines represent a powerful strategy for the preparation of vicinal diamino derivatives, but the imines always require a strong electron-withdrawing group. One way to increase the efficiency would be to break this paradigm by developing an alternative method to perform analogous Mannich reactions with unactivated imines while maintaining high levels of regio-, diastereo-, and especially enantio-selective control. Herein, we present the first example of a Pd-catalyzed asymmetric alkylation of N-allyl imines with glycinates by combining the Pd-catalyzed oxidative allylic C–H activation and chiral phosphine ligands (path c). Using glycinates as nucleophiles provides unprecedented access to 2,3-diamino carboxylic acid derivatives asymmetrically. | ||
| Fig. 1 Asymmetric allylic alkylations (AAA) involving a π-allyl complex.16 | ||
Results and discussion
We started our investigation by using N-allylbenzylidene imine (1a) with tert-butyl glycinate (2a) as the nucleophile. Initial studies with phosphine ligands were disappointing. Our previous work on Pd-catalyzed asymmetric allylic C–H alkylation9b prompted us to initiate our study with chiral phosphoramidite ligands. As shown in the ESI,† no desired product was observed with such ligands. Further studies with other common bidentate ligands such as the Trost ligand, BINAP, DuPhos and Walphos ligands did not yield any desired product. On the other hand, surprisingly, the (R,R)-DIOP ligand (L1) was able to promote this transformation in 60% yield (Scheme 1) with good diastereoselectivity (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and modest enantioselectivity (78:22 er). Notably, the N-crotylbenzylideneimine substrate remained inert under these reaction conditions. Although these results were less promising than we had hoped for, they demonstrated the theoretical potential for an asymmetric reaction involving 2-aza-π-allyl palladium intermediates with phosphine ligands.
:1 dr) and modest enantioselectivity (78:22 er). Notably, the N-crotylbenzylideneimine substrate remained inert under these reaction conditions. Although these results were less promising than we had hoped for, they demonstrated the theoretical potential for an asymmetric reaction involving 2-aza-π-allyl palladium intermediates with phosphine ligands.
 | ||
| Scheme 1 Preliminary results. | ||
Encouraged by the promising preliminary results with the (R,R)-DIOP ligand in such a catalyst system, we began to explore DIOP analogues for further optimization. One of the main advantages of DIOP is that it offers numerous options for derivatization. According to procedures reported in the literature for the simple DIOP ligand,17 several ligands based on the DIOP structure were synthesized and screened (Table 1). The major difference between these synthesized ligands and DIOP is the substitution of the aromatic moieties and structural modification via alteration of the acetal backbone, either by opening the ring (L5) or by switching it to a different cyclic acetal. As expected, (S,S)-DIOP afforded a similar level of enantioselectivity compared to (R,R)-DIOP but with opposite absolute stereochemistry. More sterically-hindered ligands like L3 and L4 resulted in either low conversion (<5%) or decreased enantioselectivity. Low conversion and poor enantioselectivity were obtained with L5, indicating that changing the bite angles of DIOP derivatives by altering the acetal backbone has a significant impact on enantioselectivity. A variety of substitutions for the acetal backbone such as phenyl, di-isopropyl, cyclopentyl, cyclohexyl and cycloheptyl groups were tested, and the highest enantioselectivity (78:22 er) was obtained with L10. Surprisingly, L8, bearing a six-membered ketal tether,16b afforded a nearly racemic product. Additionally, we were also interested in improving enantioselectivity by varying the glycinate nucleophiles. As shown in the ESI,† the two halves of the glycinate structure are of paramount importance. The highest enantioselectivity (85:15 er) was obtained using methyl glycinate (2b) as the nucleophile. Further investigation of the non-C2 symmetrical ligands (L12, L14, L15 and L16) gave product 3b in a similar yield and with the same enantioselectivity (85:15 er) and similar diastereoselectivity compared with that obtained when using L2 as the ligand. On the other hand, a decrease in enantioselectivity (75:25 er) is observed with the analogous ortho-ester phenyl ligand (L13, see Table 1, entry 13). We hypothesize that this ligand contains secondary interactions between the ester and the metal center, preventing the desired mode of reactivity.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Glycinate | Ligand | Yieldb | drc | erd |
a Reaction conditions: 1a (0.1 mmol), 2a or 2b (0.1 mmol), Pd(OAc)2 (5 mol%), ligand (6 mol%), Hunig’s base (0.1 mmol), 2,6-DMBQ (0.1 mmol), dioxane, rt, 15–20 h.
b Isolated yield.
c Determined by 1H-NMR.
d Determined by chiral HPLC.
|
|||||
| 1 | 2a | L1 | 60% | 10:1 |
78:22 |
| 2 | 2a | L2 | 62% | 10:1 |
23:77 |
| 3 | 2a | L3 | Trace | — | — |
| 4 | 2a | L4 | 65% | 8:1 |
70:30 |
| 5 | 2a | L5 | 22% | 3:1 |
57:43 |
| 6 | 2a | L6 | 60% | 10:1 |
72:28 |
| 7 | 2a | L7 | 64% | 10:1 |
71:29 |
| 8 | 2a | L8 | 25% | 10:1 |
53:47 |
| 9 | 2a | L9 | 64% | 10:1 |
74:26 |
| 10 | 2a | L10 | 67% | 11:1 |
78:22 |
| 11 | 2a | L11 | 66% | 9:1 |
76:24 |
| 12 | 2b | L12 | 65% | 5:1 |
85:15 |
| 13 | 2b | L13 | 58% | 6:1 |
75:25 |
| 14 | 2b | L14 | 62% | 5:1 |
85:15 |
| 15 | 2b | L15 | 56% | 5:1 |
85:15 |
| 16 | 2b | L16 | 62% | 6:1 |
85:15 |

Efforts to improve the enantioselectivity then shifted to examine the solvent effect, the reaction concentration, temperature and bases (see the ESI† for details). With a robust set of reaction conditions in hand, we re-examined all the previous ligands to further optimize this transformation. When (S,S)-Cy DIOP was employed,16c3b was isolated in 65% yield with 92:8 er, the highest level of enantioinduction among all the ligands examined (Scheme 2).
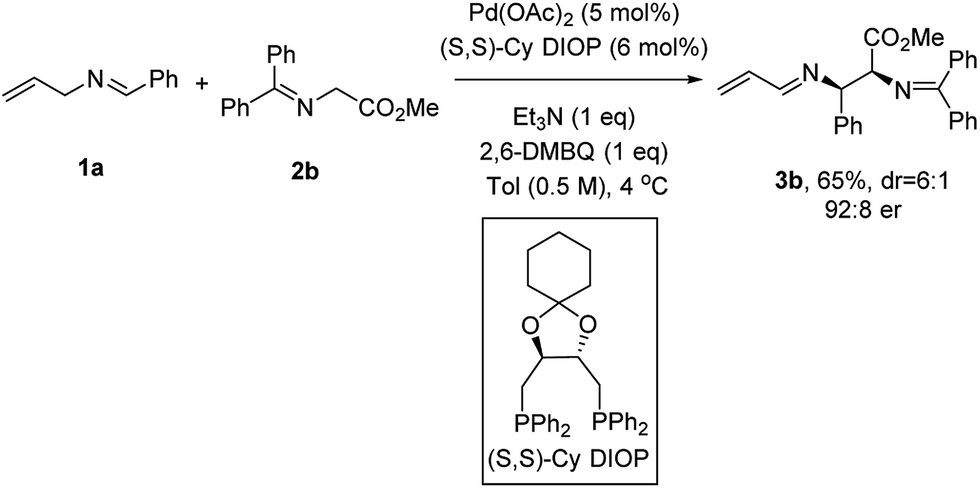 | ||
| Scheme 2 Optimized reaction conditions. | ||
Under the optimized conditions, we next explored the generality of the Pd-catalyzed asymmetric allylic alkylations involving 2-aza-π-allyl complexes. A wide selection of N-allylarylidene imines was subjected to the optimized conditions to produce adducts with methyl glycinate donors 2b (Fig. 2). Both electron-rich and electron-deficient substrates underwent efficient coupling using this protocol, though higher reaction rates were observed for substrates bearing electron-withdrawing groups. The reaction temperature for those substrates could be lowered to −4 °C in order to give higher enantioselectivity (3c, 3f and 3g–3l). The enantioselectivity is sensitive to the temperature (4 °C, −4 °C and −20 °C), as shown for nitro-substituted N-allylimine 3f: the corresponding 1-aza-1,3-diene was obtained in 66%, 64% and 70% yield and with 85:15, 88:12 and 90:10 er, respectively.18 Notably, the diastereoselectivity decreased at −20 °C after a prolonged reaction time (70 h). An electron-donating substituent (e.g. OMe or Me) on the aryl ring proved effective as well, yielding 3d and 3e with 89.5:10.5 and 86:14 er, respectively. An aryl bromide on the imine (3g) was well tolerated under the oxidative allylic C–H activation conditions. The position of the substituent on the aryl system had no influence on either the yield or enantioselectivity, though the diastereoselectivity was diminished when the substituents were moved from the para to the meta to the ortho position (3h–3j). When a cyano-substituted N-allyl imine was employed, the alkylation product (3k) was obtained in 70% yield and 11.5:88.5 er. An electron-deficient imine bearing a trifluoromethyl group was successfully employed to form the desired product (3l) in 72% yield and 91.5:8.5 er. Notably, significantly lower enantioselectivity (38% ee) was observed in the direct Mannich reaction15h for substrate p-trifluoromethyl benzaldimine with tert-butyl glycinate. The corresponding 2-naphthyl imine gave the desired product (3m) in 75% yield and 90:10 er, but no reaction was observed for the 1-naphthyl variant.
 | ||
| Fig. 2 Substrate scope for N-allyl imines. aReaction conditions: 1 (0.5 mmol), 2b (0.5 mmol), Pd(OAc)2 (5 mol%), (S,S)-Cy DIOP (6 mol%), Et3N (0.5 mmol), 2,6-DMBQ (0.5 mmol), toluene (0.5 M), 4 °C, −4 °C or −20 °C, 30–70 h. The diastereoselectivity was determined by 1H-NMR spectroscopy of the crude reaction mixture. The yields are those of the isolated products. b4 °C. c−4 °C. d−20 °C. eIt is assumed that the major diastereomer is the same, although the retention time of the two peaks is inverted. | ||
This methodology extends beyond benzenoid imines. Thus, heteroaromatic imine systems were screened (Fig. 3). 2-Substituted benzofuran and benzothiophene gave quite similar results. Further, a 3-substituted indole gave the tryptophan analogue 3p with 89:11 er. The reaction with N-allyl α-methylcinnamyl imine was significantly slower, but the corresponding adduct was isolated in 74% yield with 86.5:13.5 er.
 | ||
| Fig. 3 Substrate scope for heteroaromatic and non-aromatic imines. aReaction conditions: 1 (0.5 mmol), 2b (0.5 mmol), Pd(OAc)2 (5 mol%), (S,S)-Cy DIOP (6 mol%), Et3N (0.5 mmol), 2,6-DMBQ (0.5 mmol), toluene (0.5 M), 4 °C, 40–70 h. The diastereoselectivity was determined by 1H-NMR spectroscopy of the crude reaction mixture. The yields are those of the isolated products. | ||
With the success of the Pd-catalyzed asymmetric allylic alkylation of N-allyl imines, we considered whether a more practical one-pot process from commercially available materials would be feasible by in situ generation of the imine.19 Thus, handling water and air sensitive imines would be avoided. To a mixture of Pd(OAc)2, ligand, quinone and powdered 4 Å molecular sieves was added a toluene solution of allyl amine, aldehyde, and methyl glycinate 2b. The only modification of the allylic alkylation procedure was the addition of molecular sieves to facilitate in situ imine formation. Comparable yields, diastereoselectivity and enantioselectivity were obtained, as the substrate scope shown in Fig. 4 illustrates. p-Bromobenzaldehyde (4b) was found to perform well, giving the desired product in 70% yield and 90:10 er. Electron-deficient and electron-rich aldehydes, 4c and 4d, undergo efficient coupling using this protocol to provide adducts in 68% and 52% yield and 92:8 and 88.5:11.5 er, respectively. This three-component method could be extended to heteroaromatic systems. When benzo[b]thiophene-2-carboxaldehyde 4e was employed, the alkylation product was obtained in 60% yield and 90:10 er.
 | ||
| Fig. 4 Substrate scope of the Pd-catalyzed three-component AAA reaction. aReaction conditions: 4 (0.3 mmol), 5 (0.3 mmol), 2b (0.3 mmol), Pd(OAc)2 (5 mol%), (S,S)-Cy DIOP (6 mol%), Et3N (0.3 mmol), 2,6-DMBQ (0.3 mmol), MS powder (4 Å, 120 mg), toluene (0.5 M), 4 °C or −4 °C, 30–50 h. The diastereoselectivity was determined by 1H-NMR spectroscopy of the crude reaction mixture. The yields are those of the isolated products. b4 °C. c−4 °C. | ||
A plausible mechanism of this reaction is shown in Scheme 3. We propose that the catalytic cycle is initiated with the coordination of the PdII complex to the allyl double bond. Subsequent PdII-catalyzed allylic C–H activation generates the all-carbon π-allyl complex A, which would be in equilibrium with the 2-aza-π-allyl complex B. This hypothesis is supported by our previous study.12 Then, glycinate nucleophiles attack via a transition state involving maximum delocalization of charge to give the (S,R) or (R,S) product 3 with the reduction of the PdII complex to Pd0. The facial selectivity that preferentially generates the S,R-isomer is rationalized by the minimization of steric interactions within the palladium ligand complex and the ester of the glycinate. Finally, DMBQ-mediated oxidation regenerates PdII to complete the catalytic cycle.
 | ||
| Scheme 3 Plausible catalytic cycle. | ||
The regioselectivity of the nucleophilic addition derives from preferential attack on intermediate B compared to A. In our previous study, this regioselectivity was observed with more reactive nucleophiles. Such an observation is consistent with the reaction at the most electrophilic carbon.
Products 3 could be converted to biologically interesting chiral α,β-vicinal diamino acid derivatives (7a–7c) in high yields upon hydrolysis under mildly acidic conditions (Scheme 4). This process also provides a mechanism to establish the relative and absolute stereochemistry of this new process. A comparison of the spectral data of product 7a with reports in the literature20a,20b confirms it is either the S,R- or R,S-isomer (i.e. not the R,R- or S,S-isomer). The absolute configuration was established by its optical rotation of [α]25D = +11.0° (c = 1.0, CHCl3, 92:8 er), which agrees with that reported for the S,R-isomer, [α]25D = +16.0° (c = 1.0, CHCl3, >99% ee).20c
 | ||
| Scheme 4 Hydrolysis of products 3. | ||
To further demonstrate the utility of this reaction, we converted the products to other useful amino derivatives (Scheme 5). The adduct 3b was treated with sodium borohydride at −20 °C for 15 min to give the imine product 8a in 62% yield. The bis-benzylidene imine moiety was inert under these conditions and no epimerization was observed. Thus, the corresponding products 8b and 8c were obtained in 63% and 58% yield, respectively. Now, the two imine moieties could be easily differentiated after the reduction of 1-aza-1,3-diene. For example, product 9 was prepared in 95% yield after the hydrolysis of 8a (see Scheme 5). In addition, the allyl group of the products 8 could serve as a useful handle for further functionalization. For example, N-acylation with acryloyl chloride followed by metathesis led to the dihydropyrrolenone 10. The product could also be converted to amino alcohol 11 after a dihydroxylation, oxidation, reduction and hydrolysis sequence. Simple propargylation to enyne 12 proceeded in high yield. Subjection of the enyne to hydrolysis followed by Au(I)-catalyzed cyclization provided the tetrahydropyrazine 13. On the other hand, subjection of enyne 12 to the conditions of a Pauson–Khand reaction smoothly produced the [3.3.0]-azabicyclooctanone 14 in good yield.
 | ||
| Scheme 5 Derivatization of AAA products. Conditions: (a) 1 M HCl, acetone, rt; (b) acryloyl chloride, Et3N, DCM, rt; (c) Grubbs II (5 mol%), DCM, rt; (d) Ac2O, Et3N, DMAP (cat), DCM, rt; (e) K2OsO4·2H2O (5 mol%), NMO (2 eq.), acetone, rt; (f) NaIO4 (2 eq.), Et2O/H2O (2:1), rt; (g) NaBH4 (0.5 eq.), 0 °C; (h) propargyl bromide, K2CO3, CH3CN, 80 °C; (i) IPrAuNTf2 (5 mol%), DCM, rt; (j) Co2(CO)8 (1.05 eq.), DCM, rt; then Me3NO (6 eq.), MS powder, rt. | ||
Conclusions
In summary, we have developed the first example of Pd-catalyzed asymmetric allylic alkylation (AAA) involving a 2-aza-π-allyl intermediate with (S,S)-Cy DIOP. The effectiveness of this chiral bidentate bis-phosphine is particularly surprising given the expectation that such phosphines would be incompatible with the oxidative conditions of such a C–H activation mechanism. Further, the total failure of the phosphoramidite ligand, which was successful in our all-carbon π-allyl oxidative allylation, highlights the uniqueness of the ligand choice. When glycinate was employed as the nucleophile, a wide variety of synthetically important α,β-diamino ester derivatives were isolated in high yields, with good diastereo- and enantioselectivity and complete regioselectivity. The substrate scope was extended from aromatic imines to heteroaromatic and alkenylidene imines. This procedure is highly atom economical and it also could be performed in a simple one-pot operation under mild conditions. The utility of this method was further demonstrated with diverse functionalization of the adducts. The juxtaposition of functionality available from this process provides access to a diverse range of structures possessing an α,β-vicinal diamino acid as a common motif. Furthermore, the reaction is quite amenable to scale-up. Thus, running the reaction with N-allyl-benzylidene imine on a 5 mmol scale gave the product 3b in a slightly improved yield of 67% and with identical diastereoselectivity and enantioselectivity as on a 0.5 mmol scale.Experimental section
1. General procedure for the Pd-catalyzed asymmetric allylic alkylation involving π-allyl intermediate
An oven-dried Pyrex microwave vial was charged with Pd(OAc)2 (5.6 mg, 0.025 mmol), (S,S)-Cy DIOP (16.2 mg, 0.03 mmol) and 2,6-dimethylbenzoquinone (70 mg, 0.5 mmol), and was sealed with a rubber septum. The vial was evacuated and filled with nitrogen three times in an interval of 10 min. In a separate sealed nitrogen flushed vial, glycinate (0.5 mmol) and N-allyl imine (0.5 mmol) were taken in freshly distilled toluene (1 ml). The solution was cannulated to the microwave vial with the palladium catalyst. Et3N (50 mg, 0.5 mmol) was added to the resulting turbid solution and it was allowed to stir at 4 °C or −4 °C for 35–60 h. Upon completion (monitored by crude NMR), the solvent was removed in vacuo and the residue was purified by flash chromatography over silica gel (pre-neutralized with 3% Et3N in hexane), eluting with EtOAc/hexane/Et3N, to give the product.2. General procedure for the three-component reaction
An oven-dried Pyrex microwave vial was charged with Pd(OAc)2 (3.4 mg, 0.015 mmol), (S,S)-Cy DIOP (10.1 mg, 0.018 mmol), 2,6-dimethylbenzoquinone (42 mg, 0.3 mmol) and molecular sieves (4 Å, 120 mg), and was sealed with a rubber septum. The vial was evacuated and filled with nitrogen three times in an interval of 10 min. In a separate sealed nitrogen flushed vial, aldehyde (0.3 mmol), allyl amine (0.3 mmol) and glycinate 2e (0.3 mmol) were dissolved in freshly distilled toluene (0.6 mL). The solution was cannulated to the microwave vial with the palladium catalyst. Et3N (30 mg, 0.3 mmol) was added to the resulting turbid solution and it was allowed to stir at 4 °C or −4 °C for 35–60 h. Upon completion (monitored by crude NMR), the solvent was removed in vacuo and the residue was purified by flash chromatography over silica gel (pre-neutralized with 3% Et3N in hexane), eluting with EtOAc/hexane/Et3N, to give the product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NIH (GM-033049) and the NSF (NSF CHE-1360634) for their generous support of our programs.References
- (a) Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, in Topics in Organometallic Chemistry, ed. U. Kazmaier, Springer, 2012, vol. 38 Search PubMed; (b) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929 RSC.
- (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (c) B. M. Trost, J. Org. Chem., 2004, 69, 5813 CrossRef CAS PubMed; (d) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS PubMed; (e) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427 RSC.
- (a) H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2005, 44, 4435 CrossRef CAS PubMed; (b) A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796 CrossRef CAS PubMed; (c) C. A. Falciola and A. Alexakis, Eur. J. Org. Chem., 2008, 3765 CrossRef CAS.
- (a) G. Helmchen, A. Dahnz, P. Dübon, M. Schelwies and R. Wiehofen, Chem. Commun., 2007, 675 RSC; (b) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS PubMed; (c) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627 CrossRef CAS PubMed.
- B. M. Trost, Org. Process Res. Dev., 2012, 16, 185 CrossRef CAS PubMed.
- B. M. Trost, Science, 1991, 254, 1471 CAS.
- P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524 CrossRef CAS PubMed.
- B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292 CrossRef CAS.
- (a) D. J. Covell and M. C. White, Angew. Chem., Int. Ed., 2008, 47, 6448 CrossRef CAS PubMed; (b) B. M. Trost, D. A. Thaisrivongs and E. J. Donckele, Angew. Chem., Int. Ed., 2013, 52, 1523 CrossRef CAS PubMed; (c) P.-S. Wang, H.-C. Lin, Y.-J. Zhai, Z.-Y. Han and L.-Z. Gong, Angew. Chem., Int. Ed., 2014, 53, 12218 CrossRef CAS PubMed; (d) B. M. Trost, E. J. Donckele, D. A. Thaisrivongs, M. Osipov and J. T. Masters, J. Am. Chem. Soc., 2015, 137, 2776 CrossRef CAS PubMed; (e) P.-S. Wang, P. Liu, Y.-J. Zhai, H.-C. Lin, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2015, 137, 12732 CrossRef CAS PubMed; (f) S. E. Ammann, W. Liu and M. C. White, Angew. Chem., Int. Ed., 2016, 55, 9571 CrossRef CAS PubMed; (g) H.-C. Lin, P.-S. Wang, Z.-L. Tao, Y.-G. Chen, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2016, 138, 14354 CrossRef CAS PubMed.
- For selected reviews of C–H functionalization, see: (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (e) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (f) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed; (g) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (h) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed.
- M. J. O’Donnell, N. Chen, C. Zhou and A. Murray, J. Org. Chem., 1997, 62, 3962 CrossRef.
- B. M. Trost, S. Mahapatra and M. Hansen, Angew. Chem., Int. Ed., 2015, 54, 6032 CrossRef CAS PubMed.
- (a) N. C. Miltenburg and W. Boogerd, Cancer Treat. Rev., 2014, 40, 872 CrossRef CAS PubMed; (b) A. W. Schmidt, K. R. Reddy and H.-J. Knolker, Chem. Rev., 2012, 112, 3193 CrossRef CAS PubMed.
- (a) A. Viso, R. F. de la Pradilla, A. García and A. Flores, Chem. Rev., 2005, 105, 3167 CrossRef CAS PubMed; (b) A. Viso, R. F. de la Pradilla, M. Tortosa, A. García and A. Flores, Chem. Rev., 2011, 111, PR1 CrossRef PubMed.
- For metal- or organocatalytic Mannich reactions of glycine imines, see: (a) L. Bernardi, A. S. Gothelf, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 2003, 68, 2583 CrossRef CAS PubMed; (b) X. X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X. L. Hou and Y. D. Wu, J. Am. Chem. Soc., 2008, 130, 14362 CrossRef CAS PubMed; (c) S. Kobayashi, R. Yazaki, K. Seki and Y. Yamashita, Angew. Chem., Int. Ed., 2008, 47, 5613 CrossRef CAS PubMed; (d) J. Hernández-Toribio, R. Gómez Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2008, 130, 16150 CrossRef PubMed; (e) D. Shang, Y. Liu, X. Zhou, X. Liu and X. Feng, Chem.–Eur. J., 2009, 15, 3678 CrossRef CAS PubMed; (f) J. Hernández-Toribio, R. Gómez Arrayás and J. C. Carretero, Chem.–Eur. J., 2010, 16, 1153 CrossRef PubMed; (g) G. Liang, M. C. Tong, H. Tao and C. J. Wang, Adv. Synth. Catal., 2010, 352, 1851 CrossRef CAS; (h) J. S. Bandar and T. H. Lambert, J. Am. Chem. Soc., 2013, 135, 11799 CrossRef CAS PubMed; (i) G. A. Cutting, N. E. Stainforth, M. P. John, G. Kociok-Köhn and M. C. Willis, J. Am. Chem. Soc., 2007, 129, 10632 CrossRef CAS PubMed; (j) K. Juhl, N. Gathergood and K. A. Jørgensen, Angew. Chem., Int. Ed., 2001, 40, 2995 CrossRef CAS.
- Generally, Pd-catalyzed AAA with DIOP ligand gave poor enantioselectivity, see: (a) B. M. Trost and P. E. Strege, J. Am. Chem. Soc., 1977, 99, 1649 CrossRef CAS; (b) C. S. Marques and A. J. Burke, Tetrahedron: Asymmetry, 2007, 18, 1804 CrossRef CAS. Rh-catalyzed coupling of carboxylic acids with terminal alkynes for the preparation of branched allylic esters utilizing DIOP ligand gave excellent enantioselectivity, see: (c) P. Koschker, M. Kahny and B. Breit, J. Am. Chem. Soc., 2015, 137, 3131 CrossRef CAS PubMed.
- (a) T. P. Dang and H. B. Kagan, Chem. Commun., 1971, 481 RSC; (b) H. B. Kagan and T. P. Dang, J. Am. Chem. Soc., 1972, 94, 6429 CrossRef CAS.
- see ESI.†.
- M. M. Salter, J. Kobayashi, Y. Shimizu and S. Kobayashi, Org. Lett., 2006, 8, 3533 CrossRef CAS PubMed.
- (a) A. Viso, R. Fernández de la Pradilla, M. L. López-Rodríguez, A. García, A. Flores and M. Alonso, J. Org. Chem., 2004, 69, 1542 CrossRef CAS PubMed; (b) G. Cremonesi, P. Dalla Croce, M. Gallanti and C. La Rosa, Tetrahedron, 2014, 70, 2054 CrossRef CAS; (c) T. Arai, A. Mishiro, E. Matsumura, A. Awata and M. Shirasugi, Chem.–Eur. J., 2012, 18, 11219 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details of experimental methods and results. See DOI: 10.1039/c7sc02899g |
| This journal is © The Royal Society of Chemistry 2017 |