
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmuno-targeting of Staphylococcus aureus via surface remodeling complexes†
Mary J.
Sabulski
,
Sean E.
Pidgeon
and
Marcos M.
Pires
 *
*
Department of Chemistry, Lehigh University, 6 E Packer Ave., Bethlehem, PA 18015, USA. E-mail: map311@lehigh.edu
First published on 23rd August 2017
Abstract
Agents with novel mechanisms of action are needed to complement traditional antibiotics. Towards these goals, we have exploited the surface-homing properties of vancomycin to tag the surface of Gram-positive pathogens with immune cell attractants in two unique modes. First, vancomycin was conjugated to the small molecule hapten 2,4-dinitrophenol (DNP) to promote bacterial opsonization. Second, we built on these results by improving the tagging specificity and mechanism of incorporation by coupling it to a sortase A substrate peptide. We demonstrated, for the first time, that the surface of Staphylococcus aureus (S. aureus) can be metabolically labeled in live Caenorhabditis elegans hosts. These constructs represent a class of promising narrow-spectrum agents that target S. aureus for opsonization and establish a new surface labeling modality in live host organisms, which should be a powerful tool in dissecting features of host–pathogen interactions.
Introduction
The human immune system is extremely efficient at preventing the entry and colonization of the vast majority of pathogens it encounters.1,2 However, its utility in fighting bacterial infections after the onset of acute symptoms remains an open question. Today, there is mounting evidence that immuno-therapeutic agents can evoke selective responses to diseased tissues (e.g., cancer) and dramatically reverse disease progression.3–7 These successes have been achieved despite similarities between cancer and non-transformed cells, which provide narrow therapeutic windows. In contrast, bacterial cells are vastly different compared to patient cells in size, shape, and composition – features that are already exploited by our innate immune system.8,9 Given the urgent need for novel antibiotic modalities, non-traditional immuno-modulatory agents against bacteria could play important roles in complementing traditional strategies.Our laboratory recently described a series of synthetic agents that grafted antigens onto bacterial cell surfaces by hijacking the PG biosynthetic machinery.10–12 Despite inducing high levels of antibody recruitment against a number of Gram-positive pathogens, high concentrations were needed to compete with endogenous PG building blocks, which exist in high μM to mM concentrations (or effective concentrations). Herein, we describe an alternative strategy to install haptens using an FDA-approved antibiotic that inherently homes to bacterial cell surfaces. More specifically, we used vancomycin to decorate Gram-positive bacteria with 2,4-dinitrophenol (DNP) epitopes to re-enlist components of the immune system. In addition, a coupled sortase-A surface-remodeling strategy was used to improve activity and selectivity towards drug-sensitive and methicillin-resistant strains of S. aureus.
A number of FDA-approved antibiotics seek out bacterial surfaces to inactivate vital cellular processes. Vancomycin binds lipid II molecules at the cytoplasmic membrane to halt PG biosynthesis but can also associate with D-Ala-D-Ala dipeptide units within the full length pentapeptide found on mature PG scaffolds (Fig. 1A and B). The amount of intact pentapeptide units, which retain the vancomycin target D-Ala-D-Ala dipeptides, found on mature PG scaffolds is highly dependent on the phylogenetic and phenotypic differences among bacteria. For the problematic human pathogen S. aureus, PG D-Ala-D-Ala content is ∼20%, with the rest being composed of shorter stem peptides processed by carboxypeptidases or transpeptidases.13 Given the display of D-Ala-D-Ala on PG scaffolds of Gram-positive bacteria, we reasoned that hapten-conjugated vancomycin should effectively graft antigenic epitopes onto bacterial cell surfaces (Fig. 1C).
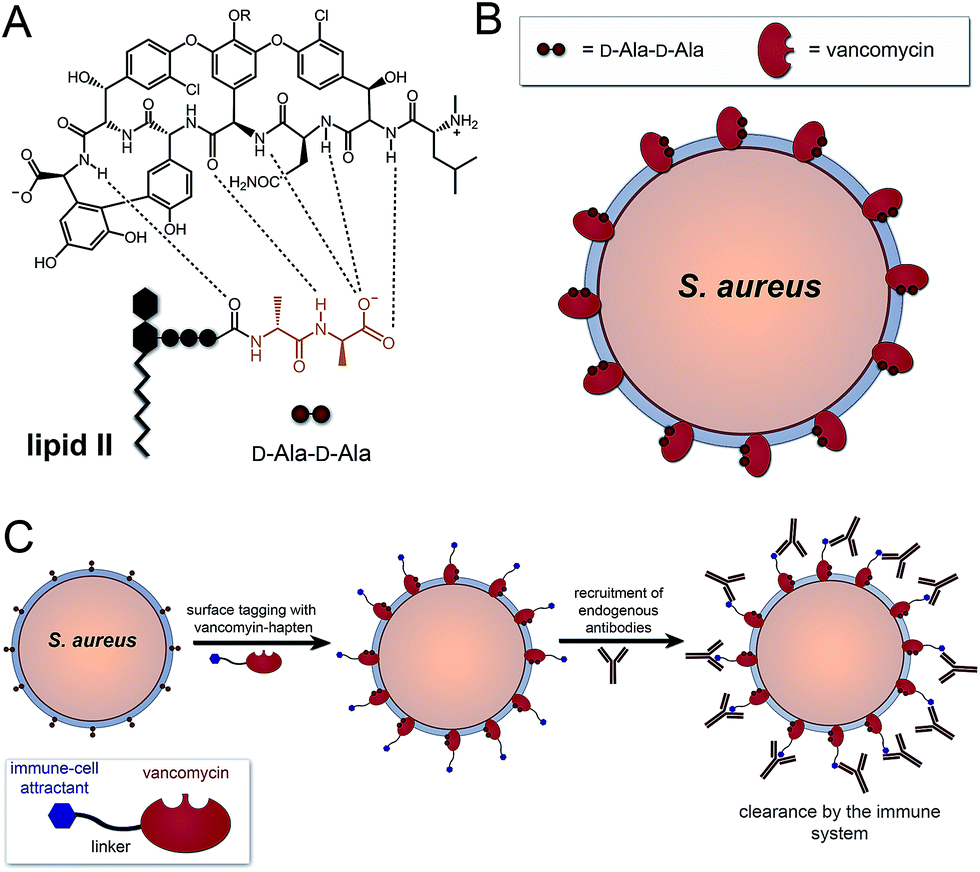 | ||
| Fig. 1 (A) Association of vancomycin with the D-Ala-D-Ala motif on lipid II. (B) Binding of vancomycin to PG scaffold on cell surface. (C) Strategy to graft immune-cell attracts on the surface of S. aureus based on PG targeting by vancomycin. | ||
Results and discussion
Vancomycin-based design
Semi-synthetic derivatives of vancomycin have proven to be potent second generation glycopeptide antibiotics (e.g., telavancin14) and valuable chemical probes. Fluorescent vancomycin derivatives have been shown to label at the septal region of Bacillus subtilis (B. subtilis), which has a higher density of lipid II molecules, and also throughout the bacterial sidewalls.15–19 Vancomycin can also target Gram-positive bacteria in live hosts as recently demonstrated by a near-IR fluorophore–vancomycin conjugate, which labeled S. aureus infections in mice.20,21 Likewise, we repeated the labeling experiments using BODIPY® FL vancomycin in B. subtilis, S. aureus, and Enterococcus faecium (E. faecium) and analyzed by confocal microscopy imaging (Fig. S1†). Cells were found to be labeled both at the septal region and the sidewalls. Based on these results and the well-established ability of vancomycin to target Gram-positive bacteria, we anticipated that hapten-modified vancomycin derivatives would effectively tag bacterial cell surfaces. Accordingly, we synthesized two derivatives of vancomycin, VanNdnp and VanCdnp that incorporated DNP epitopes (Fig. 2A) at the amino group and carboxylic acid sites, respectively. S. aureus cells were incubated with both vancomycin derivatives, exposed to FITC-conjugated anti-DNP antibodies, and antibody recruitment was measured by flow cytometry (Fig. 2B). Due to the nature of our assay, it was important to minimize non-specific binding of antibodies by surface anchored protein A. By using a protein A-deletion mutant S. aureus strain, fluorescence levels should reflect the specific recruitment of anti-DNP antibodies onto cell surfaces. Treatment of S. aureus cells with VancCdnp and VancNdnp resulted in 10.3-fold and 2.7-fold increases in fluorescence levels, respectively (Fig. 2C). Similarly, VancCdnp led to greater antibody recruitment in E. faecium and B. subtilis. These results indicated that anti-DNP recruitment can be specifically induced by DNP-modified vancomycin.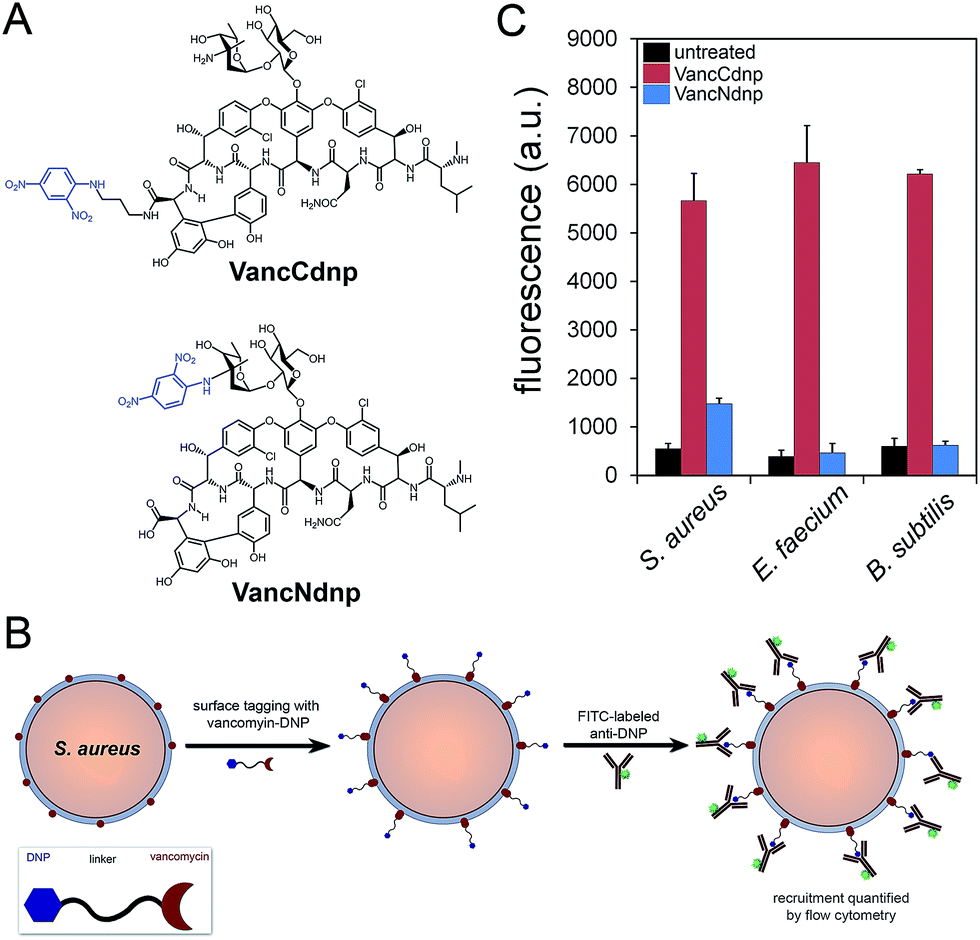 | ||
| Fig. 2 (A) Chemical structures of VancCdnp and VancNdnp. (B) Scheme of the assay to measure anti-DNP recruitment. (C) S. aureus cells were incubated for 30 min with 10 μM of either VancCdnp or VancNdnp followed by incubation with FITC-conjugated anti-DNP antibodies and analyzed using flow cytometry. Data are represented as mean + SD (n = 3). | ||
Sortase–vancomycin conjugates
Next, we redesigned VancCdnp to (1) increase surface display of haptens in pathogens that have reduced D-Ala-D-Ala content on mature PG scaffolds and (2) covalently incorporate haptens within the PG scaffold. Towards these goals, vancomycin–DNP constructs were conjugated to the substrate peptide sequence from S. aureus sortase A (SrtA). SrtA transpeptidase is a surface-bound enzyme that attaches bacterial proteins onto the PG scaffold (Fig. 3A).22,23 This mode of surface modification is critically important for entry and colonization of the host organism. The transpeptidase recognizes a SrtA specific peptide sequence (LPXTG motif, where X is any amino acid) and catalyzes the acyl-transfer onto lipid II of S. aureus.24 As the PG monomeric unit from lipid II is loaded onto the existing PG scaffold, so will the anchored protein.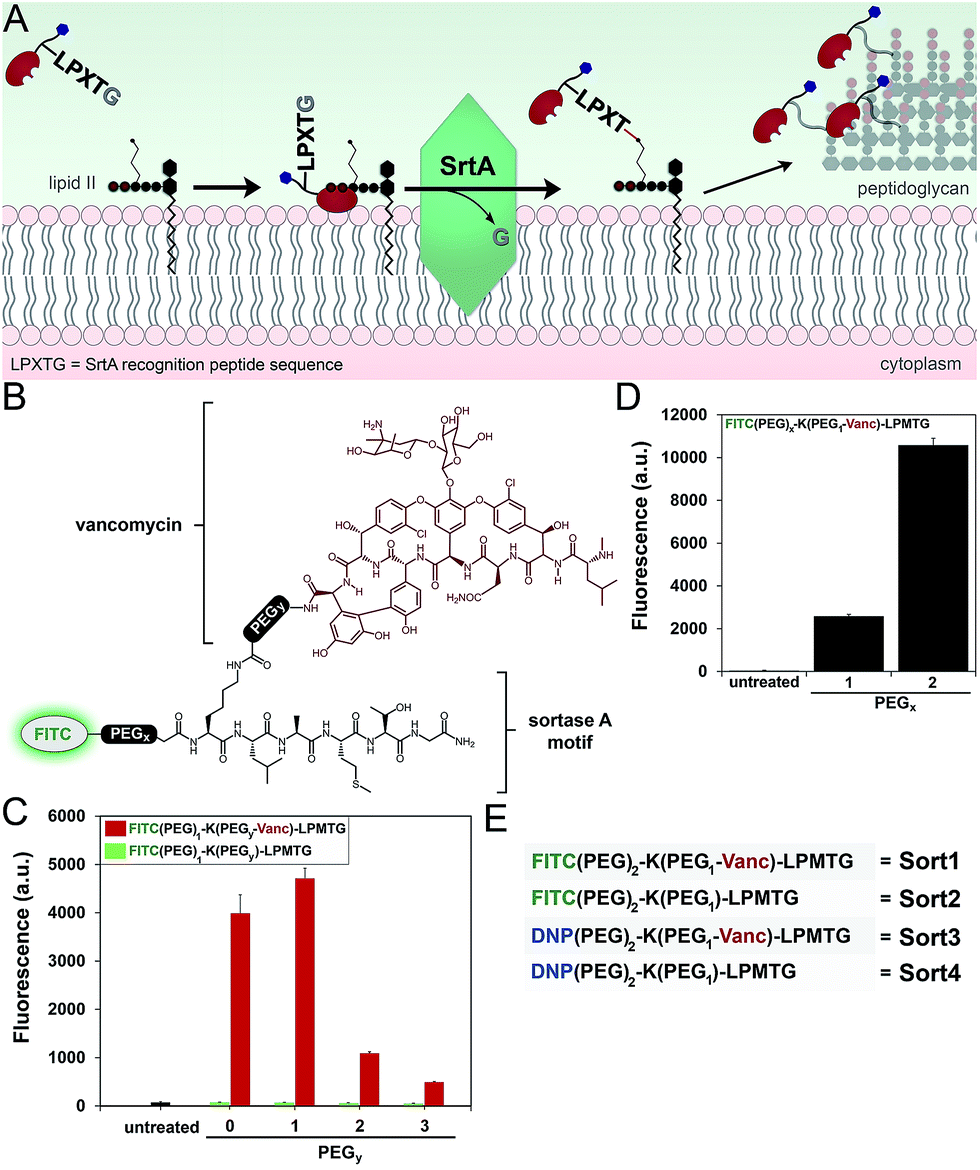 | ||
| Fig. 3 (A) Scheme showing the combination of vancomycin with SrtA to anchor DNP epitopes onto bacterial PG. (B) Basic chemical structure of FITC-labeled constructs. S. aureus cells were treated with FITC-based constructs with variable PEG spacers at Y position (C) or the X position (D) and analyzed using flow cytometry. Data are represented as mean + SD (n = 3). (E) Designation of variants with the common SrtA substrate recognition peptide. | ||
Previous reports by Spiegel and co-workers established that live S. aureus cells treated with fluorescently labeled LPXTG peptides (overnight incubation at 0.5–1 mM) resulted in the labeling of bacterial surfaces.25,26 We hypothesized that the covalent conjugation of vancomycin to LPXTG would bring the substrate peptide to its partner (lipid II) based on the ability of vancomycin to associate with the neighboring D-Ala-D-Ala dipeptide on lipid II (Fig. 3A). The higher effective concentration should result in covalent PG labeling at physiologically relevant concentrations.27 Most importantly for our design, the combination of SrtA and vancomycin will decouple the tagging step from availability of D-Ala-D-Ala on the PG scaffold. Therefore, high tagging levels should be achievable irrespective of the levels of D-Ala-D-Ala on surface exposed PG.
We first repeated the cell labeling experiments using SrtA sequences alone to establish baseline values. The three primary peptide sequences were: K(FITC)LPETG, K(FITC)LPMTG, and K(FITC)MGTLP. Bacterial cells were separately treated with all three peptides (incubated overnight at 1 mM). S. aureus cells displayed a 20-fold increase in fluorescence at 1 mM using K(FITC)LPETG, relative to unlabeled cells (Fig. S2†). Consistent with previous reports,28 the methionine containing K(FITC)LPMTG peptide resulted in an additional ∼2-fold increase in surface labeling. Attenuated cellular fluorescence was observed for the control scrambled sequence peptide. We next assembled molecules that combined SrtA substrate sequence and vancomycin (Fig. 3B). The linker between LPMTG and vancomycin was systematically varied to establish the optimum linker length to bridge binding to D-Ala-D-Ala and SrtA. The tether length was empirically established by evaluating a small panel of polyethylene glycol (PEG) spacers. From our results, it is clear that shorter linkers are preferred (Fig. 3C). Most importantly, the covalent attachment of vancomycin to each one of these constructs led to a major increase in surface tagging at 5 μM (up to 200-fold increase). Similarly, the PEG spacer between LPMTG and FITC was sampled at two different lengths (Fig. 3D). A longer PEG linker was preferred, which should lead to improved hapten availability to antibodies.
Confocal microscopy imaging was performed to delineate the localization of cellular labelling of Sort1 (Fig. S3†). As expected with the proposed mode of installation, fluorescence was observed throughout the entire cell surface with pronounced labeling at the septal region. The role of vancomycin within Sort1 in mediating cellular labeling was also probed. The co-incubation of S. aureus cells with Sort2 and vancomycin led to fluorescence signals near background levels (Fig. S4†). These results suggest that it is not enough to treat cells with both vancomycin and Sort2. The covalent conjugation is necessary to induce a large increase in cellular labeling. Interestingly, the addition of free vancomycin to cells treated with Sort1 resulted in a 34% increase in cellular fluorescence. This finding is in agreement with previously reported increase in cellular fluorescence of cells treated with fluorescent derivatives of vancomycin.15 Moreover, it also points to the potential of co-treatment with vancomycin in future in vivo testing, which should target bacterial cells in two complementary ways.
To evaluate the labeling across different bacterial species, cellular labeling was measured in S. aureus, Staphylococcus epidermidis (S. epidermidis), Listeria monocytogenes (L. monocytogenes), B. subtilis, and Escherichia coli (E. coli). Fluorescence levels were highest for S. aureus treated with Sort1 followed by S. epidermidis (Fig. 4A). These results are not surprising given the similarity between these two bacteria and it was recently suggested that S. epidermidis proteins displaying the sequence LPXTG are important in biofilm formation.29B. subtilis were sensitive to Sort1 presumably due to the presence of vancomycin. Gram-negative E. coli showed background fluorescence levels, which is consistent with the mode of incorporation and lack of vancomycin accessibility to the PG layer. L. monocytogenes labeled ∼20-fold less than S. aureus, which may reflect a difference in utilization of SrtA. In fact, the ortholog SrtB has been shown to be important for anchoring surface proteins in L. monocytogenes.30 It should be the case that the preferred substrate sequence (NAKTN) for SrtB should also label the surface of L. monocytogenes cells.31 To test this idea, FITC(PEG)-NAKTN was synthesized and incubated with L. monocytogenes cells, which led to high levels of cellular fluorescence (Fig. S5†). Taken together, these results suggest that our constructs label the surface of bacteria with defined specificity and can potentially be generalized based on sortase substrate sequence.
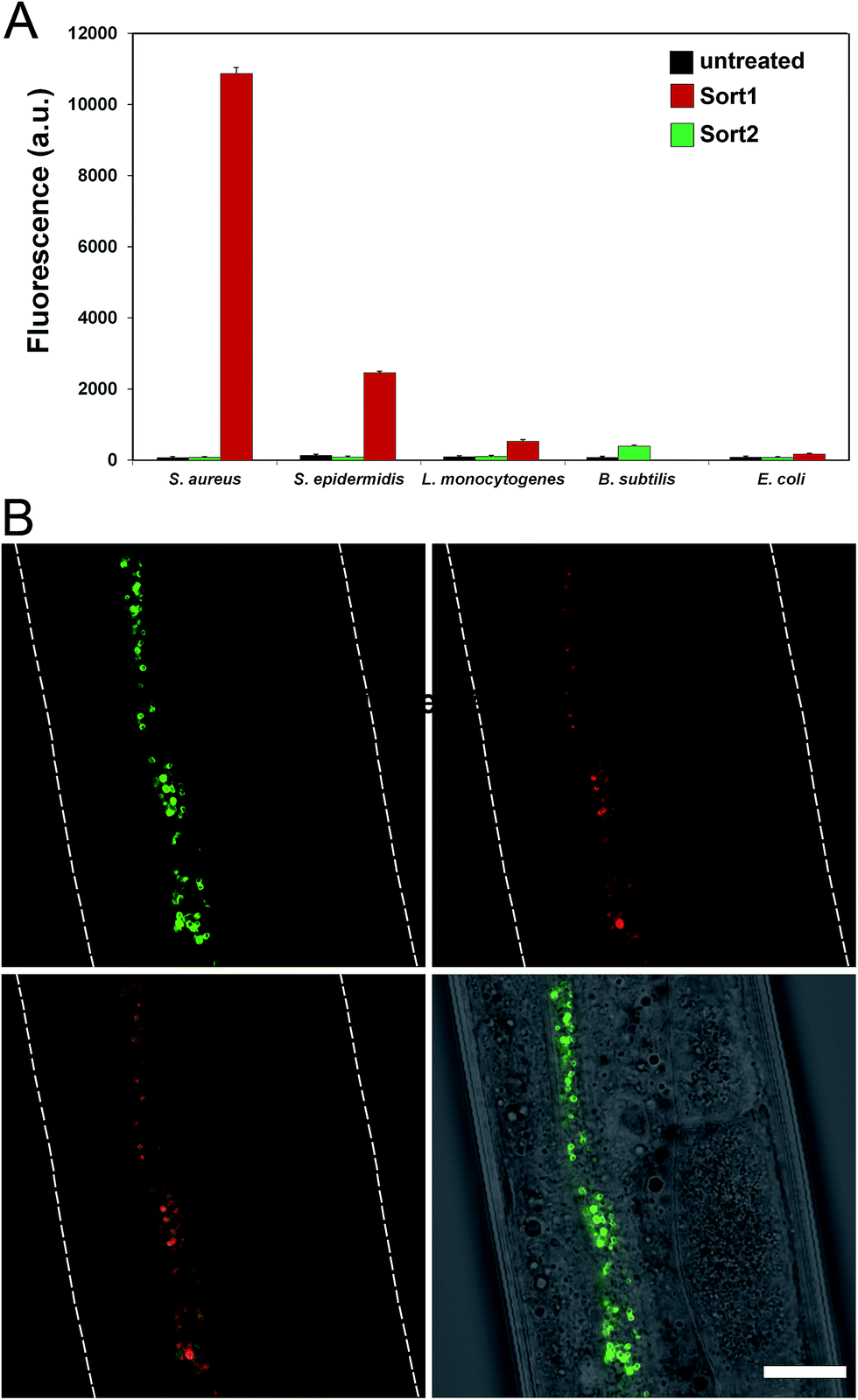 | ||
| Fig. 4 (A) Specified bacteria were treated with Sort1 and Sort 2 (5 μM) overnight and fluorescence was measured using flow cytometry. Data are represented as mean + SD (n = 3). (B) C. elegans infected with S. aureus (expressing mCherry) were treated with Sort1 (50 μM), washed, anesthetized, mounted on a bed of agarose, and imaged using confocal microscopy. Scale bar presents 20 μm. | ||
Labeling in live host animal
With a construct in hand that operates at low μM concentrations, we focused on establishing bacterial surface labeling in Caenorhabditis elegans (C. elegans), which is a powerful model animal for bacterial pathogenesis.32–34C. elegans (L4 larval stage) were incubated with S. aureus to establish bacterial colonization.After removing residual bacteria, S. aureus infected C. elegans were treated with Sort1. Remarkably, bacteria were clearly labeled in vivo post infection (Fig. 4B and S6†). This represents the first example of metabolic labeling of bacterial PG in vivo. These results establish that exogenous epitopes can be selectively grafted onto the surface of S. aureus in a live host. Distinguishing PG features of Sort1 treated S. aureus cells were readily visible in the green channel consistent with the mode of incorporation. Localization of S. aureus cells was accomplished under the constitutive cytosolic expression of mCherry.35 Co-localization of green and red signals were consistently observed in all the samples analyzed. Treatment of S. aureus-infected C. elegans with Sort2 (lacking the vancomycin moiety) resulted in no observable green signal (Fig. S7†). Taken together, the combined results demonstrated the ability to target bacterial cells in live hosts.
A construct composed of DNP in place of FITC (Sort3) was synthesized using the same synthetic route. The hapten DNP was chosen due to the naturally high abundance of endogenous anti-DNP antibodies in human serum.36–38S. aureus cells were exposed to Sort3 and anti-DNP recruitment was analyzed similar to VanCdnp (Fig. 5A). Gratifyingly, a clear increase in cellular fluorescence was observed at sub-micromolar concentrations indicative of anti-DNP recruitment. At 5 μM, fluorescence levels were 14.8-fold above untreated cells. Cell treatment with Sort4 led to background fluorescence levels, again suggesting that vancomycin is critical for surface labeling. Recruitment of anti-DNP was also observed directly from pooled human serum, indicating that Sort3 can potentially operate in physiologically relevant conditions (Fig. S8†). Next, we shifted our focus to the pathogenic and widely disseminated methicillin-resistant S. aureus (MRSA). Protein A on the surface of MRSA can be disruptive to our assay read-out. To circumvent this, MRSA cells were pre-treated with mock IgGs (lacking a FITC label) to mimic the anticipated occupancy of protein A by antibodies from serum. Satisfyingly, treatment of MRSA cells with Sort3 led to similar fluorescent levels to protein A-deleted strains (Fig. S9†). In addition, pre-incubation of MRSA cells with pooled human IgGs, which include anti-DNP antibodies, effectively blocked binding of FITC-labeled anti-DNP.
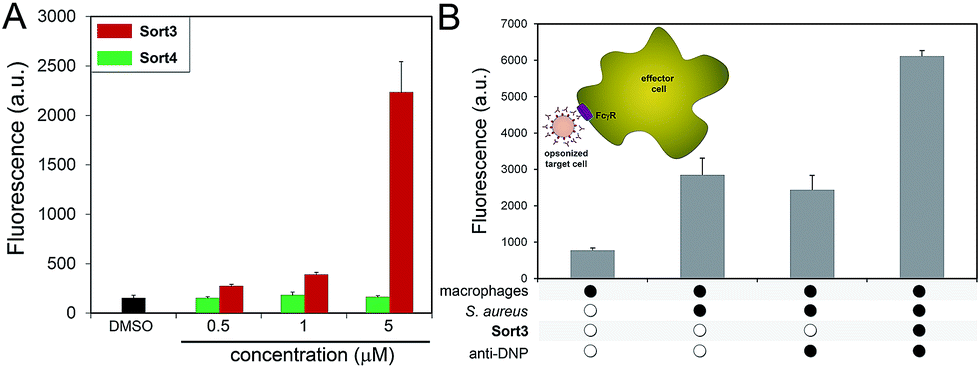 | ||
| Fig. 5 (A) S. aureus cells were incubated overnight with 5 μM of Sort3 followed by incubation with FITC-conjugated anti-DNP antibodies and analyzed using flow cytometry. Data are represented as mean + SD (n = 3). (B) Phagocytosis of bacterial cells was evaluated by treating S. aureus cells in the presence or absence of 5 μM of Sort3. Untreated or opsonized cells (with anti-DNP antibody) were incubated with J774A.1 macrophages for 30 min in the absence or the presence of calcein-AM labeled S. aureus cells. Fluorescence was measured by flow cytometry. Data are represented as mean + SD (n = 3). | ||
The inherent toxicity of Sort3 towards S. aureus cells was evaluated next. Surprisingly, exposing S. aureus to concentration > 100 μM led to no significant change in cell density (Fig. S10†). We hypothesize that the covalent anchoring of vancomycin within the stem peptide leads to segregation from its lethal lipid II target. In the future, we will explore the incorporation of vancomycin at the C-terminus of the SrtA substrate peptide sequence, which should release vancomycin upon anchoring of the construct within the stem peptide. Toxicity was also minimal towards mammalian cells at all concentrations tested, a finding that was expected based on the polarity and size of Sort3 and lack of cognate binding partners (Fig. S11†). Finally, we set out to recapitulate the recognition and phagocytosis of opsonized bacteria by macrophages. Bacteria treated with Sort3 and exposed to anti-DNP antibodies led to a 2-fold increase in phagocytosis compared to treatment of Sort3 alone and anti-DNP alone (Fig. 5B and S12†). This result demonstrates the surface of bacterial cells remodeled with Sort3 display antigenic epitopes that are available to engage with immune cells.
Conclusions
In conclusion, we have described two classes of vancomycin conjugates that tag S. aureus cell surfaces by non-covalent association with the PG scaffold and covalent integration within bacterial PG. Fluorescent-based constructs were synthesized to optimize incorporation levels. We showed, for the first time, that the surface of S. aureus can be metabolically tagged with unnatural epitopes in live C. elegans hosts. Hapten-based constructs were synthesized and bacterial opsonization was demonstrated, which resulted in the induction of phagocytosis by macrophages. Combined, we anticipate that this class of agents represents a promising immune-modulatory strategy to combat bacterial infections.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Lehigh University (M. P.). We would like to thank Dr Horswill (University of Iowa) for kindly sharing the mCherry-expressing S. aureus strain.Notes and references
- B. B. Finlay and R. E. Hancock, Nat. Rev. Microbiol., 2004, 2, 497–504 CrossRef CAS PubMed.
- R. E. Hancock, A. Nijnik and D. J. Philpott, Nat. Rev. Microbiol., 2012, 10, 243–254 CrossRef CAS PubMed.
- D. Killock, Nat. Rev. Clin. Oncol., 2014, 11, 562 CrossRef PubMed.
- K. N. Masihi, Expert Opin. Biol. Ther., 2001, 1, 641–653 CrossRef CAS PubMed.
- S. L. Topalian, F. S. Hodi, J. R. Brahmer, S. N. Gettinger, D. C. Smith, D. F. McDermott, J. D. Powderly, R. D. Carvajal, J. A. Sosman, M. B. Atkins, P. D. Leming, D. R. Spigel, S. J. Antonia, L. Horn, C. G. Drake, D. M. Pardoll, L. Chen, W. H. Sharfman, R. A. Anders, J. M. Taube, T. L. McMiller, H. Xu, A. J. Korman, M. Jure-Kunkel, S. Agrawal, D. McDonald, G. D. Kollia, A. Gupta, J. M. Wigginton and M. Sznol, N. Engl. J. Med., 2012, 366, 2443–2454 CrossRef CAS PubMed.
- S. A. Rosenberg, J. C. Yang and N. P. Restifo, Nat. Med., 2004, 10, 909–915 CrossRef CAS PubMed.
- S. L. Topalian, J. D. Wolchok, T. A. Chan, I. Mellman, K. Palucka, J. Banchereau, S. A. Rosenberg and K. Dane Wittrup, Cell, 2015, 161, 185–186 CrossRef PubMed.
- J. M. Blander and L. E. Sander, Nat. Rev. Immunol., 2012, 12, 215–225 CrossRef CAS PubMed.
- R. Medzhitov, Nat. Rev. Immunol., 2001, 1, 135–145 CrossRef CAS PubMed.
- J. M. Fura, S. E. Pidgeon, M. Birabaharan and M. M. Pires, ACS Infect. Dis., 2016, 2, 302–309 CrossRef CAS PubMed.
- J. M. Fura and M. M. Pires, Biopolymers, 2015, 104, 351–359 CrossRef CAS PubMed.
- J. M. Fura, M. J. Sabulski and M. M. Pires, ACS Chem. Biol., 2014, 9, 1480–1489 CrossRef CAS PubMed.
- S. Boyle-Vavra, H. Labischinski, C. C. Ebert, K. Ehlert and R. S. Daum, Antimicrob. Agents Chemother., 2001, 45, 280–287 CrossRef CAS PubMed.
- M. R. Leadbetter, S. M. Adams, B. Bazzini, P. R. Fatheree, D. E. Karr, K. M. Krause, B. M. Lam, M. S. Linsell, M. B. Nodwell, J. L. Pace, K. Quast, J. P. Shaw, E. Soriano, S. G. Trapp, J. D. Villena, T. X. Wu, B. G. Christensen and J. K. Judice, J. Antibiot., 2004, 57, 326–336 CrossRef CAS PubMed.
- R. A. Daniel and J. Errington, Cell, 2003, 113, 767–776 CrossRef CAS PubMed.
- K. Tiyanont, T. Doan, M. B. Lazarus, X. Fang, D. Z. Rudner and S. Walker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11033–11038 CrossRef CAS PubMed.
- D. Morales Angeles, Y. Liu, A. M. Hartman, M. Borisova, A. de Sousa Borges, N. de Kok, K. Beilharz, J. W. Veening, C. Mayer, A. K. Hirsch and D. J. Scheffers, Mol. Microbiol., 2017, 104, 319–333 CrossRef CAS PubMed.
- J. M. Monteiro, P. B. Fernandes, F. Vaz, A. R. Pereira, A. C. Tavares, M. T. Ferreira, P. M. Pereira, H. Veiga, E. Kuru, M. S. VanNieuwenhze, Y. V. Brun, S. R. Filipe and M. G. Pinho, Nat. Commun., 2015, 6, 8055 CrossRef CAS PubMed.
- P. Reed, M. L. Atilano, R. Alves, E. Hoiczyk, X. Sher, N. T. Reichmann, P. M. Pereira, T. Roemer, S. R. Filipe, J. B. Pereira-Leal, P. Ligoxygakis and M. G. Pinho, PLoS Pathog., 2015, 11, e1004891 Search PubMed.
- M. van Oosten, T. Schafer, J. A. Gazendam, K. Ohlsen, E. Tsompanidou, M. C. de Goffau, H. J. Harmsen, L. M. Crane, E. Lim, K. P. Francis, L. Cheung, M. Olive, V. Ntziachristos, J. M. van Dijl and G. M. van Dam, Nat. Commun., 2013, 4, 2584 Search PubMed.
- C. Yang, C. Ren, J. Zhou, J. Liu, Y. Zhang, F. Huang, D. Ding and B. Xu, Angew. Chem., Int. Ed. Engl., 2017, 56, 2356–2360 CrossRef CAS PubMed.
- A. P. Hendrickx, J. M. Budzik, S. Y. Oh and O. Schneewind, Nat. Rev. Microbiol., 2011, 9, 166–176 CrossRef CAS PubMed.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS PubMed.
- L. A. Marraffini, A. C. Dedent and O. Schneewind, Microbiol. Mol. Biol. Rev., 2006, 70, 192–221 CrossRef CAS PubMed.
- J. W. Nelson, A. G. Chamessian, P. J. McEnaney, R. P. Murelli, B. I. Kazmierczak and D. A. Spiegel, ACS Chem. Biol., 2010, 5, 1147–1155 CrossRef CAS PubMed.
- S. Gautam, T. Kim, E. Lester, D. Deep and D. A. Spiegel, ACS Chem. Biol., 2016, 11, 25–30 CrossRef CAS PubMed.
- S. Hansenova Manaskova, K. Nazmi, W. van't Hof, A. van Belkum, N. I. Martin, F. J. Bikker, W. J. van Wamel and E. C. Veerman, PLoS One, 2016, 11, e0147401 Search PubMed.
- R. G. Kruger, B. Otvos, B. A. Frankel, M. Bentley, P. Dostal and D. G. McCafferty, Biochemistry, 2004, 43, 1541–1551 CrossRef CAS PubMed.
- L. Khodaparast, M. Shahrooei, B. Stijlemans, R. Merckx, P. Baatsen, J. P. O'Gara, E. Waters, L. Van Mellaert and J. Van Eldere, PLoS One, 2016, 11, e0146704 Search PubMed.
- H. Bierne, C. Garandeau, M. G. Pucciarelli, C. Sabet, S. Newton, F. Garcia-del Portillo, P. Cossart and A. Charbit, J. Bacteriol., 2004, 186, 1972–1982 CrossRef CAS PubMed.
- J. F. Mariscotti, F. Garcia-del Portillo and M. G. Pucciarelli, J. Biol. Chem., 2009, 284, 6140–6146 CrossRef CAS PubMed.
- J. Garcia-Lara, A. J. Needham and S. J. Foster, FEMS Immunol. Med. Microbiol., 2005, 43, 311–323 CrossRef CAS PubMed.
- C. D. Sifri, J. Begun, F. M. Ausubel and S. B. Calderwood, Infect. Immun., 2003, 71, 2208–2217 CrossRef CAS PubMed.
- D. A. Garsin, J. M. Villanueva, J. Begun, D. H. Kim, C. D. Sifri, S. B. Calderwood, G. Ruvkun and F. M. Ausubel, Science, 2003, 300, 1921 CrossRef CAS PubMed.
- C. L. Malone, B. R. Boles, K. J. Lauderdale, M. Thoendel, J. S. Kavanaugh and A. R. Horswill, J. Microbiol. Methods, 2009, 77, 251–260 CrossRef CAS PubMed.
- Y. Lu, E. Sega and P. S. Low, Int. J. Cancer, 2005, 116, 710–719 CrossRef CAS PubMed.
- C. E. Jakobsche, C. G. Parker, R. N. Tao, M. D. Kolesnikova, E. F. Douglass Jr and D. A. Spiegel, ACS Chem. Biol., 2013, 8, 2404–2411 CrossRef CAS PubMed.
- R. T. Sheridan, J. Hudon, J. A. Hank, P. M. Sondel and L. L. Kiessling, ChemBioChem, 2014, 15, 1393–1398 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02721d |
| This journal is © The Royal Society of Chemistry 2017 |