
Influence of mesopore size and peptide aggregation on the adsorption and release of a model antimicrobial peptide onto/from mesoporous silica nanoparticles in vitro†
Katharina
Braun
,
Alexander
Pochert
,
Marlena
Gerber
,
Heinz Fabian
Raber
and
Mika
Lindén
 *
*
Inorganic Chemistry II, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: mlinden@uni-ulm.de
First published on 1st September 2017
Abstract
Mesoporous silica is a promising carrier for cationic peptides, as such peptides can be adsorbed onto silica through attractive electrostatic interactions. However, concentration-dependent peptide aggregation could influence the adsorption process itself, and may also influence the kinetics of peptide release. In order to shed more light on these processes, we chose to study the adsorption and release of a model antimicrobial peptide, salmon calcitonin, using three silica nanoparticles that are different in terms of pore size. It is shown that pronounced adsorption of calcitonin at low calcitonin solution concentrations was only observed when the mesopore size was in the range of the size of the peptide. When the mesopore size clearly exceeded the size of the peptide, very limited peptide adsorption was observed. In this case, pronounced peptide adsorption was only observed when the solution concentration of the peptide was so high that the peptide formed oligomeric aggregates in solution. The results clearly indicate that electrostatic attraction is not the only driver for peptide adsorption, but that the possibility for multivalent peptide–silica interactions in the case of the smaller-pore silica strongly enhances peptide adsorption. This conclusion is supported by results obtained for calcitonin adsorption to non-porous Stöber particles of similar size, where an adsorption behavior almost identical to that seen for the large-mesopore silica nanoparticles was observed. Interestingly, the release rates were also highly influenced by the nature of the adsorption, and very fast peptide release was observed for calcitonin loaded into the small-mesopore silica nanoparticles, while very slow release was observed for calcitonin adsorbed to the large-mesopore and the non-porous silicas. Such an adsorption–desorption behavior is opposite to that normally observed for small molecule–drug–silica nanoparticle combinations, and highlights the importance of taking peptide aggregation into account when designing mesoporous silica-based carrier systems for delivery of therapeutic peptides.
Design, System, ApplicationMesoporous silica nanoparticles, MSNs, have emerged as a promising drug delivery platform for a variety of drug candidates, including small molecule drugs, peptides, and proteins. The current study focuses on the influence of peptide aggregation on the adsorption of peptides onto purely siliceous MSNs as a function of mesopore diameter. Calcitonin is used as a model peptide for antimicrobial peptides. It is shown that dimers of calcitonin adsorb strongly onto MSNs if the pore size allows for multidirectional peptide–silica interactions, but if the pore size is large enough that multidirectional interactions are not operative, pronounced adsorption does not occur unless larger peptide aggregates are present in solution. This also strongly influences the kinetics of peptide release, as slow release of the peptide is only observed in the latter case. The results thus introduce a new parameter for controlling peptide release in biological media in the case of self-assembling peptides keeping the chemical composition of the carrier identical. Thus, the results are thought to be of importance for the optimization of carrier properties to tune the peptide release rates for a given application. |
1. Introduction
Antimicrobial peptides, AMPs, are positively charged, amphiphilic peptides which are capable of associating with membranes.1 Their antimicrobial activity is normally associated with selective lysis of bacterial membranes without affecting membranes of mammalian cells, but can also act on intracellular targets. AMPs are therefore attractive alternatives to antibiotics which often are associated with resistance issues.2 AMPs typically contain 12–50 amino acids, and the positive charge is provided by arginine and lysine residues. However, as for many peptides, the blood half-life of AMPs may be limited due to enzymatic degradation processes.3,4 Furthermore, as AMPs are amphiphilic, they also bind to serum proteins and are rapidly cleared from the bloodstream circulation. Therefore, the development of carriers for AMPs which would provide shielding against peptidase activity could be designed to fit the requirements of a specific administration route, and maybe even provide the possibility for targeted delivery of peptides which would be highly attractive. In addition, intracellular bacterial localization, as is the case for some infections, including tuberculosis, poses a challenge on how to reach the intracellular bacteria without lysing and killing the host macrophages,5 and also here a carrier system allowing for intracellular AMP delivery would be attractive. However, despite these evident beneficial effects, studies related to carrier-mediated AMP delivery are scarce. We have recently shown that negatively charged, mesoporous all-silica nanoparticles with a pore diameter of 3–4 nm are capable of carrying a high payload of the antimicrobial peptide LL-37, and that the peptide is shielded against enzymatic degradation as long as the peptide remains inside the pore system of the carrier particles.6 Furthermore, the peptide also remained active post release, and the loaded particles showed a low level of hemolysis. More positively charged, amino-functionalized mesoporous silica nanoparticles, on the other hand, showed lysis activity even in the absence of LL-37 due to their positive charge, and the lysis activity was naturally further enhanced upon loading of these particles with LL-37. However, we noted that the loading capacity of the amino-functionalized particles was lower than that of the all-silica particles due to differences in their surface charge. Non-porous silica nanoparticles used as a control did also adsorb LL-37, but due to the absence of intra-particulate mesopores, the peptide was located on the outer surface of the particles. In this case, the loaded particles showed antimicrobial activity even at low levels of peptide release, which is related to the peptide coating on the particles, rendering the particles themselves antimicrobially active. However, in this case, no shielding is provided against enzymatic degradation. These results highlight the importance of surface charge and the presence of mesoporosity for efficient antimicrobial delivery using mesoporous silica carriers.Many peptides are known to form aggregates in a concentration-dependent manner.7 In order for LL-37 to efficiently fit into the pores of the mesoporous silica host having a pore size of 3–4 nm, it is assumed that the peptide preferentially adsorbed in its monomeric form, while it could be assumed that larger peptide aggregates could adsorb on the outer surface of the particles. As the degree of peptide aggregation could have an influence on the antimicrobial efficiency of the peptide, knowing if the peptide is located inside the mesopore system or preferentially adsorbed onto the surface of the carrier particles is important in order to estimate its stability against enzymatic degradation. Furthermore, the pore size could influence the distribution of such aggregates between the inner and outer parts of the particles, which in turn could influence the release kinetics of the peptide. On this note, the mesopore size has also been shown to influence the kinetics of protein adsorption onto mesoporous silica nanoparticles8 and also the location and conformation of the adsorbed proteins.9 In order to shed some more light on these processes, we studied the adsorption and release of salmon calcitonin onto and from 3 silica nanoparticles which were comparable in terms of particle size and surface charge but different in terms of mesopore size and porosity. Salmon calcitonin is a cationic peptide with a 32 amino acid sequence Cys–Ser–Asn–Leu–Ser–Thr–Cys–Val–Leu–Gly–Lys–Leu–Ser–Gln–Glu–Leu–His–Lys–Leu–Gln–Thr–Tyr–Pro–Arg–Thr–Asn–Thr–Gly–Ser–Gly–Thr–Pro–NH2 with a pI value of 8.96 and a molecular weight of 3.4 kDa, and thus resembles antimicrobial peptides in terms of both charge and size, but can be obtained at a much lower cost. This allows the use of higher peptide concentrations in the experiments and thus also the determination of adsorption isotherms over all the relevant concentration ranges. However, despite being used here as a model antimicrobial peptide, calcitonin is a drug candidate in its own right, as it is used to treat Paget's disease, phantom limb pain and osteoporosis.10–12 Our results imply a key role of mesopore size and peptide aggregation in both peptide adsorption and release kinetics and contribute to a better understanding of peptide–silica interactions, which are important for the design of mesoporous silica-based carriers for antimicrobial peptides.
2. Materials and methods
2.1. Materials
The following chemicals were used in the synthesis of silica particles: tetramethyl orthosilicate (TMOS, purum, ≥98%, Fluka Analytical/Sigma Aldrich), tetraethyl orthosilicate (TEOS, purum, ≥99%, Sigma Aldrich), cetyltrimethylammonium bromide (CTAB, ≥98%, Sigma Aldrich), (3-aminopropyl)trimethoxysilane (APTMS, 97%, Aldrich Chemistry), methanol (MeOH, technical, VWR), ethanol (EtOH, 99.5% denatured with 1% MEK, VWR), sodium hydroxide (NaOH, ≥98%, Sigma Aldrich), ammonium hydroxide solution (NH3, 28%, VWR), mesitylene (TMB, 98%, Sigma Aldrich), phosphate buffered saline (PBS, Life Technologies), sodium chloride (NaCl, reagent-grade, Merck KGaA), sodium bicarbonate (NaHCO3, reagent-grade, Merck KGaA), potassium chloride (KCl, reagent-grade, Merck KGaA), potassium phosphate dibasic trihydrate (K2HPO4·3H2O, reagent-grade, Merck KGaA), magnesium chloride hexahydrate (MgCl2·6H2O, reagent-grade, Merck KGaA), calcium chloride dihydrate (CaCl2·2H2O, purum, Merck KGaA), sodium sulfate (Na2SO4, ≥99%, Merck KGaA), tris(hydroxymethyl)aminomethane (TRIS, reagent-grade, Merck KGaA), salmon calcitonin (sCt, PolyPeptide) and Milli-Q water. All chemicals were used as-received.2.2. Synthesis of different silica materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.74 NH3:113.48 ethanol:39.71 water. The as-obtained particles were washed twice in ethanol and subsequently dried under vacuum.
:0.14 APTMS:1.46 CTAB:1353.49 MeOH:2612.56 H2O:8.04 NaOH. The as-synthesized particles were washed twice with EtOH, dried under vacuum, and subsequently calcined for 6 h at 550 °C with a heating rate of 1 °C min−1.
:4.74 NH3:113.48 ethanol:39.71 water. The as-obtained particles were washed twice in ethanol and subsequently dried under vacuum.
:0.14 APTMS:1.46 CTAB:1353.49 MeOH:2612.56 H2O:8.04 NaOH. The as-synthesized particles were washed twice with EtOH, dried under vacuum, and subsequently calcined for 6 h at 550 °C with a heating rate of 1 °C min−1.
2.3. Peptide experiments
High-resolution mass spectra (HRMS) were measured using a Fourier Transform Ion Cyclotron Resonance (FT-ICR) solariX mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a 7.0 T superconducting magnet and interfaced to an Apollo II Dual ESI/MALDI source. The spectra were acquired in positive MALDI mode using the ftms control software version 2.1.0. The Peptide Calibration Standard (Bruker Daltonics, Bremen, Germany) was used for external calibration. The processing and peak selection of the spectra were carried out automatically using Bruker Compass Data Analysis version 4.4 software.
3. Results
In order to understand the effects of particle porosity on salmon calcitonin adsorption and release behavior, non-porous silica nanoparticles (NSN) were combined with calcined mesoporous silica nanoparticles (MSNc) as well as with pore-expanded silica nanoparticles (MSNpe). The scanning electron microscopy (SEM) images reveal a relatively uniform particle size distribution as shown in Fig. 1. The MSNpe were hollow after the hydrothermal treatment step, which is clearly observed in the SEM image taken for these particles, also supported by transmission electron microscopy (TEM) images taken for the microtomed particles. The hollow nature of all MSNpe is not as clearly seen though in the TEM images as in the SEM images, which is attributed to the fact that the cut through the particles upon microtoming will not always be located in the middle of the particles. The MSNc exhibit limited long-range ordering of the mesopores, which is typical for particles having a radial mesopore arrangement, while the MSNpe do not show any indication of long-range mesopore ordering. | ||
| Fig. 1 SEM images of NSN (A), MSNc (B), and MSNpe (C). Microtomed TEM images of NSN (D), MSNc (E), and MSNpe (F). The hollow structure of the MSNpe is clearly seen. | ||
The nitrogen adsorption isotherms measured for the different particles are shown in Fig. 2. The calculated specific BET surface areas were ∼1000 m2 g−1 for MSNc, ∼130 m2 g−1 for MSNpe, and ∼10 m2 g−1 for NSN. The corresponding mesopore diameters calculated using the NLDFT-kernel (adsorption branch) were ∼3 nm for MSNc and 8 nm and 27 nm (bimodal) for MSNpe. The pore size distribution curves are shown in Fig. S1.† We note that the relative pressure range corresponding to filling of intra- and interparticulate mesopores overlaps for MSNpe, but the broadened hysteresis observed for these particles gives evidence for filling of intraparticulate pores also in these particles in addition to interparticulate porosity. Furthermore, the relatively broad hysteresis in the high relative pressure range suggests the presence of some additional mesopores with a smaller diameter than that of the main portion of the mesopores. As expected, the ζ-potential values measured in 10−3 M KCl at a pH of 5.5 were similar, in the range of −20 to −30 mV, for all particles.
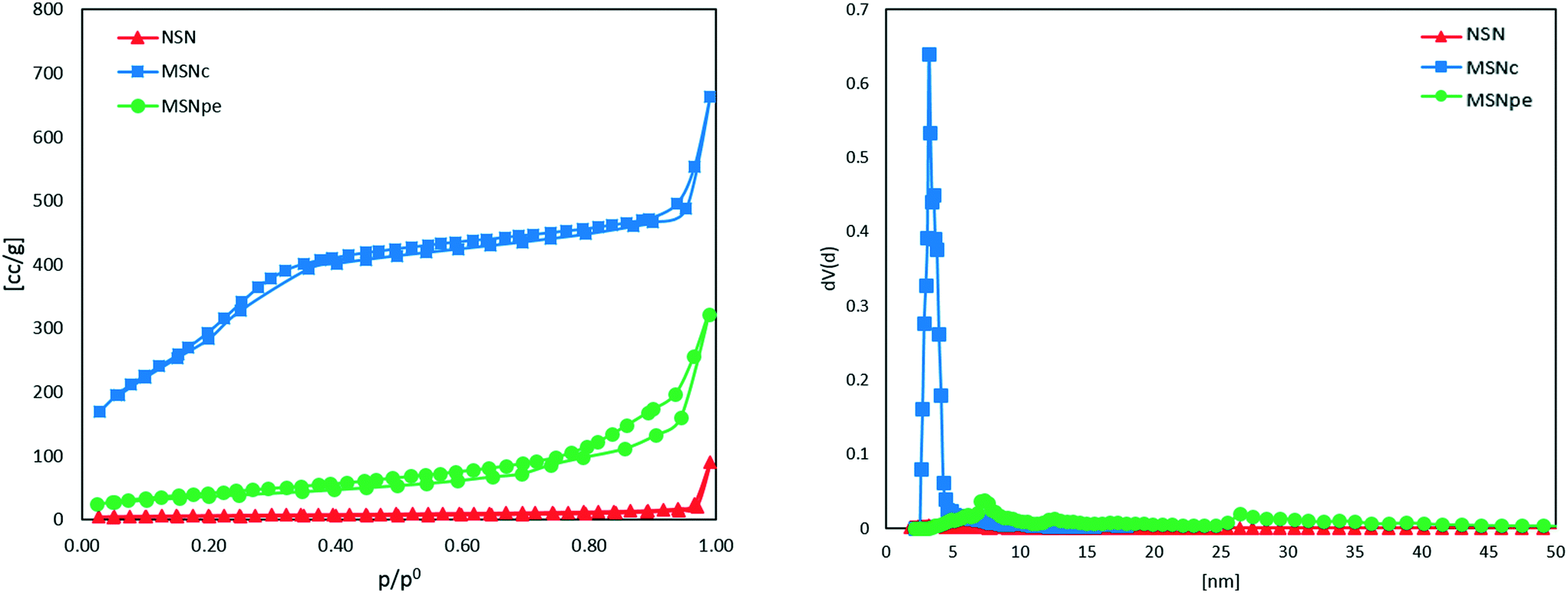 | ||
| Fig. 2 Nitrogen sorption isotherms and pore size distribution measured at 77 K of NSN (red ▲), MSNc (blue ■) and MSNpe (green ●). | ||
The main physicochemical characteristics of the three particles are summarized in Table 1. The particles differ mainly in terms of porosity and pore size.
The adsorption of salmon calcitonin to the different particles in PBS at room temperature was studied as a function of peptide concentration. The determined adsorption isotherms are shown in Fig. 3.
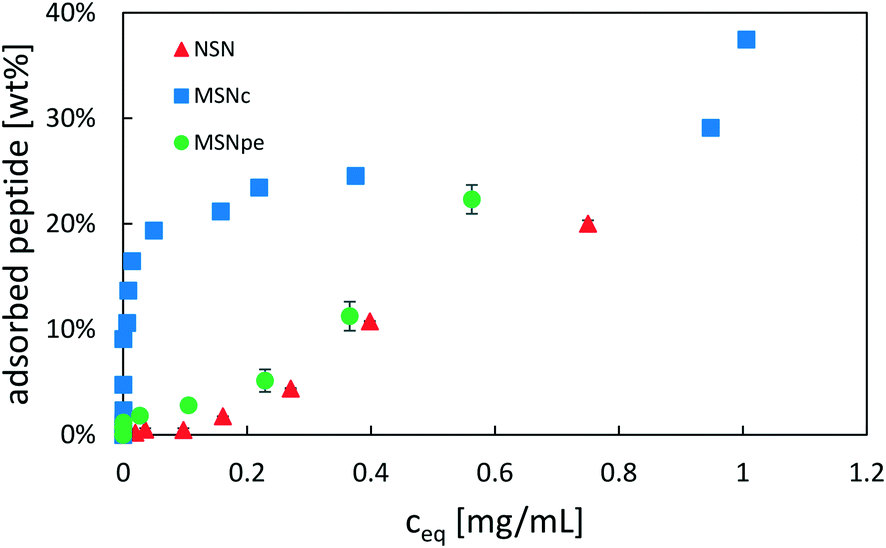 | ||
| Fig. 3 Adsorption isotherms of salmon calcitonin on NSN (red ▲), MSNc (blue ■) and MSNpe (green ●) in phosphate buffered saline (PBS) at a pH of 7.4 and a particle concentration of 5 mg mL−1. | ||
Calcitonin adsorbed very strongly to MSNc as evidenced by the steep initial part of the adsorption isotherm, reaching a plateau at about 25 wt%. A second step of pronounced calcitonin adsorption was observed at high solution equilibration concentrations exceeding about 1 mg mL−1, suggesting that the overall adsorption cannot be described by simple Langmuir-type adsorption. For the MSNpe, a completely different shape of the adsorption isotherm was observed, with only a small portion of calcitonin adsorbing strongly reaching calcitonin loadings of about 2 wt%, after which a close-to-linear increase in the amount of calcitonin with increasing solution concentration was observed at solution equilibrium concentrations exceeding 0.15 mg mL−1. This adsorption behavior was similar to that observed for the non-porous silica nanoparticles, with the exception that there was no initial adsorption of calcitonin at low solution concentrations. No plateau was observed even at the highest peptide concentrations studied (starting peptide concentration of 2 mg mL−1, equilibrium peptide concentration of ∼0.75 mg mL−1). Under the investigated conditions, all particles showed a maximum adsorption capacity of ∼20–25 wt%, but the different shapes of the adsorption isotherms suggest a strongly particle-dependent peptide adsorption mechanism.
In order to obtain more information about possible concentration-dependent peptide aggregation in solution, SDS-PAGE measurements were performed at relevant concentrations. The results obtained for salmon calcitonin in PBS are shown in Fig. 4. A single band centered at 6.8 kDa corresponding to dimers of calcitonin is observed up to a concentration of 0.25 mg mL−1, after which additional bands corresponding to trimers and tetramers are observed, which become more pronounced and show higher masses with increasing peptide concentration.
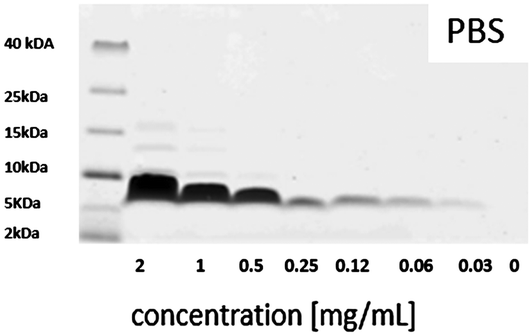 | ||
| Fig. 4 SDS-PAGE of different salmon calcitonin dilutions in PBS with a pH of 7.4 after staining with Coomassie Brilliant Blue. | ||
The formation of trimers and tetramers in solution corresponds roughly to the transition towards a linear increase in adsorption of the peptide with increasing concentration in the adsorption isotherms, if the starting peptide concentrations are taken as a measure. This could suggest that the change in adsorption behavior observed towards a linear concentration behavior could be connected to the presence of potentially kinetically stable trimers and tetramers in solution which show a higher affinity for the outer silica surfaces or the pore walls if the mesopore diameters are larger than those of the dimeric units. The strong adsorption at low peptide concentrations observed for the MSNc but not to the same extent for the other particles having either larger mesopores or no intra-particulate pores could then be explained by the possibility for multivalent interactions with the silica pore walls in the case of the dimers if the mesopore size corresponds roughly to the size of the peptide dimers, which was estimated to be about 2.5 nm.17 This is a much more relevant measure than the hydrodynamic radius of the peptide dimers, as the dimer is anisotropic, and thus also the length of the aggregate will influence the value of the hydrodynamic diameter as it is approximated to be a sphere in the calculations. We note, however, that we do not have evidence for the presence of dimers in the pores, and thus cannot exclude that even adsorption as monomers could be active for this small-pore silica. However, clearly, peptide adsorption is highly sensitive both to the mesopore size and to the degree of aggregation of the peptide in solution.
In order to obtain more information about the location of the peptide, we performed ζ-potential measurements as a function of peptide concentration. If the peptide is preferentially located inside the particles or if the positive peptide charges are fully neutralized by the negative surface charges of the particles, a smaller change in ζ-potential towards more positive values is to be expected. The results are shown in Fig. 5 and the data is plotted against the peptide loading degree.
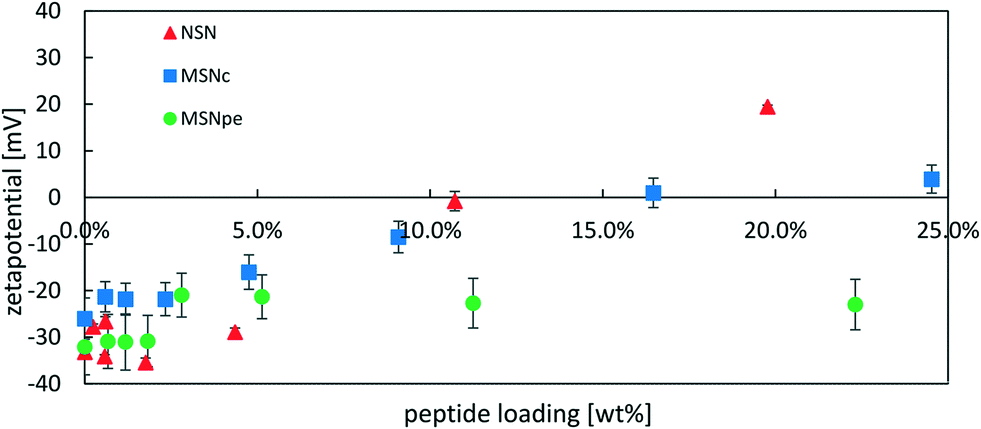 | ||
| Fig. 5 Zeta-potential of NSN (red ▲), MSNc (blue ■) and MSNpe (green ●) after loading with different amounts of salmon calcitonin measured at a particle concentration of 0.1 mg mL−1 in 1 mM KCl solution. | ||
The ζ-potential of NSN remained negative at approximately −30 mV at loading degrees up to 4 wt% after which an almost linear change in the ζ-potential values in the direction of more positive values was observed and the ζ-potential was strongly positive at the highest loading level. For the MSNpe, a slight initial change in the ζ-potential values was observed at very low peptide loadings, after which a virtually constant negative ζ-potential value was recorded regardless of the loading level. The MSNc showed intermediate behavior, with virtually constant negative ζ-potential values up to a loading degree of about 4 wt% after which a steady change in the direction of more positive values was observed with increasing peptide loading, but the change in the ζ-potential with increasing peptide loading was clearly lower than that observed for the NSN. The results seem to indicate that the absolute majority of the calcitonin is adsorbed inside the pore system of the MSNpe, while some peptides are adsorbed on the outer surface of the MSNc and even more so naturally for the NSN lacking mesoporosity. As shown below, the zeta-potentials recorded at low calcitonin loadings are insensitive to the location of calcitonin due to some fast release of calcitonin from MSNc and NSN at low loading levels, which is why the zeta-potential values recorded for such particles closely resemble those of the native silica particles. Finally, we also want to emphasize that no sedimentation of peptide–particle aggregates was observed, which excludes any possibility of particle aggregation-induced effects on the results.
Peptide release was studied in simulated blood plasma to mimic physiological conditions, i.e. at a higher ionic strength as compared to the conditions used for peptide loading. Thus, one would expect that the increased salt concentration would lead to a decrease in the affinity of calcitonin for the silica surface if the interaction is mainly electrostatic in nature. The results are summarized in Fig. 6.
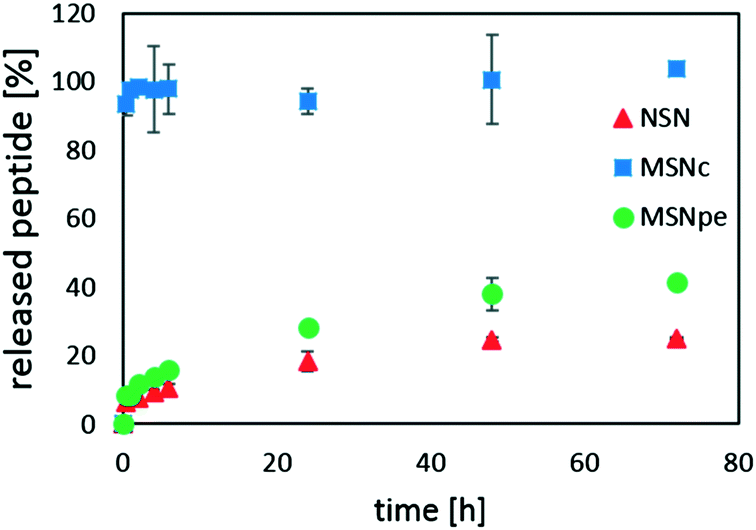 | ||
| Fig. 6 Release kinetics of salmon calcitonin from NSN (red ▲, initial loading = 23.1 wt%), MSNc (blue ■, initial loading = 24.4 wt%) and MSNpe (green ●, initial loading = 23.2 wt%). The experiment was carried out in simulated blood plasma, at 37 °C and at a particle concentration of 0.5 mg mL−1. | ||
For the MSNc, very rapid burst release of almost all of the peptide was observed. NSN and MSNpe released the calcitonin with an initial burst release of about 5% of the adsorbed peptide after which slow release of clearly less than half of the peptide was observed within 48 h. Thus, our results suggest that the contribution of the initial burst release and also the extent of peptide release are inversely proportional to the mesopore diameter for the mesoporous particles, and that the release of calcitonin from the MSNpe resembles that from the NSN.
In order to ensure that the difference in the release behavior was not related to peptide degradation influences, MALDI-TOF spectra were taken after a release time of 1 h in all cases. For these experiments, the particle concentration was increased to 5 mg mL−1 in order to obtain good quality spectra. In all cases, the MALDI-TOF spectra measured for the released peptide were identical to those of the starting peptide (see ESI† 3), suggesting that peptide degradation did not occur in any of the studied cases.
4. Discussion
The strong dependence of mesopore size and the degree of peptide aggregation in solution on the calcitonin adsorption and release behavior is interesting and suggests that the adsorption is controlled by both parameters. If the mesopore size is in the range of the size of the peptide aggregates, in this case, there is a good match between the size of the calcitonin dimers and the mesopore size of the MSNc, there is a possibility for more peptide–silica interactions in confinement, and there is strong adsorption of the peptide into the mesoporous system of the particles. On the other hand, if the mesopores are much larger than the size of the calcitonin aggregates, like in the case of MSNpe, the total attractive electrostatic silica-peptide interactions are smaller in magnitude, and larger peptide aggregates are needed in this case in order to enhance adsorption. Here, the adsorption process is similar to that observed for a non-porous particle and suggests that confinement effects are not dominating in this case. For the MSNc, a similar increase in the adsorbed amount was observed at higher calcitonin concentrations, but the calcitonin concentrations needed for the second adsorption step were clearly higher than those observed for the MSNpe and NSN. This is not surprising, as the MSNc were already highly loaded with calcitonin at this stage, and thus the surface chemistry of these particles were radically different as compared to the MSNpe and NSN. Interestingly, the release kinetics was highly dependent on the type of aggregate adsorbing to the silica surface; the larger the aggregates, the slower the release of the peptide. This may sound counterintuitive, as the strongest adsorption of calcitonin was observed for dimer adsorption onto MSNc, although here the fastest release kinetics was also recorded. This may be a kinetic effect related to a kinetic barrier for disintegration of larger adsorbed peptide aggregates in addition to the evidently high total adsorption energy of larger aggregates. It is known that peptide adsorption can occur as formation of mono- or multilayers, depending on the relative contributions of peptide–surface as well as peptide–peptide interactions. Monolayer adsorption is typically observed when attractive peptide–peptide interactions are weak, while peptide–surface interactions are strong enough to promote adsorption on the surface.18,19 Multilayers are formed when attractive peptide–peptide interactions are strong enough to form aggregates on the surface.20,21 In our case, we cannot fully distinguish between a cooperative adsorption process being active for the MSNpe and NSN, where the adsorption would be driven by the initial low-energy adsorption of smaller peptide aggregates triggering subsequent formation of larger calcitonin aggregates on the surface and direct adsorption of preformed larger peptide aggregates in solution. However, whatever the exact reason is for the observations, it is highly interesting that the release kinetics can be tuned simply through variation of the mesopore size. This provides a simple means for optimization of mesoporous silica nanoparticles for a given application involving macromolecular therapeutic agents, in which a fast or a slow release profile could be optimum depending on the application. Current research in our laboratory is focused on determining to which extent these conclusions are general in nature and to which extent peptide-specific contributions play a role.5. Conclusions and outlook
The present study shows that the adsorption and release kinetics of salmon calcitonin is strongly influenced by the pore size of the carrier particles. If there is a close match between the size of the peptide and the mesopore diameter of the silica, confinement effects can lead not only to very strong adsorption of the peptide, but also to fast release kinetics. Peptide aggregation, either on the surface of the silica or in solution, can lead not only to enhanced adsorption, but also to slower peptide release. Thus, understanding the interplay between peptide self-aggregation and adsorption seems to be key to developing carrier systems based on mesoporous silica nanoparticles where the release kinetics can be easily tuned. However, as enzymatic degradation of the peptide can be supposed to be faster if the mesopore diameter is much larger than the peptide even if the peptide is adsorbed inside the carrier, future studies will focus also on this aspect.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Cornelia Egger for nitrogen sorption measurements, Markus Wunderlin for MALDI-TOF measurements, the Institute of Electron Microscopy for microtome sample preparation, Dario Stanic for peptide adsorption experiments, and Margit Lang for ICP-OES analysis. The research was funded by the European Union's Seventh Framework Programme (FP7/2007-2013) under Grant Agreement No. 604182, FORMAMP Innovative Nanoformulation of Antimicrobial Peptides to Treat Bacterial Infectious Diseases (http://ec.europa.eu.research).Notes and references
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- R. E. W. Hancock and H. G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315–329 CrossRef CAS PubMed.
- A. Schmidtchen, M. Pasupuleti, M. Mörgelin, M. Davoudi, J. Alenfall, A. Chalupka and M. Malmsten, J. Biol. Chem., 2009, 284, 17584–17594 CrossRef CAS PubMed.
- B. C. VanderVen, R. J. Fahey, W. Lee, Y. Liu, R. B. Abramovitch, C. Memmott, A. M. Crowe, L. D. Eltis, E. Perola, D. D. Deininger, T. Wang, C. P. Locher and D. G. Russell, PLoS Pathog., 2015, 11(2), e1004679 Search PubMed.
- K. Braun, A. Pochert, M. Lindén, M. Davoudi, A. Schmidtchen, R. Nordström and M. Malmsten, J. Colloid Interface Sci., 2016, 475, 161–170 CrossRef CAS PubMed.
- E. T. Maggio, J. Excipients Food Chem., 2010, 1, 40–49 CAS.
- Z. Ma, J. Bai, Y. Wang and X. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 2431–2438 CAS.
- J. Deere, E. Magner, J. G. Wall and B. K. Hodnett, J. Phys. Chem. B, 2002, 106, 7340–7347 CrossRef CAS.
- J. Y. Reginster, A. Albert and P. Franchimont, Calcif. Tissue Int., 1985, 37, 577–580 CrossRef CAS PubMed.
- G. Thamsborg, T. L. Storm, E. Sykulski-R, H. Brinch, K. Nielsen and O. H. Sorensen, Calcif. Tissue Int., 1991, 48(5), 302–307 CrossRef CAS PubMed.
- G. C. Wall and C. A. Heyneman, Ann. Pharmacother., 1999, 33, 499–501 CrossRef CAS PubMed.
- R. Sato-Berrú, J. M. Saniger, J. Flores-Flores and M. Sanchez-Espíndola, J. Mater. Sci. Eng. A, 2013, 3, 237–242 Search PubMed.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, ACS Nano, 2009, 3, 197–206 CrossRef CAS PubMed.
- M. R. C. Marques, R. Loebenberg and M. Almukainzi, Dissolution Technol., 2011, 15–28 CrossRef CAS.
- A. Sridharan and K. Prakash, Geotech. Test. J., 2001, 24, 330–333 CrossRef.
- Calculated using the database http://www.rcsb.org/pdb/home/home.do based on data in G. Andreotti and A. Motta, J. Biol. Chem., 2004, 279, 6364–6370 CrossRef CAS PubMed.
- P. Facci, D. Alliata, L. Andolfi, B. Schnyder and R. Kötz, Surf. Sci., 2002, 504, 282–292 CrossRef CAS.
- T. J. Su, J. R. Lu, R. K. Thomas and Z. F. Cui, J. Phys. Chem. B, 1999, 103, 3727–3736 CrossRef CAS.
- A. N. Asanov, L. J. Delucas, P. B. Oldham and W. W. Wilson, J. Colloid Interface Sci., 1997, 196, 62–73 CrossRef CAS PubMed.
- F. Höök, M. Rodahl and B. Kasemo, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12271–12276 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00059f |
| This journal is © The Royal Society of Chemistry 2017 |