
Design of SERS nanotags for multiplexed lateral flow immunoassays†
Maria
Sánchez-Purrà
 a,
Biel
Roig-Solvas
b,
Alice
Versiani
cd,
Cristina
Rodriguez-Quijada
a,
Helena
de Puig
e,
Irene
Bosch
dg,
Lee
Gehrke
df and
Kimberly
Hamad-Schifferli
*ae
a,
Biel
Roig-Solvas
b,
Alice
Versiani
cd,
Cristina
Rodriguez-Quijada
a,
Helena
de Puig
e,
Irene
Bosch
dg,
Lee
Gehrke
df and
Kimberly
Hamad-Schifferli
*ae
aDepartment of Engineering, University of Massachusetts Boston, Boston, MA, USA. E-mail: kim.hamad@umb.edu
bDepartment of Electrical and Computer Engineering, Northeastern University, Boston, MA, USA
cDepartment of Microbiology, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil
dInstitute of Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA, USA
eDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
fDepartment of Microbiology and Immunology, Harvard Medical School, Boston, MA, USA
gDepartment of Medicine, Mount Sinai School of Medicine, New York, NY 10029, USA
First published on 27th September 2017
Abstract
Surface enhanced Raman spectroscopy (SERS) has been attractive for enhancing the sensitivity of lateral flow immunoassays (LFA). A format that has enabled specific detection of biomarkers is to use Raman reporter molecules linked to gold nanoparticles (NPs), which are conjugated to laboratory made antibodies specific for the target of interest. Many factors such as the NP and Ab properties and the method of signal readout impact the sensitivity of a SERS based immunoassay. To understand how to optimize assay sensitivity, we studied SERS readouts of multiplexed sandwich immunoassays for the zika and dengue non-structural protein 1 (NS1) biomarkers as a test case. We investigated the effect of NP shape on the SERS enhancement of the reporter molecules 1,2-bis(4-pyridyl)ethylene (BPE) and 4-mercaptobenzoic acid (MBA). We also performed SERS imaging of test lines to map the spatial distribution of signal in test lines on the nitrocellulose. Finally, we used a modified least squares analysis to differentiate reporter contributions.
Design, System, ApplicationPaper-based immunoassays have been attractive for point of care rapid diagnostics for infectious disease. However, their sensitivity can be limited as they are typically read visually via gold nanoparticles attached to the capture antibody. One exciting route for improving their sensitivity that has gained much interest is to use surface enhanced Raman spectroscopy (SERS) by exploiting the gold nanoparticle in the immunoassay as a SERS “nanotag.” However, using SERS nanotags for immunoassays poses unique challenges. While optimization of visually read immunoassays is well understood, equivalent information does not yet exist for SERS, as its use in immunoassays is still emerging. For example, nanoparticle shape can strongly influence SERS signal enhancement, so how it impacts immunoassay sensitivity needs to be explored. Paper-based immunoassays have migrating samples, and thus binding is not at equilibrium, resulting in non-uniform signal distribution. Additionally, a powerful implementation of paper-based immunoassays is a multiplexed format that can provide a differential diagnosis, which requires multiple reporter molecules. However, differentiation of SERS signals for multiple reporter molecules needs to be robust. These results could help scientists and engineers optimally leverage the advantages of SERS for better point of care devices for diagnosing and detecting viral disease biomarkers. |
Introduction
Surface enhanced Raman spectroscopy (SERS) is an attractive technique because of its high sensitivity in detecting analytes. The enhancement of the Raman spectrum of an analyte when it is in proximity to a roughened or nanoscale metallic surface can reach as high as 10,9 which has led to detection of molecules down to attomole levels. Because of this, there has been great interest in engineering gold and silver structures to detect analytes by SERS.1However, detection of biological species is not as straightforward with SERS compared to other applications, as proteins have complex vibrational spectra compared to small molecules, so defining a unique fingerprint of a protein is difficult. One solution to this problem is a format of SERS that specifically recognizes the target of interest by utilizing antibodies, peptides, or DNA conjugated to a gold nanoparticle with a reporter molecule that is detected by SERS, or a “nanotag.” The reporter can be a small molecule with a narrow Raman spectrum, facilitating detection. SERS nanotags2,3 have been used successfully for biological imaging in cells. In addition, by using multiple reporters, it can enable multicolour SERS imaging.4
One promising area for SERS nanotags is in sandwich immunoassays, which are used for biomarker sensing in paper-based point of care diagnostics for infectious disease.5,6 Here, a target antigen is detected at the test line, which has immobilized antibodies specific for the antigen. In addition, an antibody tagged with a nanoparticle binds to the target, resulting in the formation of a sandwich (Fig. 1). Thus, antigen can be detected by the presence of the nanoparticle at the test line, which results in a red band due to the gold nanoparticles.7
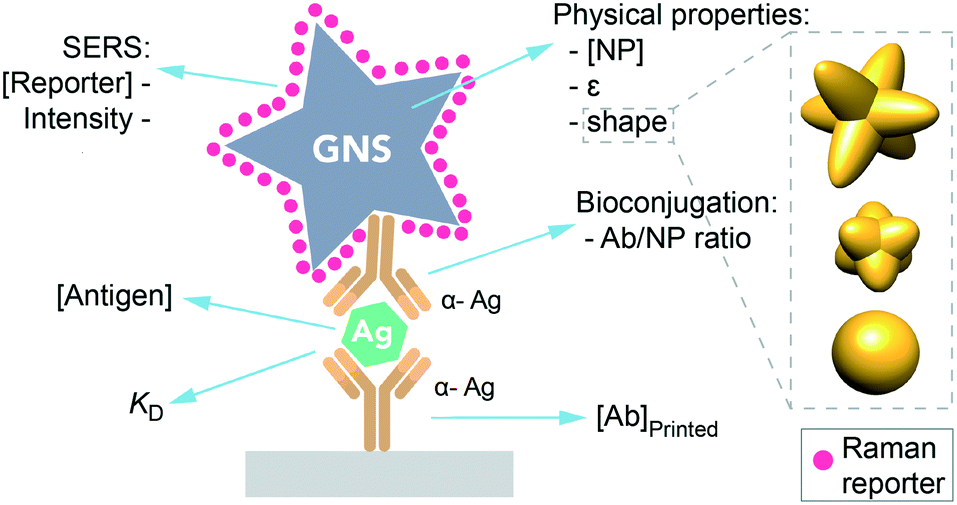 | ||
| Fig. 1 Scheme of a sandwich immunoassay in a SERS-based LFA. Factors influencing the immunoassay sensitivity. | ||
For a readout using SERS, the gold nanoparticle conjugated to the antibody is tagged with a small Raman reporter molecule, and thus the nanoparticle-reporter-antibody can act as a nanotag, exhibiting the characteristic spectrum of the reporter when shining a 785 nm-laser on the test line.5,6 Using SERS to detect binding at the test line instead of a visual readout exploits the high sensitivity of SERS, which can be improved by 1000 fold compared to ELISA.6 For zika, increasing sensitivity is critical, as NS1 levels are estimated to be very low based on initial reports of low viremia,8 and expected to be below the typical LFA limit of detection when read out by eye. Thus, a SERS readout could also aid in detecting co-infections of zika and dengue and other flavivirus, where viral NS1 levels are lower than in single infections,8 in early and late stages of the diseases, when NS1 concentration drops down some days after the onset of symptoms, or on secondary infections.9,10
In addition, using SERS in a multiplexed format can be used for a multiplexed biomarker assay, where multiple nanotags can be used to recognize different biomarkers of multiple diseases. This is especially interesting for flavivirus co-infections because a single patient can be co-infected with both zika and dengue as they share the same transmission vector.11 A multiplexed platform could allow the detection of both infections, reducing the required amount of sample.
However, using SERS nanotags for paper-based immunoassays poses unique challenges. While there is an abundance of information on how to optimize LFAs and dipstick assays that use a visual readout, equivalent information does not exist for SERS, as its use in LFAs is still emerging. For instance, what are the optimal nanotag or immobilized Ab properties for SERS in immunoassays? LFA readout when using SERS depends on multiple factors in addition to those typical of LFA, such as the NP physical properties (shape, extinction coefficient), the reporter's concentration and its inherent Raman intensity (Fig. 1), having an impact on the final test intensity and thus on the analyte detection.
There have been many efforts to engineer lithographically patterned plasmonic shapes for SERS hot spots12–15 but these strategies are not applicable for soluble nanoparticles. In addition, because SERS has a high sensitivity, background signal can become a problem, as it can obscure the analyte spectra when present at very low concentrations. Paper-based immunoassays also pose unique challenges as the sample is migratory and binding is not at equilibrium, resulting in non-uniform signal distribution in the strips. Finally, the most powerful implementation of LFAs is a multiplexed POC assay16 that can provide a differential diagnosis, and which would require multiple reporter molecules. However, differentiation of signals as the number of reporter molecules increases needs to be robust.
These issues need to be understood for optimal utilization of SERS for LFAs. Here, we investigate different parameters influencing the behavior of a SERS dipstick immunoassay for multiplexed detection of biomarkers, demonstrated by detection of zika and dengue antigens. We synthesized NPs with different shapes (spherical NPs and star-shaped particles with two different arm lengths), and compared their behavior in SERS immunoassays. We also examined the test line by SERS mapping, and developed algorithms to optimally separate signal in a multiplexed test. From the results, we determine a set of guidelines for optimal implementation of SERS in paper-based immunoassays.
Results and discussion
Effect of GNP shape on SERS enhancement
The detection of the biomarker is read out by the Raman signal of the reporter molecule. Because of this, the sensitivity of the assay is impacted by how much the signal is enhanced by the NP. Consequently, we examined the effect of NP shape on SERS enhancement. First, gold nanoparticles (GNPs) of three different morphologies were synthesized: gold nanostars with long spikes (GNS900), nanostars with short spikes (GNS400) and gold nanospheres (GNSph). Gold Nanostars (GNS) were synthesized in HEPES buffer as previously described,17–19 and gold nanospheres were synthesized by reducing chloroauric acid (HAuCl4) with sodium citrate, as described by Frens.20 Reporter molecules 1,2-bis(4-pyridyl)ethylene (BPE) and 4-mercaptobenzoic acid (MBA) were either adsorbed onto the GNS NPs (BPE) or chemically conjugated via the thiol (MBA). BPE was used as a reporter to detect zika, thus it was used for GNPs conjugated to anti-zika antibodies, resulting in GNP–BPE-anti-zika or “Z-nanotags.” MBA was used as a reporter to detect dengue, and attached to GNPs conjugated to anti-dengue antibodies, yielding GNP–MBA-anti-dengue or “D-nanotags.”Dynamic light scattering showed that the hydrodynamic diameter (DH) was higher for nanostars with longer spikes (GNS900), and decreased for nanostars with shorter arms (GNS400) and GNSph, a trend that was observed for both BPE (Fig. 2a) and MBA (Fig. S1a†). Zeta potential (ζ) (Fig. 2b, S1b†) of the particles were all negative. GNPs exhibited shape-dependent optical properties. The nanostar SPR maxima red-shifted with increasing HEPES concentration,17 with the peak at 722 and 589 nm for GNS900 and GNS400, respectively, for BPE samples (Fig. 2f), and 735 and 592 nm for GNS900 and GNS400, respectively, for MBA samples (Fig. S1f†). GNSph showed a lower SPR maximum, at 531 for both BPE and MBA (Fig. 2f, S1f†).
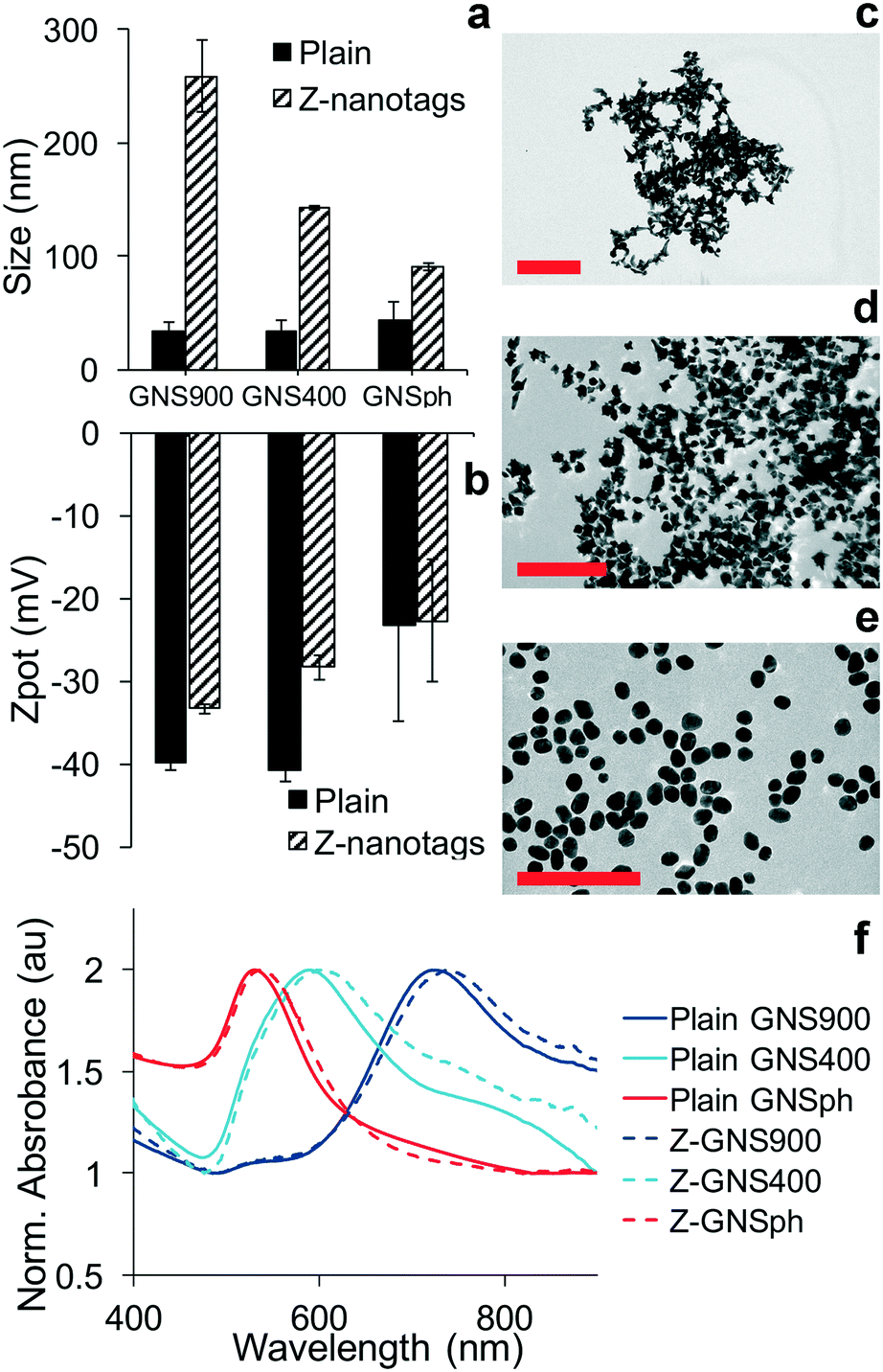 | ||
| Fig. 2 GNP characterization of Z-nanotags. a) Hydrodynamic size of GNS900, GNS400 and GNSph. b) Zeta potential of GNS900, GNS400 and GNSph. TEM images of c) GNS900, d) GNS400, and e) GNSph, scale bar = 200 nm. f) UV-vis spectra of plain GNPs (solid line) and Ab-conjugated GNPs (dashed line). | ||
After antibody conjugation, the SPR peak red shifted for all samples due to changes in the local refractive index upon bioconjugation to the GNS.21 The shift observed was of 20, 9 and 7 nm for Z-nanotags of GNS900, GNS400 and GNSph, and 7, 6 and 7 nm for D-nanotags of GNS900, GNS400 and GNSph. These differences are most likely due to the fact that the particles had varying antibody coverages. Zeta potential changed upon bioconjugation for all samples, showing lower values for most of the samples, and values depended on their antibody coverage (Fig. S2†) and PEG backfill.
GNP morphology of the conjugated samples was characterized by TEM, which showed star-shaped gold cores of ∼51.4 ± 8.8 nm for Z900-nanotags (Fig. 2c), 31.8 ± 4.5 nm (Fig. 2d) for Z400-nanotags, 55 ± 7.5 nm for D900-nanotags (Fig. S1c†) and 28.3 ± 4 nm for D400-nanotags (Fig. 2d). GNSph showed round-shaped cores of 27.9 ± 4.7 nm for ZSph-nanotags (Fig. 2e) and 26.8 ± 4.6 nm for DSph-nanotags (Fig. S1e†). GNS900 had sharper spines compared to GNS400, confirming an expected morphology difference resulting from the synthesis conditions.
It has been observed that GNP geometry can influence the Raman signal enhancement of reporter molecules in solution22 and in cancer imaging.2,23 Therefore, SERS enhancement of the different nanoparticle shapes were measured in the sandwich immunoassays. Raman spectral intensities of the plain reporter molecules were compared to their respective SERS intensities with 785 nm excitation (Fig. 3).
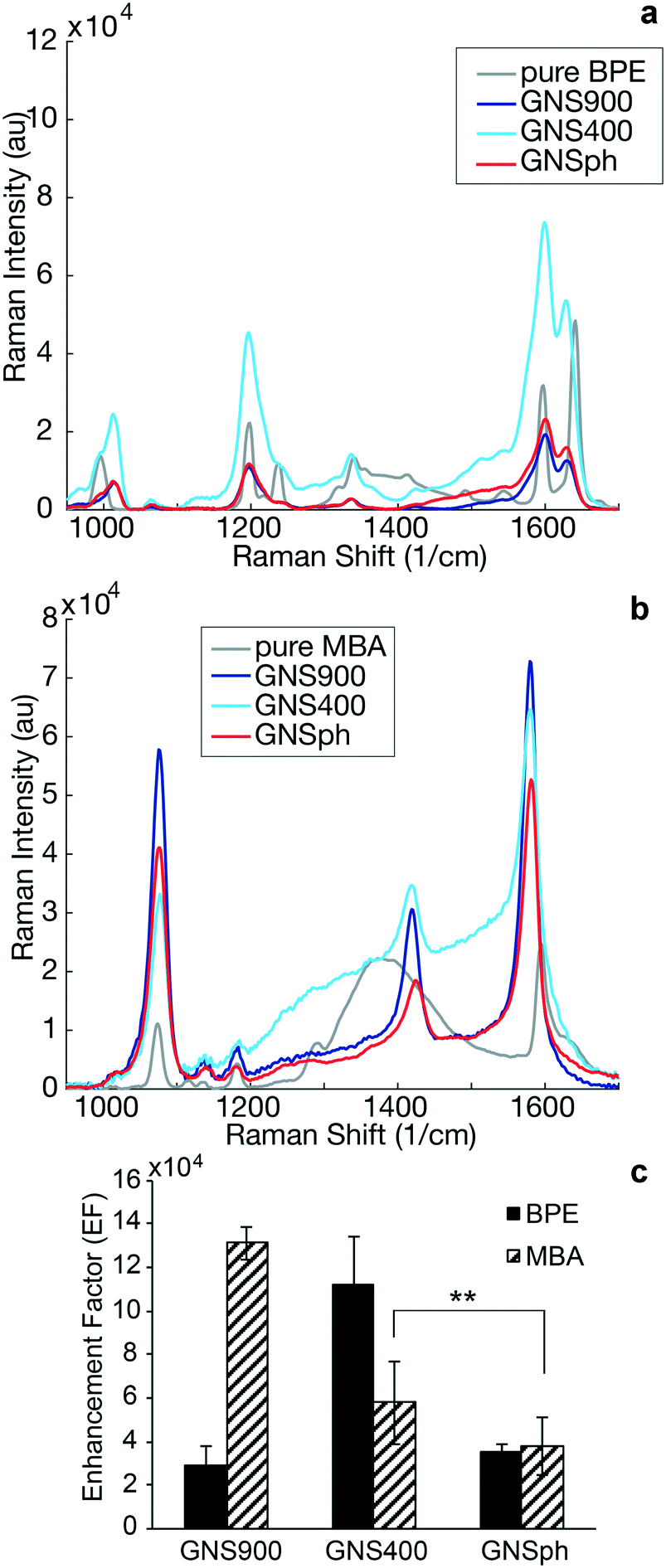 | ||
| Fig. 3 SERS enhancement by nanospheres and nanostars. a) Raman spectrum of pure BPE (gray) and SERS spectra of BPE-encoded samples: GNS900 (dark blue), GNS400 (light blue) and GNSph (red). b) Raman spectrum of pure MBA (gray) and SERS spectra of MBA-encoded samples: GNS900 (dark blue), GNS400 (light blue) and GNSph (red). c) Enhancement factor for GNS900, GNS400 and GNSph encoding BPE (solid) and MBA (hashed), error bars represent mean ± SD, n = 25, **p < 0.01, two-sample Student's t-test. | ||
SERS enhancement factors (EF) were calculated using the highest intensity peaks characteristic of each reporter. In the case of BPE, the C–C/N stretch at 1609 cm−1 was used,24,25 whereas for MBA, it was the ν8a vibrational mode of phenyl ring-stretching motion at 1584 cm−1.26 The EF was calculated using standard equations (ESI,† eqn (1))13,27 assuming monolayer coverage of the reporter on the nanoparticle surfaces. Calculated EFs of GNP–BPE (Fig. 3c) varied from 104–106, which are within the range for similar reporter molecules on GNPs found in the literature.14,28,29 GNS900 EF values with both BPE and MBA, were 2.95 × 104 and 1.31 × 105, respectively, while GNS400 had values of 1.12 × 105 and 5.79 × 104. GNSph EF values were calculated to be 3.55 × 104 and 3.8 × 104. However, EFs were found to vary between batches, and we sometimes encountered EF for GNS900 that were as high as 6.32 × 105 and 8.66 × 105 for BPE and MBA (Fig. S3†), showing that the SERS intensities can be variable depending on measurement conditions. We expected that GNS900 would have the highest EFs due to the smaller radius of curvature of their arms compared to GNS400 and GNSph, which increases the induced electric fields, thus increasing SERS enhancement.30–32 This trend was observed for MBA-encoded GNPs, but did not always occur for BPE samples, where GNS400 showed the highest enhancement for some samples (Fig. 3c).
Because SERS enhancement is due to the high electric fields around curved or roughened surfaces, we quantified the radii of curvature of the GNS900, GNS400 and GNSph by measuring tip curvature of nanostars and nanospheres in TEM images (Fig. 4a).
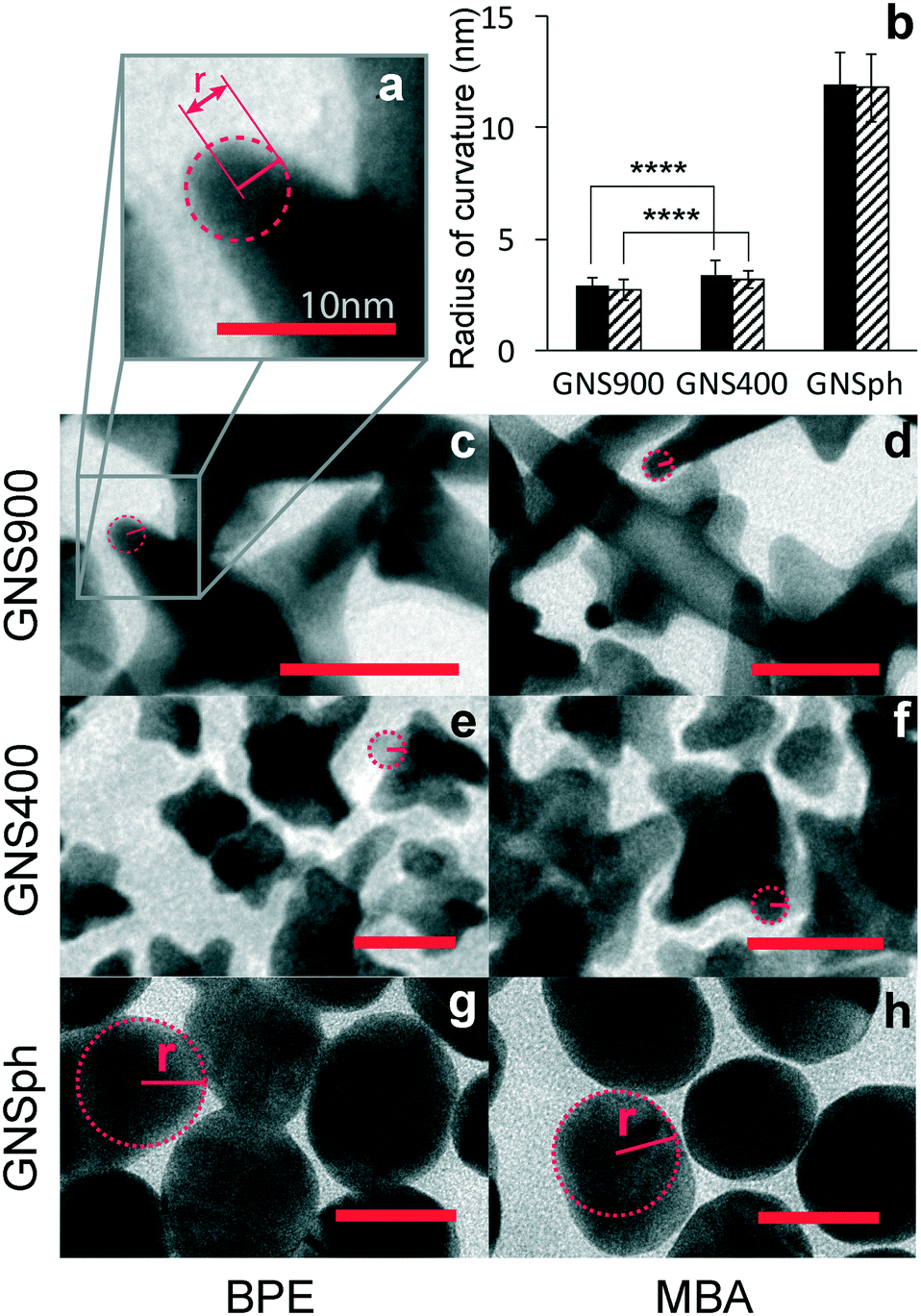 | ||
| Fig. 4 Radii of curvature. a) Scheme of the radius of curvature of a GNS900 tip. b) Calculated radii of curvature, n = 44, ****p < 0.0001, two-sample Student's t-test. TEM images used for the calculation of the radius of curvature of c) GNS900, e) GNS400 and g) GNSph with BPE, and d) GNS900, f) GNS400 and h) GNSph with MBA. Scale bar = 25 nm. | ||
Average radii of curvature for GNS900 were 2.88 ± 0.35 and 2.72 ± 0.45 nm, with BPE and MBA, for GNS400 were 3.41 ± 0.62 and 3.17 ± 0.68 nm, and for GNSph were 11.96 ± 1.38 and 11.8 ± 1.52 nm (Fig. 4b). As expected, the radii of curvature for GNS900 was the smallest of the GNPs tested, followed by GNS400 and GNSph, for both BPE and MBA samples. However, the measured difference between the radii of curvature of the two types of nanostars was found to be insignificant, which could explain the variability of their EFs between them (Fig. 3c). In addition, the reporters on the GNS are most likely distributed all over the GNS surface, where some are not near the particle tips where the enhancement is the strongest. These results show that all the GNPs can enhance the reporter molecule signals, and that GNS with longer spires tend to exhibit higher EFs. For this reason, GNS900 were selected for this SERS-based immunoassays.
SERS signal processing
Although the reporter molecules used in this work were chosen based on the minimal overlap of their principal Raman peaks, both BPE and MBA have non-negligible Raman scattering contributions at the wavelength of the other reporter's main peak. This means that the combined Raman scattering at the main peaks cannot simply be regarded as having a contribution from only one of the reporters. Moreover, the presence of other components in the Raman signal, like that of the glass sample holder or the nitrocellulose support can further alter the Raman intensity on these main peaks.To overcome these problems, we used a broad region of the spectra (from 800 to 1800 cm−1, ∼2000 measured wavelengths) in our Raman measurements. We investigated the use of a non-negative least squares (NNLS) algorithm that separates an input signal into a non-negative linear combination of pure components in an optimal way with respect to the mean squared error.33,34 The pure components used were BPE (Z-nanotag), MBA (D-nanotag), nitrocellulose and glass (Fig. S6†). From now on, ZIKV-NNLS and DENV-NNLS refer to the components of each reporter obtained by the described NNLS analysis.
Effect of GNP shape on spatial distribution of SERS signal
In LFA and dipstick assays, non-uniform test lines can sometimes be observed.35 Typically, the intensity is higher at the bottom edge of the test line band where the fluid containing the NP–Ab conjugate first encounters the immobilized antibody. We investigated the spatial distribution of the SERS signal in the assays and compared it to bright field imaging (i.e., visual). First, tests were run with ZIKV NS1 using the three different GNPs with BPE (Fig. 5a–f).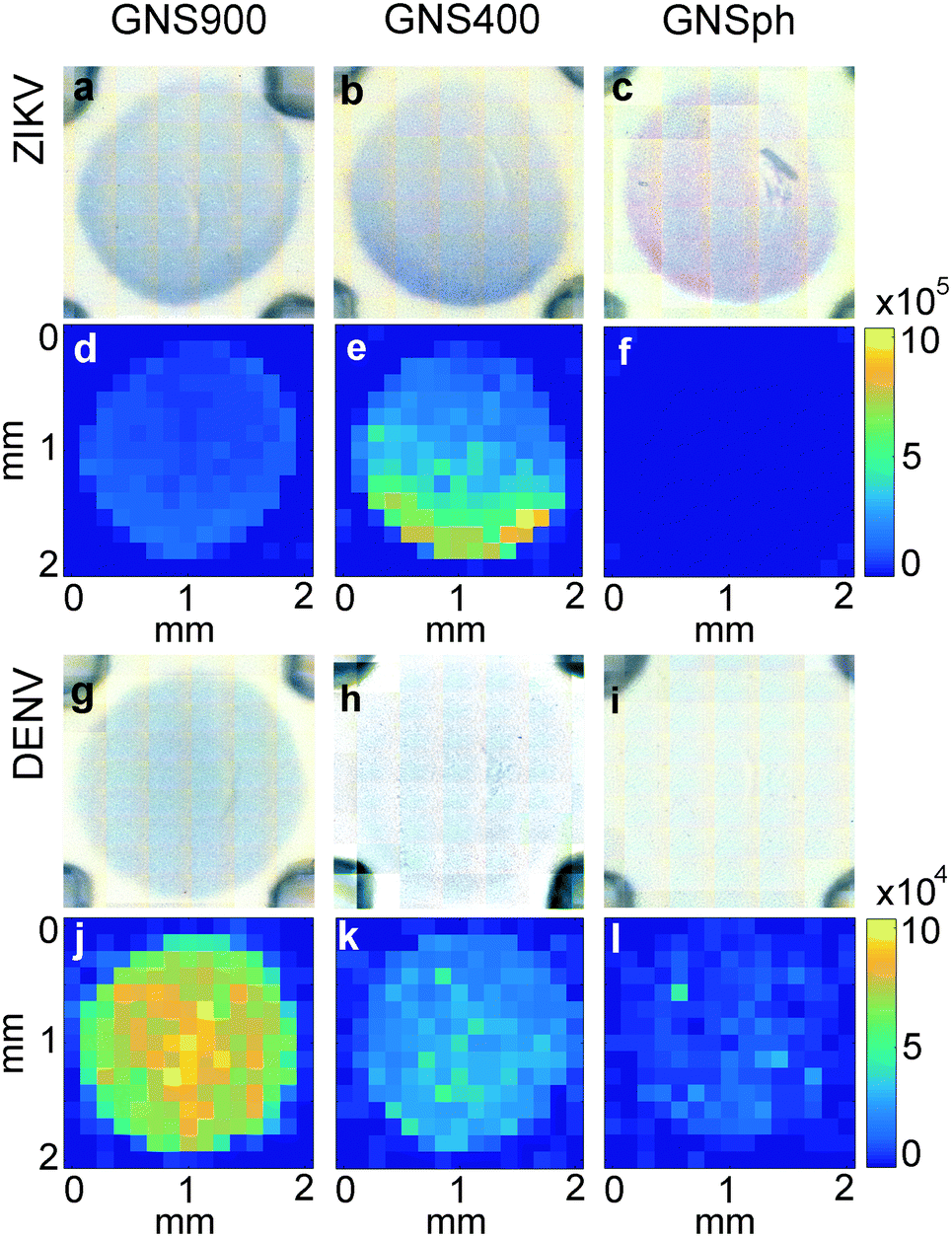 | ||
| Fig. 5 Distribution of signal in sandwich immunoassays. Bright field images of test area for a) GNS900-ZIKV, b) GNS400-ZIKV, c) GNSph-ZIKV, g) GNS900-DENV, h) GNS400-DENV, i) GNSph DENV. SERS mapping of the ZIKV-NNLS component at 1609 cm−1 of d) GNS900-ZIKV, e) GNS400-ZIKV, f) GNSph ZIKV and DENV-NNLS component at 1584 cm−1 of j) GNS900-DENV, k) GNS400-DENV, l) GNSph DENV. Color bars show Raman intensity (au). | ||
Bright field images showed positive tests for all samples (Fig. 5a–c). SERS mapping of ZIKV test lines when run with GNS900 (Fig. 5d) and GNS400 (Fig. 5e) nanotags exhibited images similar to bright field images, with higher GNSs concentration and SERS signal at the lower part of the test line, which is the area where GNS–Ab-antigen complexes first encounter the immobilized Ab. We observed that the signal intensity varied across the same spot for GNS900 ZIKV by a factor of 4 and 2.4 in bright field (Fig. S5a†) and SERS image (Fig. S5c†), respectively. Profile plots for all samples are shown in ESI† (Fig. S5). GNS400 (Fig. 5e) exhibited higher intensity than GNS900 (Fig. 5d), which is in good agreement with EFs estimated in Fig. 3c. On the contrary, SERS mapping of GNSph ZIKV did not exhibit a significant Raman signal (Fig. 5f), despite the fact that their bright field image and visual readout showed a positive test. This discrepancy between readouts suggested that SERS enhancement by GNSph was not high enough to show signal on the image (Fig. 3c).
Then, tests were run with DENV NS1 using the three different GNPs with MBA (Fig. 5g–l). Both GNS900 (Fig. 5g) and GNS400 (Fig. 5h) showed positive tests, although intensity of the GNS400 assay was much lower, probably due to a lower Ab coverage on those particles. This was also the case of GNSph, which showed a negative test in the bright field (Fig. 5i). MBA-encoded GNS exhibited similar signal distributions in the test line for both bright field and SERS (Fig. 5j and k) images compared to BPE samples. However, overall signal for GNS900 was higher than for GNS400, which correlates with the EFs shown in Fig. 3c. Visually, GNSph DENV did not provide a bright field signal (Fig. 5i), but did exhibit SERS signal with a spatial distribution fairly uniform across the test area (Fig. 5l). It should be of note that the SERS spectrum of 4-MBA is pH dependent, so variations in pH across the strip could also contribute to the intensity variation.
In order to investigate the non-uniformity of the signal observed in ZIKV samples (Fig. 5a–c), immobilized Ab was line-dispensed on the nitrocellulose using an automated dispenser system so that the entire eluting front, and thus all nanotags, would reach the Ab area at the same time. The resulting signal from ZIKV samples and compared spotted and line-dispensed Ab by bright field and SERS mapping for the same sample, GNS400-Ab with ZIKV. The overall intensity was lower when the Ab was line-dispensed, both in bright field (Fig. 6b) and by SERS (Fig. 6d) mapping for the same concentration of immobilized Ab. However, distribution of the SERS signal was much more homogeneous when it was line-dispensed, and more concentrated towards the center of the test line (Fig. 6d), demonstrating that a horizontal front aids in a more homogeneous Ab sandwich formation and minimizes inhomogeneous signal distribution.
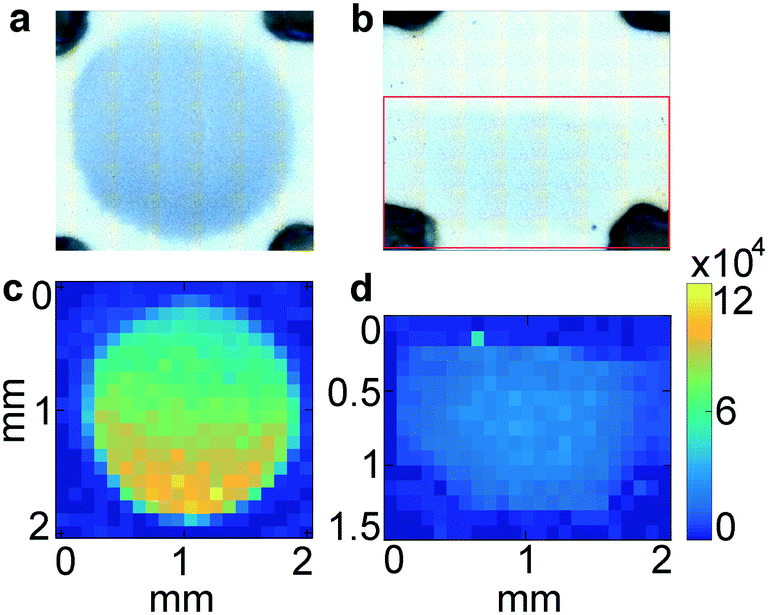 | ||
| Fig. 6 Diffusion of GNP depending on the immobilized Ab area. BPE-encoded GNS400 spotted with micropipette (a) and painted with inject printer (b) at the same immobilized Ab concentration. ZIKV-NNLS component from the SERS mapping of GNS400-ZIKV for spotted Ab (c) and printed Ab (d). Color bars show Raman intensity (au). | ||
These results show that the spatial distribution of the SERS signal is non-uniform, even when the bright field image appears to be homogeneous. One potential explanation is an interparticle effect, where nanostars in proximity with each other can further increase reporter signal. This can have important consequences on quantifying the amount of antigen, as signal varies with location. In addition, it suggests that the highest signal is at the front edge of the spot, and that the back edge of the spot does not contribute significantly to the signal, so that judicious choice of test line spotting can result in economic use of the immobilized antibody.
SERS mapping in multiplexed tests
Multiplexed LFAs and dipstick immunoassays provide differential diagnosis by using multiple lanes, or spatial separation of test lines on a given strip.16 However, multiplexing within a single test line would enable increased miniaturization, which could minimize the sample volume required for the assay. In fact, multiplexed LFA has been achieved using multiple colors of nanoparticles when they are co-localized at the same test line,36,37 where RGB analysis of the test line is critical for signal differentiation. Because the signal is the spectra of the individual reporter molecules, the ability to distinguish between different spectra is key for multiplexed detection. Thus, we investigated the ability to differentiate between the BPE vs. MBA SERS signals in a multiplexed test by using the Z-nanotags and D-nanotags together.To test the accuracy of the SERS-based immunoassay, two different multiplexed experiments were performed. Anti-ZIKV and anti-DENV antibodies were mixed and immobilized onto the same test line. Strips were run with a mixture of ZIKV and DENV NS1 and both nanotags.
First, the NS1 concentration was fixed at 500 ng ml−1 and 5000 ng ml−1, for ZIKV and DENV respectively, while the Z-nanotag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D-nanotag ratio was varied from 1:0 to 0:1 (Fig. 7a–e). Bright field images showed a uniform gray spot, indicating that nanotags were distributed over the entire test area (Fig. 7a–e). NNLS algorithm was used to differentiate the nanotag signals in SERS mapping images. The test area had only BPE signal (blue) for the Z-nanotag:D-nanotag ratio of 1:0 (Fig. 7f), which turned purple as BPE amount decreased and MBA increased (0.8:0.2, 0.5:0.5 and 0.2:0.8) (Fig. 7g–i), and showed only MBA (red) for Z-nanotag:D-nanotag of 0:1 (Fig. 7j), as expected.
:D-nanotag ratio was varied from 1:0 to 0:1 (Fig. 7a–e). Bright field images showed a uniform gray spot, indicating that nanotags were distributed over the entire test area (Fig. 7a–e). NNLS algorithm was used to differentiate the nanotag signals in SERS mapping images. The test area had only BPE signal (blue) for the Z-nanotag:D-nanotag ratio of 1:0 (Fig. 7f), which turned purple as BPE amount decreased and MBA increased (0.8:0.2, 0.5:0.5 and 0.2:0.8) (Fig. 7g–i), and showed only MBA (red) for Z-nanotag:D-nanotag of 0:1 (Fig. 7j), as expected.
 | ||
| Fig. 7 Mapping of test lines in a multiplexed test for fixed ZIKV and DENV NS1 concentration varying Z-nanotag: D-nanotag ratio in a–e) bright field and f–j) SERS. Mapping of assays for varying ZIKV NS1: DENV NS1 ratio at fixed Z- and D-nanotag concentration in k–o) bright field and p–t) SERS mapping. Blue: ZIKV-NNLS component red: DENV-NNLS component. | ||
Then, we investigated whether the nanotag signal would vary when the NS1 concentration was changed (Fig. 7k–t). ZIKV and DENV NS1 concentrations were varied from 1:0 to 0:1 ZIKV:DENV for a fixed Z-nanotag and D-nanotag concentration (0.05 nM). Bright field imaging exhibited gray spots for all ratios, as expected (Fig. 7k–o). The resulting signal in SERS mapping (Fig. 7p–t) also changed from a high BPE contribution (blue) (Fig. 7p) to the signal to a high MBA contribution (red) (Fig. 7t). This shows that the spectral contributions exhibit an expected trend for when the biomarker concentration is varied, though the relative BPE:MBA contribution does not vary.
Quantitative spectral analysis
To estimate the NS1 concentration using the Raman scans, signal intensity based on a Langmuir curve was estimated because GNP intensities have been observed to exhibit Langmuir-like concentration dependences.38,39 Experiments were run at 5 different NS1 concentrations (Fig. S5†). Then, the total reporter contribution in each assay is taken from the Raman scanning measurements (Fig. S6 and S7†), and a Langmuir curve is estimated by relating these contributions with the initial NS1 concentration. It should be of note that the modified Langmuir isotherm is an equilibrium binding model, and thus makes several approximations to describe a binding event in a dipstick flow format.39Afterwards, the total reporter contribution for each reporter in the multiplex assay (Fig. 7p–t) is computed and their apparent NS1 concentration was estimated using the aforementioned Langmuir curve. The agreement between the real concentration of each multiplex assay versus the estimated concentration using this method is shown for each biomarker, in Fig. 8, where ideal concentration measurements would lie in the 1:1 dashed diagonal line.
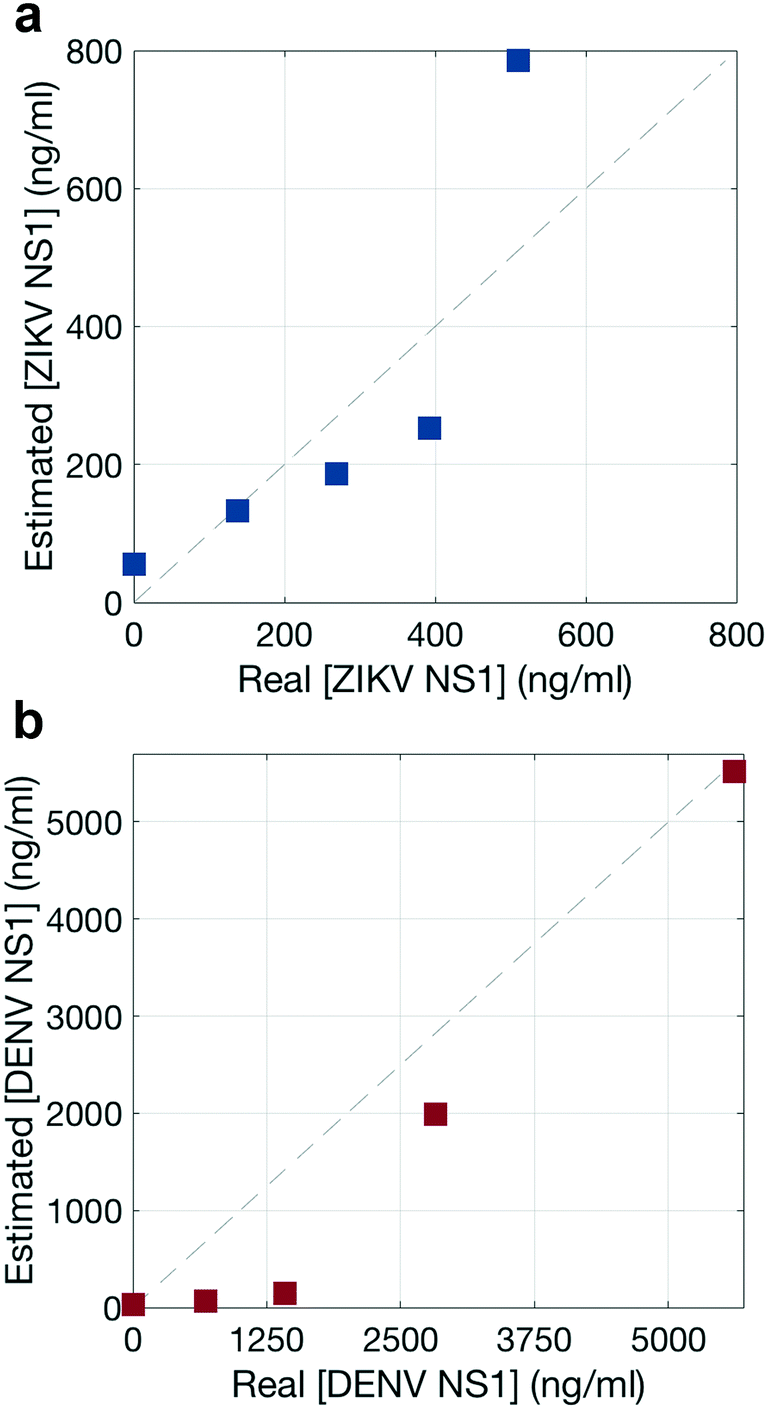 | ||
| Fig. 8 Quantitative spectral analysis. Performance of the NNLS algorithm to estimate the concentration of a) ZIKV NS1 and b) DENV NS1 present in the test line. The dashed line shows the theoretical 100% good prediction of the algorithm. | ||
The NNLS algorithm used to estimate NS1 concentration showed a good performance, with a maximum deviation from the real concentration by a factor of 1.6 for ZIKV NS1 (Fig. 8a) and 9.6 for DENV NS1 (Fig. 8b), and thus was considered acceptable. Nevertheless, this approach is intended to give a false-negative result, indicating whether the virus is present or not, and which one of the viruses is, either ZIKV or DENV. These results indicate that the quantitative contributions of the nanotags to the SERS spectra agree with specific recognition of each biomarker. Thus, quantification of the biomarker using NNLS would allow distinguishing reporters in mixtures with multiple reporters.
Summary and conclusions
In summary, several factors need to be considered for design of nanoparticle–antibody conjugates for SERS detection of sandwich immunoassays. For the nanotag, nanoparticles with sharp tips such as nanostars and other similar morphologies can have higher enhancement factors compared to spherical particles due to the strong electric fields at the tips. This can be used to increase immunoassay sensitivity and potentially enable detection of lower antigen levels and/or earlier detection when antigen concentrations are still low.Secondly, results here illustrate the non-homogeneity of the test line signal, which is often greater compared to bright field/visual imaging. This non-uniform distribution could be due to interparticle enhancement effects, which impacts the SERS intensity distribution much more than that of the bright field intensity. Because the SERS signal is concentrated in the area where the fluid first encounters the immobilized antibody, strategic design of the paper strip or antibody spotting could potentially economize the use of the biological reagents. These factors can be used to decrease strip cost, which is a significant parameter in strip deployability.
Finally, for multiplexed SERS immunoassays, the multiple reporter signals need to be spectrally distinguished if the nanotags are not spatially separated. The method here used, enables the detection and distinction between both viral biomarkers tested. Multiplexing within a test area on a single strip is attractive because it could help decrease the required sample volume for the assay, which is an important factor for patient samples. This is key for when several reporter molecules are used in a single assay have overlapping Raman spectra.
All of these studies show that optimization of the properties of the NP–Ab conjugate and understanding their properties can be used to optimize SERS signal when used in LFAs. These factors can be used to economize the use of antibodies or increase the sensitivity of the assays.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the UMB Engineering Department for use of the Raman microscope. Work was supported by funding from the NIH NIAID (AI100190). We would like to thank Dr. Tom Tague and Dr. Peng Wang from Bruker Optics for technical assistance, and Dr. Justina Tam from the US FDA.HdP was supported by the MIT/SUTD International Design Centre, a Broshy Fellowship and a MIT Tata Center for Technology and Design Fellowship. Both HdP and CRQ were supported by Rafael del Pino Fellowships.
Some of the figure graphics were generated using the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
References
- L. Fabris, J. Opt., 2015, 17, 114002 CrossRef.
- L. Rodríguez-Lorenzo, Z. Krpetic, S. Barbosa, R. A. Alvarez-Puebla, L. M. Liz-Marzán, I. A. Prior and M. Brust, Integr. Biol., 2011, 3, 922–926 RSC.
- J. Xie, Q. Zhang, J. Y. Lee and D. I. C. Wang, ACS Nano, 2008, 2, 2473–2480 CrossRef CAS PubMed.
- M. Vendrell, K. K. Maiti, K. Dhaliwal and Y.-T. Chang, Trends Biotechnol., 2013, 31, 249–257 CrossRef CAS PubMed.
- X. Fu, Z. Cheng, J. Yu, P. Choo, L. Chen and J. Choo, Biosens. Bioelectron., 2016, 78, 530–537 CrossRef CAS PubMed.
- J. Hwang, S. Lee and J. Choo, Nanoscale, 2016, 8, 11418–11425 RSC.
- D. Quesada-González and A. Merkoçi, Biosens. Bioelectron., 2015, 73, 47–63 CrossRef PubMed.
- J. J. Waggoner, L. Gresh, M. J. Vargas, G. Ballesteros, Y. Tellez, K. J. Soda, M. K. Sahoo, A. Nuñez, A. Balmaseda, E. Harris and B. A. Pinsky, Clin. Infect. Dis., 2016, 63, 1584–1590 CrossRef PubMed.
- R. W. Peeling, H. Artsob, J. L. Pelegrino, P. Buchy, M. J. Cardosa, S. Devi, D. A. Enria, J. Farrar, D. J. Gubler, M. G. Guzman, S. B. Halstead, E. Hunsperger, S. Kliks and H. S. Margolis, Nat. Rev. Microbiol., 2010, 8, S30–S38 CrossRef CAS PubMed.
- M. G. Guzman, S. B. Halstead, H. Artsob, P. Buchy, J. Farrar, D. J. Gubler, E. Hunsperger, A. Kroeger, H. S. Margolis, E. Martínez, M. B. Nathan, J. L. Pelegrino, C. Simmons, S. Yoksan and R. W. Peeling, Nat. Rev. Microbiol., 2010, 8, S7–S16 CrossRef CAS PubMed.
- J. J. Waggoner and B. A. Pinsky, J. Clin. Microbiol., 2016, 54, 860–867 CrossRef CAS PubMed.
- N. H. Kim, S. J. Lee and M. Moskovits, Nano Lett., 2010, 10, 4181–4185 CrossRef CAS PubMed.
- M. Chirumamilla, G. Das, A. Toma, A. Gopalakrishnan, R. P. Zaccaria, C. Liberale, F. De Angelis and E. Di Fabrizio, Microelectron. Eng., 2012, 97, 189–192 CrossRef CAS.
- M. Chirumamilla, A. Gopalakrishnan, A. Toma, R. P. Zaccaria and R. Krahne, Nanotechnology, 2014, 25, 235303 CrossRef PubMed.
- M. R. Gonçalves, F. Enderle and O. Marti, J. Nanotechnol., 2012, 2012, 173273 Search PubMed.
- J. Li and J. Macdonald, Biosens. Bioelectron., 2016, 83, 177–192 CrossRef CAS PubMed.
- H. de Puig, J. O. Tam, C.-W. Yen, L. Gehrke and K. Hamad-Schifferli, J. Phys. Chem. C, 2015, 119, 17408–17415 CAS.
- J. Xie, J. Y. Lee and D. I. C. Wang, Chem. Mater., 2007, 19, 2823–2830 CrossRef CAS.
- D. H. M. Dam, J. H. Lee, P. N. Sisco, D. T. Co, M. Zhang, M. R. Wasielewski and T. W. Odom, ACS Nano, 2012, 6, 3318–3326 CrossRef CAS PubMed.
- G. Frens, Nature, 1973, 241, 20–22 CAS.
- S. Kumar, J. Aaron and K. Sokolov, Nat. Protoc., 2008, 3, 314–320 CrossRef CAS PubMed.
- L. Rodríguez-Lorenzo, R. A. Alvarez-Puebla, F. J. G. de Abajo and L. M. Liz-Marzán, J. Phys. Chem. C, 2010, 47, 7336–7340 Search PubMed.
- X. Xia, W. Li, Y. Zhang and Y. Xia, Interface Focus, 2013, 3, 20120092 CrossRef PubMed.
- H. Kearns, N. C. Shand, W. E. Smith, K. Faulds and D. Graham, Phys. Chem. Chem. Phys., 2015, 17, 1980–1986 RSC.
- S. P. Mulvaney, M. D. Musick, C. D. Keating and M. J. Natan, Langmuir, 2003, 19, 4784–4790 CrossRef CAS.
- A. J. Tatem, Int. Health, 2014, 6, 5–11 CrossRef PubMed.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CAS.
- E. Nalbant Esenturk and A. R. Hight Walker, J. Raman Spectrosc., 2009, 40, 86–91 CrossRef.
- A. Garcia-Leis, A. Torreggiani, J. V. Garcia-Ramos and S. Sanchez-Cortes, Nanoscale, 2015, 7, 13629–13637 RSC.
- S. Atta, T. V. Tsoulos and L. Fabris, J. Phys. Chem. C, 2016, 120, 20749–20758 CAS.
- L. Osinkina, T. Lohmüller, F. Jäckel and J. Feldmann, J. Phys. Chem. C, 2013, 117, 22198–22202 CAS.
- J. I. Gersten, J. Chem. Phys., 1980, 72, 5779–5780 CrossRef CAS.
- D. M. Haaland and R. G. Easterling, Appl. Spectrosc., 1980, 34, 539–548 CrossRef CAS.
- C. L. Zavaleta, B. R. Smith, I. Walton, W. Doering, G. Davis, B. Shojaei, M. J. Natan and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13511–13516 CrossRef CAS PubMed.
- J. Li and J. Macdonald, Lab Chip, 2016, 16, 242–245 RSC.
- C.-W. Yen, H. de Puig, J. O. Tam, J. Gómez-Márquez, I. Bosch, K. Hamad-Schifferli and L. Gehrke, Lab Chip, 2015, 15, 1638–1641 RSC.
- S. Lee, S. Mehta and D. Erickson, Anal. Chem., 2016, 88, 8359–8363 CrossRef CAS PubMed.
- J. O. Tam, H. de Puig, C. Yen, I. Bosch, J. Gomez-Marquez, C. Clavet, K. Hamad-Schifferli and L. Gehrke, J. Immunoassay Immunochem., 2017, 38, 355–377 CrossRef CAS PubMed.
- H. de Puig, I. Bosch, M. Carré-Camps and K. Hamad-Schifferli, Bioconjugate Chem., 2017, 28, 230–238 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00052a |
| This journal is © The Royal Society of Chemistry 2017 |