Biradical character in the ground state of [Mn@Si12]+: a DFT and CASPT2 study†
Received
23rd May 2016
, Accepted 8th August 2016
First published on 8th August 2016
Abstract
Both density functional theory and multi-configurational ab initio (CASPT2) calculations are used to explore the potential energy surface of the hexagonal prismatic cluster [Mn@Si12]+. Unlike isoelectronic Cr@Si12, the ground state is a biradical, with triplet and open-shell singlet states lying very close in energy. The results are discussed in the context of recent experimental studies using infra-red multiple photon dissociation spectroscopy and X-ray MCD spectroscopy.
Introduction
The study of small clusters containing both silicon and a transition element can be traced back to two papers published by Beck in the late 1980s1,2 which described the formation of M@Sin clusters in a supersonic beam. Hiura, Miyazaki and Kanayama later used a quadrupole ion trap to isolate [M@SinHx]+ and show that the tungsten series [W@SinHx]+, for example, terminates at n = 12.3 Moreover, high-resolution spectra showed that the hydrogen content of the clusters, x, is reduced as n increases, and in the limit of n = 12 the most abundant tungsten cluster was completely devoid of hydrogens. The interpretation of this data is that the metal atom is completely encapsulated in M@Si12, binding to all twelve silicon atoms and hence eliminating the need for additional hydrogens to saturate their valence. In the same paper, Hiura and co-workers also used density functional theory to explore the structure of W@Si12, initially reporting a ‘basketlike’ structure where the metal is encapsulated within the cluster and each silicon atom is bonded to three others. However, a note added in proof identified a more stable hexagonal prismatic structure (D6h point symmetry, see Fig. 1), which Khanna, Rao and Jena subsequently confirmed to be the global minimum for Cr@Si12.4 Experimental support for this proposal came from the photo-detachment spectrum of the corresponding anion, [Cr@Si12]−,5 which shows a peak at 3.18 eV compared to Khanna's computed value of 3.11 eV. Later scanning tunneling microscopy experiments by Uchida and co-workers showed features consistent with intact hexagonal prismatic Ta@Si12 clusters on reconstructed Si(111) surfaces.6 XANES and EXAFS spectroscopy at the W L3 edge of W@Six clusters deposited on silica also suggest that the hexagonal prismatic structure is retained.7 The structure of Cr@Si12 and W@Si12 is remarkable, simply because it is so very different from the deltahedral geometries that dominate the cluster chemistry of electron-deficient species such as the boranes (icosahedral [B12H12]2−, for example). These deltahedra typically have 4- or 5-connected vertices: 3-connected structures such as the hexagonal prism, in contrast, are typically the preserve of electron-precise clusters such as tetrahedral P4 or prismane, the unstable isomer of benzene. Over the past decade, however, a number of 3-connected structures have emerged in the cluster chemistry of germanium, including pentagonal prismatic [Co/Fe@Ge10]3−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8–12 and D2d-symmetric [Ru@Ge12]3−.13 The fact that the endohedral metals are all from the middle of the transition series suggests that 3-connected structural motifs require filled metal d orbitals that are relatively high in energy. The origin of the stability of Cr@Si12 has been debated by a number of groups including our own. One explanation is that the Cr or W atom is simply too large to fit inside an icosahedral Si12 cage, and so the prismatic structure, with its larger volume, is preferred.14,15 Jellium-type models can also be applied in this context, and configurations of 1s21p61d10 (18 electrons) and 1s21p61d102s2 (20 electrons) have been invoked to account for the relatively high dissociation energies of Cr@Si12 and isostructural Fe@Si12.16–18 In their initial report,3 Hiura et al. suggested that the stability of W@Si12 could be rationalised in terms of an 18-electron rule: donation of 12 electrons from the Si12 cage to the metal raises the electron count to 18, leaving three electrons per Si center to form the three Si–Si bonds. The consensus now, however, is that the electronic configuration about the metal is best described as 16-electron, with a vacant dz2 orbital directed towards the centre of the two 6-membered rings (11a1g in Fig. 1).8,18 Despite the debate about the qualitative description of bonding in Cr@Si12, the fact that it is a singlet with a perfectly D6h-symmetric structure, as proposed by Hiura and Khanna, has not been seriously questioned in the literature. The apparently conflicting results of recent experimental observations on the isoelectronic cationic manganese cluster cation, [Mn@Si12]+, therefore appear rather surprising. Ngan et al. have reported the infra-red multiple photon dissociation spectroscopy of [Mn@Si12]+·Xe, and based on comparison of their spectrum to DFT-computed values (with the B3P86 functional), have argued that the ground state is in fact a triplet rather than a singlet. Moreover, the structure is not perfectly hexagonal prismatic, but rather distorted to Ci point symmetry, suggesting that it is descended from a state which is orbitally degenerate in D6h symmetry.19 Silicon-based clusters which retain some or all of the characteristic magnetic moment of the endohedral transition metal have potential applications in the spintronics field,20–22 and so the apparent emergence of a triplet state represents a significant finding. In direct contrast, however, Lau and co-workers have measured the X-ray MCD spectrum of [Mn@Si12]+ and concluded that the cluster is in fact diamagnetic.23 Their DFT calculations (using the PBE0 functional) identified a D6h-symmetric singlet ground state, although more recent computational work from the same group suggests that a distorted triplet is in fact more stable.24 The purpose of this work is to explore the potential energy surface using a variety of theoretical tools, and hence to establish the factors that lead to the close proximity of singlet and triplet states in [Mn@Si12]+ but not in isoelectronic Cr@Si12. We also address the relationship between structure and spin state, and how this impacts on the vibrational spectrum. We touched briefly on related issues in a previous paper8 which focused on the energy landscape for a range of M@E12 clusters. Specifically, we showed that the relative stabilization of the metal d orbitals in [Mn@Si12]+ compared to Cr@Si12 stabilises a charge-transfer state where an electron is promoted from the 8a2u orbital localised exclusively on the Si12 cage (Fig. 1) into the 11a1g LUMO with dominant Mn dz2 character. For reasons that we discuss in detail below, this charge transfer state is a singlet rather than a triplet and moreover is not orbitally degenerate, and so on both counts appears inconsistent with Ngan et al.'s interpretation of the IR data.19 What we did not consider previously is the fact that the 8a2u orbital is almost degenerate with another orbital localised primarily on the Si12 cluster, 8e2g in Fig. 1, and so the manifold of charge transfer states is in fact much richer than previously suspected. Moreover, the degeneracy of the 8e2g orbital provides a natural driving force for distortions from the hexagonal prismatic limit in charge transfer states where it is partially occupied.
8–12 and D2d-symmetric [Ru@Ge12]3−.13 The fact that the endohedral metals are all from the middle of the transition series suggests that 3-connected structural motifs require filled metal d orbitals that are relatively high in energy. The origin of the stability of Cr@Si12 has been debated by a number of groups including our own. One explanation is that the Cr or W atom is simply too large to fit inside an icosahedral Si12 cage, and so the prismatic structure, with its larger volume, is preferred.14,15 Jellium-type models can also be applied in this context, and configurations of 1s21p61d10 (18 electrons) and 1s21p61d102s2 (20 electrons) have been invoked to account for the relatively high dissociation energies of Cr@Si12 and isostructural Fe@Si12.16–18 In their initial report,3 Hiura et al. suggested that the stability of W@Si12 could be rationalised in terms of an 18-electron rule: donation of 12 electrons from the Si12 cage to the metal raises the electron count to 18, leaving three electrons per Si center to form the three Si–Si bonds. The consensus now, however, is that the electronic configuration about the metal is best described as 16-electron, with a vacant dz2 orbital directed towards the centre of the two 6-membered rings (11a1g in Fig. 1).8,18 Despite the debate about the qualitative description of bonding in Cr@Si12, the fact that it is a singlet with a perfectly D6h-symmetric structure, as proposed by Hiura and Khanna, has not been seriously questioned in the literature. The apparently conflicting results of recent experimental observations on the isoelectronic cationic manganese cluster cation, [Mn@Si12]+, therefore appear rather surprising. Ngan et al. have reported the infra-red multiple photon dissociation spectroscopy of [Mn@Si12]+·Xe, and based on comparison of their spectrum to DFT-computed values (with the B3P86 functional), have argued that the ground state is in fact a triplet rather than a singlet. Moreover, the structure is not perfectly hexagonal prismatic, but rather distorted to Ci point symmetry, suggesting that it is descended from a state which is orbitally degenerate in D6h symmetry.19 Silicon-based clusters which retain some or all of the characteristic magnetic moment of the endohedral transition metal have potential applications in the spintronics field,20–22 and so the apparent emergence of a triplet state represents a significant finding. In direct contrast, however, Lau and co-workers have measured the X-ray MCD spectrum of [Mn@Si12]+ and concluded that the cluster is in fact diamagnetic.23 Their DFT calculations (using the PBE0 functional) identified a D6h-symmetric singlet ground state, although more recent computational work from the same group suggests that a distorted triplet is in fact more stable.24 The purpose of this work is to explore the potential energy surface using a variety of theoretical tools, and hence to establish the factors that lead to the close proximity of singlet and triplet states in [Mn@Si12]+ but not in isoelectronic Cr@Si12. We also address the relationship between structure and spin state, and how this impacts on the vibrational spectrum. We touched briefly on related issues in a previous paper8 which focused on the energy landscape for a range of M@E12 clusters. Specifically, we showed that the relative stabilization of the metal d orbitals in [Mn@Si12]+ compared to Cr@Si12 stabilises a charge-transfer state where an electron is promoted from the 8a2u orbital localised exclusively on the Si12 cage (Fig. 1) into the 11a1g LUMO with dominant Mn dz2 character. For reasons that we discuss in detail below, this charge transfer state is a singlet rather than a triplet and moreover is not orbitally degenerate, and so on both counts appears inconsistent with Ngan et al.'s interpretation of the IR data.19 What we did not consider previously is the fact that the 8a2u orbital is almost degenerate with another orbital localised primarily on the Si12 cluster, 8e2g in Fig. 1, and so the manifold of charge transfer states is in fact much richer than previously suspected. Moreover, the degeneracy of the 8e2g orbital provides a natural driving force for distortions from the hexagonal prismatic limit in charge transfer states where it is partially occupied.
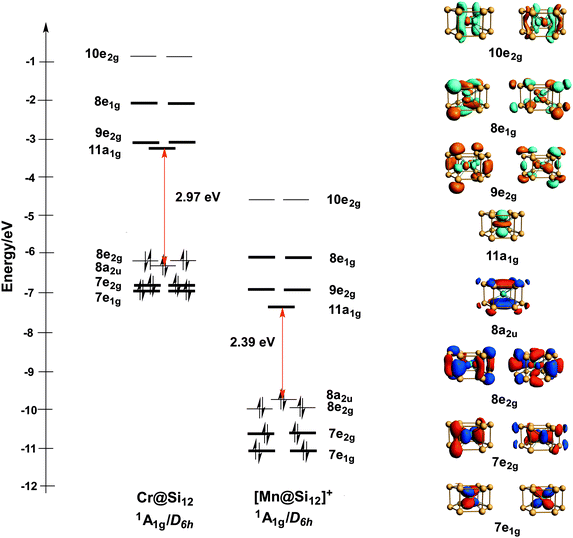 |
| | Fig. 1 Comparison of the Kohn–Sham MO diagrams for Cr@Si12 and [Mn@Si12]+ (B3LYP functional, isosurface value = 0.04 (e a.u.−3)1/2). Orbitals with substantial metal 3d character are shown in bold. | |
Computational methods
All DFT calculations described in this paper were performed with the Amsterdam Density Functional package (ADF2013.01)25–27 using the gradient-corrected (GGA) functional proposed by Becke and Lee, Yang and Parr (BLYP)28,29 and the hybrid functional B3LYP.30,31 An all electron Slater-type basis sets of triple-ζ quality, extended with a single polarisation function (TZP), was used to describe all the atoms (Cr, Mn and Si). All structures were optimised using the gradient algorithm of Versluis and Ziegler.32 For numerical frequency calculations the number of fit functions was increased by adding the subkey ‘FitType QZ4P’ of the key BASIS and a fine grid was used for numerical integration (keyword ‘Becke good’). Single point CASSCF/CASPT2 calculations were performed with the MOLCAS 8.0 package.33 Atomic natural orbitals optimised for relativistic corrections and core correlation (ANO-RCC) basis sets were used to expand the orbitals.34 The large primitive set of functions is contracted to [5s4p2d1f] for Si, [7s6p5d3f2g1h] and [7s7p5d3f2g1h] for Mn and Cr, respectively. Scalar relativistic effects were included using the Douglas–Kroll–Hess Hamiltonian. The computational cost of evaluating the two-electron integrals was reduced by the Cholesky decomposition technique (threshold 10−6 a.u.).35 We used the standard zeroth-order Hamiltonian and an imaginary shift of 0.1 a.u. to avoid the appearance of weak intruder states in the CASPT2 calculations.
Results and discussion
Symmetry considerations and the manifold of states
To set the scene for the quantitative discussion to follow, we first consider the symmetries of low-lying states that could, in principle, be candidates for the ground state of [Mn@Si12]+. The single determinant configuration shown in Fig. 1 (for both [Mn@Si12]+ and Cr@Si12) and also in Scheme 1 corresponds to a closed-shell singlet with 1A1g symmetry. The charge transfer states alluded to in the introduction all involve the promotion of a single electron into the 11a1g orbital (which carries dominant metal 3dz2 character), the donor orbital being either the 8a2u orbital or 8e2g. Within this charge-transfer manifold, both singlet and triplet configurations are possible, giving rise to states of 1/3A2u and 1/3E2g symmetry (we note from the outset that the open-shell singlets are intrinsically multi-determinantal in nature). The orbitally degenerate 1/3E2g states are necessarily unstable with respect to low-symmetry distortions, and indeed imaginary frequencies always appear for these states. In principle, any distortion that removes the 3-fold rotation axis will lift the degeneracy of the 1/3E2g states, and here we have considered two possibilities: a C2h-symmetric distortion leading to puckering of the hexagonal prism in states of 1/3Ag/Bg symmetry and a Cs-symmetric distortion where one edge of the hexagonal prism is elongated, giving states of 1/3A′/A′′ symmetry. The former is closely related to the Ci-symmetric structure reported by Ngan et al.19 while the latter is similar to the structure of Hf@Si12 reported by Uchida et al.36 In practice, it seems that the precise nature of the distortion has little impact on the computed energies (vide infra). Before moving to a discussion of these energies, we note one further symmetry-related issue that has significant implications for the subsequent discussion. The two donor orbitals of interest here are 8a2u and 8e2g, Fig. 1. The former is localised rigorously on the Si12 cage, the only contribution from the metal coming from 4pz. In contrast the 8e2g orbital retains some residual contribution from metal 3dx2−y2/xy (∼15% in the 1A1g state), and this difference proves to be important when we consider the exchange interaction between the two unpaired electrons, and hence the singlet/triplet separation (vide infra).
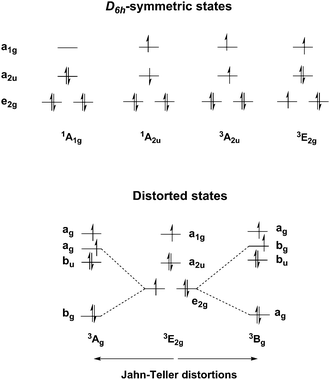 |
| | Scheme 1 Manifold of single-determinant configurations for [Mn@Si12]+ and Cr@Si12. | |
Energy landscape using density functional theory
The relative energies and optimised structures of the various configurations summarised in Scheme 1 are collected in Fig. 2: full details of the energies and Mulliken spin densities are summarised in Table 1. Given the well-known dependence of spin states on the chosen functional, we report values for two distinct and contrasting functionals – a typical GGA functional, BLYP and its hybrid analogue, B3LYP. Corresponding data for isoelectronic Cr@Si12 are presented in ESI† (Table S1). Before discussing [Mn@Si12]+ in detail, we note first that the identity of the ground state in Cr@Si12 is not in doubt: the 1A1g state lies substantially (∼0.8 eV) below any other, irrespective of functional, precisely as has been reported by Khanna,4,16,18 us,8 and others.14 In [Mn@Si12]+, however, the relative stabilization of the metal-based orbitals closes the HOMO–LUMO gap (2.39 eV vs. 2.97 eV in Cr@Si12 in Fig. 1) and as a result the 1A2u state is in fact more stable than 1A1g, only marginally so with the BLYP functional but substantially (0.26 eV) with the hybrid B3LYP. The values of 〈S2〉 in the 1A2u state (1.22/2.11 for BLYP/B3LYP) are indicative of strong spin contamination, and the Mulliken spin densities, ρ(Mn) = +1.62/+2.69 and ρ(Si12) = −1.62/−2.69, confirm a high degree of biradical character. The fact that the 1A2u state is more stable than the corresponding triplet, 3A2u, is at first sight surprising as it is apparently in conflict with Hund's rule. The preference for the singlet stems from the fact alluded to earlier that the two singly occupied orbitals, 11a1g and 8a2u, are localised in completely different regions of the molecule: 11a1g is almost entirely Mn dz2 in character while 8a2u is almost entirely localised on the silicon cage. The direct exchange interaction between the electrons in these two singly occupied orbitals, which would favour a triplet, is therefore very small. The dominant exchange interaction is instead between the single electron in 11a1g and those in the Mn–Si bonding orbitals (7e1g, 7e2g in Scheme 2). Within the unrestricted Kohn–Sham ansatz, the result is that the spin-α electrons in these orbitals are polarised towards the metal, giving Mulliken spin densities far in excess of 1.0 (+1.62/+2.69 for BLYP/B3LYP). As a corollary, the spin-β electrons are polarised in the opposite direction, towards the Si12 cage, stabilizing the 1A2u state, where the electron in the 8a2u orbital is also spin-β, over the triplet (3A2u) where the spin densities arising from the two orbitals are opposed. This is certainly a relatively minor effect simply because repulsions between electrons delocalised over the Si12 cage are in any case not severe, but it is clearly just large enough to tip the balance in favour of the singlet.
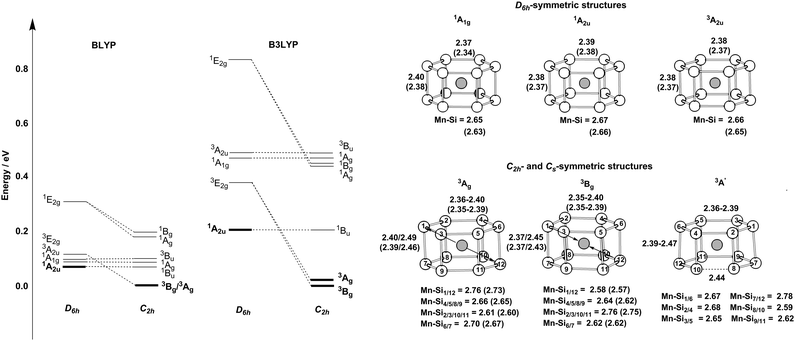 |
| | Fig. 2 The energy landscape for [Mn@Si12]+ obtained with both BLYP and B3LYP functionals and the corresponding optimised structural parameters (bond lengths obtained with B3LYP are shown in parentheses). | |
Table 1 Relative energies, structural data, Mulliken spin densities and values of 〈S2〉 for various states of [Mn@Si12]+ computed at the BLYP level of theory. Data for B3LYP are shown in parenthesis
| |
D
6h-symmetric states |
C
2h-symmetric states |
C
s-symmetric states |
|
1A1g |
1A2u |
3A2u |
1E2g |
3E2g |
3Ag |
3Bg |
3A′ |
|
The degenerate 3E2g state is defined using fractional occupation numbers: 0.5 electrons in each of the two spatial components of the 8e2g orbital with spin-β. The value of 〈S2〉 is therefore not defined.
|
| DFT:BLYP (B3LYP energies in parenthesis) |
| Energy/eV |
+0.09 (+0.47) |
+0.07 (+0.21) |
+0.10 (+0.49) |
+0.31 (+0.84) |
+0.12 (+0.38) |
0.00 (+0.02) |
0.00 (+0.00) |
0.00 |
| Mn–Si/Å |
2.65 (2.63) |
2.67 (2.66) |
2.66 (2.65) |
2.65 (2.64) |
2.66 (2.65) |
2.61–2.76 (2.60–2.73) |
2.58–2.76 (2.57–2.75) |
2.59–2.78 |
|
ρ(Mn) |
0.00 (0.00) |
1.62 (2.69) |
1.66 (2.56) |
1.51 (2.70) |
2.14 (3.13) |
2.11 (3.00) |
2.10 (3.09) |
2.09 |
|
ρ(Si12) |
0.00 (0.00) |
−1.62 (−2.69) |
0.34 (−0.56) |
−1.51 (−2.70) |
−0.14 (−1.13) |
−0.11 (−1.00) |
−0.10 (−1.09) |
−0.09 |
| 〈S2〉 |
0.00 (0.00) |
1.22 (2.11) |
2.10 (2.62) |
|
|
2.17 (2.65) |
2.17 (2.74) |
2.16 |
|
|
| CASPT2@BLYP geometry (CASPT2@B3LYP geometry in parenthesis) |
| Energy/eV |
+0.17 (+0.14) |
+0.07 (+0.09) |
+0.16 (+0.13) |
|
|
+0.06 (0.05) |
0.00 (0.00) |
|
|
ρ(Mn) |
|
|
1.15 (1.15) |
|
|
2.43 (2.42) |
2.41 (2.44) |
|
|
ρ(Si12) |
|
|
0.85 (0.85) |
|
|
−0.43 (−0.42) |
−0.41 (−0.44) |
|
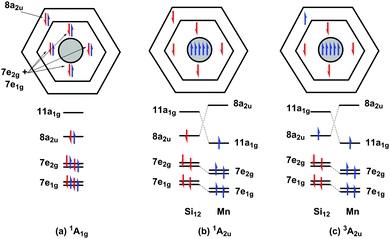 |
| | Scheme 2 Qualitative picture of the polarization of the Mn–Si electrons in the 1A2u and 3A2u states as a result of promotion of one electron into the dz2 orbital. | |
The tendency of hybrid functionals to stabilise states of high local spin density is well known, and it is therefore no surprise that the B3LYP functional favors the 1A2u state over 1A1g and also amplifies the biradical character as measured both by the value of 〈S2〉 and also the spin densities. The limit of complete localization of all four spin-α electrons in the 7e1g and 7e2g orbitals would correspond to formation of a half-filled 3d shell at the Mn center, and the B3LYP functional clearly approaches this limit much more closely than does BLYP. Finally, we note in passing that the polarization of the electrons in the Mn–Si bonding orbitals is, in principle, possible even without the preceding transfer of an electron into the dz2 orbital (i.e. in the 1A1g state). We have been unable to locate such a broken-symmetry state with the BLYP functional or even B3LYP, but the BHandHLYP functional, with 50% Hartree Fock exchange, does lead to symmetry breaking. The greater ease of symmetry breaking in the 1A2u state can be viewed as ‘seeding’ of the polarization by the unpaired electron in the dz2 orbital.
Turning now to the orbitally degenerate D6h-symmetric states, 1/3E2g, we note first that the electron density distribution shown in Table 1 is qualitatively rather similar to that in 1/3A2u: both singlet and triplet have spin densities at the metal far in excess of 1.0, particularly with the hybrid B3LYP functional. The key difference between the 1/3A2u and 1/3E2g pairs is that in the latter it is the triplet that lies lowest (i.e. the normal Hund order is restored). We emphasised above the point that the stability of the 1A2u state can be traced, ultimately, to the almost complete localization of the two singly-occupied orbitals in different regions of physical space which causes the direct exchange interaction between them to become negligibly small. In the 1/3E2g pair the situation is subtly different because the donor orbital (8e2g) carries some residual Mn 3dx2−y2/xy character, as a result of which the direct exchange interaction with the electron in 11a1g is non-negligible, favouring the triplet. Regardless of the chosen functional, however, the 1A2u state lies below 3E2g, so the most stable D6h-symmetric structure is a singlet, not a triplet, although the energies are close.
The Jahn–Teller theorem tells us that the 3E2g states must necessarily be unstable with respect to a low-symmetry distortion, and given the proximity of 1A2u and 3E2g, only a relatively small stabilization would be required to invert the singlet/triplet ordering. As noted in the discussion of symmetry-related features, any distortion that annihilates the 3-fold rotation axis will lift the degeneracy of the 3E2g state, and we have considered explicitly two possibilities, a puckering of the 6-membered rings (C2h symmetry) and an elongation of one Si–Si edge (Cs). With the BLYP functional, we can locate three distinct states of this type, C2h-symmetric 3Ag and 3Bg‡ and Cs-symmetric 3A′, which differ subtly in the distribution of Mn–Si bond lengths (Fig. 2). With B3LYP, we have been able to locate only the C2h-symmetric structures, 3Ag and 3Bg. These low-symmetry triplet states are, however, almost degenerate, suggesting that the precise nature of the distortion is not critical. More important in the context of the singlet/triplet debate is the fact that the distorted triplet states now lie below the singlet, 1A2u, albeit only marginally, irrespective of the functional. Thus the emergence of a triplet ground state for [Mn@Si12]+ is intimately linked to the distortion from perfect D6h symmetry: without this distortion, which is rather minor in structural terms and rather ill defined (in the sense that C2h- and Cs-symmetric structures are almost identical in energy), the singlet would be more stable.
CASSCF/CASPT2 studies
In light of the functional dependence of the spin state energetics, and also the strong spin contamination in the open-shell states indicated by the values of 〈S2〉, we have turned to multi-configurational SCF methodologies (CASPT2) for an alternative perspective on the bonding. In all cases we used a (10,15) active space (10 electrons in 15 orbitals). The active space includes the two singly occupied orbitals in the 1A2u state (one of a2u symmetry, the other a1g symmetry), the M–Si bonding orbitals of e2g and e1g symmetries, their antibonding counterparts and the five 4d orbitals. A qualitatively similar (14,15) active space was used by Pierloot and co-workers In their study of the closely-related [Mn@Si14]+ cluster.37 The importance of the 4d ‘double shell’ in complexes of the first transition series has been demonstrated on numerous occasions,38,39 but in this case they have very small natural occupation numbers and a parallel series of calculations without the 4d orbitals (a (10,10) active space) yields very similar results – see ESI† (Table S2). Full geometry optimisations at the CASPT2 level of theory are not feasible in systems of this size, and so we have considered the energies of the key states at both the BLYP and B3LYP-optimised structures. The B3LYP functional gives Mn–Si bond lengths that are ∼0.01–0.02 Å shorter than their BLYP analogues, and this difference leads to an ∼0.1 eV lowering of the CASPT2 energies of all states at the B3LYP geometries. Manual optimizations where all twelve Mn–Si bond lengths are uniformly decreased in 0.01 Å steps has allowed us to locate approximate minima for the 1A2u and 3Bg states a further 0.07–0.09 eV lower in energy with Mn–Si bond lengths ∼0.02 Å shorter than the B3LYP-optimised ones (see ESI† for a full discussion). However, in the context of the present discussion the key point is that the singlet–triplet separation (1A2u–3Bg) is essentially independent of the chosen geometry, with the triplet lying lower by 0.07–0.09 eV. The CASPT2 results therefore concur with both BLYP and B3LYP in identifying a distorted triplet ground state.§ The dispersion of energies at the CASPT2 level is relatively narrow, with the 1A1g, 1A2u and 3Bg states separated by only 0.17 eV: the results are therefore more consistent with the BLYP functional than B3LYP, which appears to overestimate the stability of the open-shell states, and of the triplets in particular. In the discussion of the DFT spin densities we highlighted the significance of the polarisation of the Mn–Si bonding electrons, particularly in the charge-transfer states where the presence of an unpaired electron in the dz2 orbital ‘seeds’ the localisation. We can identify related phenomena in the occupation numbers of the natural orbitals in the CAS wavefunction, which are summarised in Fig. 3 for five key states of [Mn@Si12]+: D6h-symmetric 1A1g, 1A2u and 3A2u and C2h-symmetric 3Bg and 3Ag (using the corresponding B3LYP geometry in each case). In the closed-shell singlet (1A1g), the dz2 orbital is vacant (natural orbital occupation = 0.05) while in the 1A2u state it is singly occupied (0.99), as is the a2u orbital (1.00). These changes are accompanied by more subtle but nevertheless significant variations in the occupations of the e1g- and e2g-symmetric Mn–Si bonding and antibonding orbitals. The occupations of the bonding orbitals decrease from ∼1.86 in the 1A1g state to ∼1.78 in 1A2u, while the occupations of the antibonding orbitals show the opposite trend: ∼0.12 in 1A1g but ∼0.22 in 1A2u. The correlation of the Mn–Si bonding electrons is clearly significantly enhanced by the transfer of an electron into the a1g orbital. Thus the ‘seeding’ effect that leads to polarisation of the spin density in the unrestricted density functional ansatz also emerges as enhanced multi-configurational character in the CASSCF wavefunction.
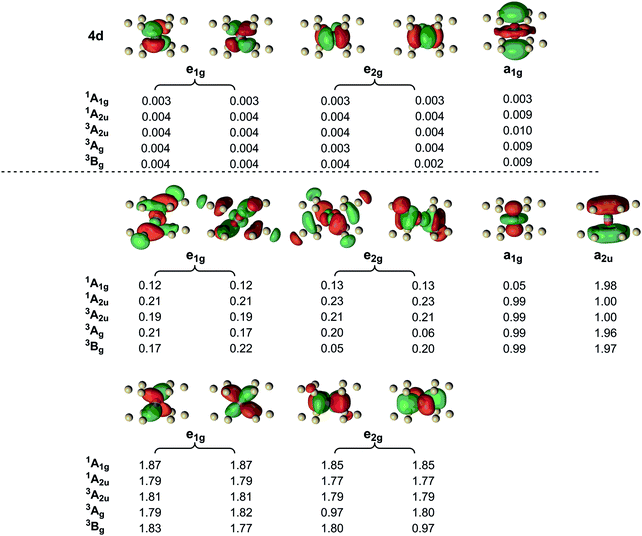 |
| | Fig. 3 Orbitals in the (10,15) active space and their occupations in the various states of [Mn@Si12]+ (B3LYP geometry). Orbitals were generated for the 1A1g state using an isosurface value of 0.04. | |
Analysis of the vibrational spectrum
Ngan et al. have argued that their measured vibrational spectrum, which shows two broad absorptions centered at ∼260 cm−1 and ∼330 cm−1 (Fig. 4, top), is consistent with a distorted Ci-symmetric triplet state.19 In order to establish the degree to which we can confidently exclude the D6h-symmetric singlets (1A1g or 1A2u) based on a poorer match to experiment, we have computed vibrational spectra for the three key, closed-shell 1A1g, open-shell 1A2u (D6h symmetry) and 3Bg (C2h symmetry). We have used the B3LYP functional as it is in generally good agreement with experimental frequencies for transition metal compounds.40,41 The computed spectrum of the 1A1g state (Fig. 4a) shows an intense feature at 222 cm−1 (e1u symmetry) corresponding to motion of the Mn atom in the xy plane and a weaker one at 333 cm−1 (a2u) corresponding to motion along the 6-fold axis (coupled to an a2u-symmetric vibration of the Si12 cage). This spectrum compares rather poorly with the experimental one: the separation of the two most intense bands is 111 cm−1 (222 and 333 cm−1) compared to a measured separation of only ∼70 cm−1. In the open-shell singlet state (1A2u), the same two intense bands (e1u and a2u) are apparent in the spectrum, but the e1u band is shifted 16 cm−1 to higher frequency, appearing at 238 cm−1, while the a2u mode is shifted to lower frequency, 310 cm−1. We note immediately that this spectrum affords a qualitatively better match to experiment in so much as the separation of the two dominant features is now only ∼70 cm−1. We can relate these shifts in vibrational frequencies to the very clear differences in electronic structure between the closed-shell 1A1g state on one hand and the open-shell 1A2u on the other. In the former, the electron density about the metal is highly anisotropic: the occupied Mn–Si bonding orbitals (7e1g and 7e2g) are localised primarily in the xy plane while the dz2 orbital is vacant. The result is that the metal can move in the xy plane with little energetic penalty (i.e. the frequency is low) but motion along the z axis is less facile. In the 1A2u state, in contrast, the dz2 orbital is singly occupied, as are, to a first approximation, dx2−y2, dxy, dxz and dyz due to the strong polarization of the bonding orbitals. The more isotropic electron density distribution in the 1A2u state is reflected in the smaller separation between vibrational modes corresponding to motion in the xy plane (e1u) and along the z axis (a2u). All our results (DFT and CASPT2) suggest that the electron density distributions in the 3Bg and 1A2u states are qualitatively very similar (both can be regarded as biradicals) and the general features of the vibrational spectra are also strikingly similar, Fig. 4b and c (we also note that our computed spectrum for 3Bg state is almost identical to that reported by Ngan et al.19). In particular, the two intense features remains centered around 240 cm−1 and 310 cm−1, and on this basis we believe that the separation of the e1u and a2u bands in the infra-red spectrum distinguishes clearly between closed-shell (1A1g) and biradical (1A2u/3Bg) configurations (we note that unfortunately no spectrum of Cr@Si12 has been reported to test this prediction). The distinction between open-shell singlet (1A2u) and triplet (3Bg) is, however, more difficult to make simply because the electron density distributions are so very similar, as are the computed vibrational spectra. The only clear difference between the two comes from the splitting of the e1u band in the 3Bg state, which should give rise to a shoulder to high frequency of the intense band at 250 cm−1. The resolution of the spectrum is, however, insufficient to establish whether such a band is present or not. Moreover, it is important to emphasise that the IRMPD experiment relates not to the cluster in isolation but rather to one with a Xe tag ([Mn@Si12]+·Xe), and the reduction in symmetry associated with the weak binding of the tag could also lead to a small splitting of the e1u peak, in either singlet or triplet. The IR spectrum therefore appears to be equally consistent with either the 1A2u or the 3Bg state, and the former cannot be dismissed as a candidate for the ground state based on this data alone.
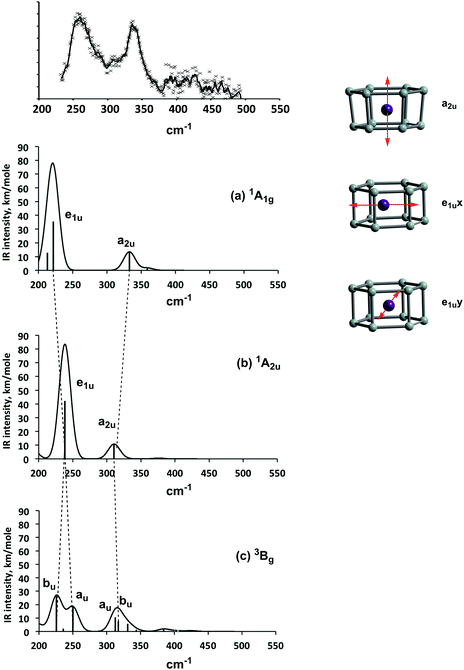 |
| | Fig. 4 Experimental Infra-red multi-photon dissociation (IRMPD) spectrum of [Mn@Si12]+ and simulated vibrational spectra of the 1A1g, 1A2u and 3Bg states. | |
Conclusions and the relationship between theory and experiment
Our interest in the [Mn@Si12]+ cluster was originally driven by the conflicting claims for singlet and triplet ground states in the experimental literature. A detailed exploration of the potential energy surface has confirmed that open-shell singlets and triplets are indeed very close in energy, in marked contrast to isoelectronic Cr@Si12 where the closed-shell singlet is energetically isolated. The triplet is found to be the ground state of [Mn@Si12]+ at all levels of theory considered here (DFT and CASPT2), and so, on face value, our calculations appear consistent with Ngan et al.'s interpretation of the IRMPD experiments: i.e. that the ground state is a triplet. Our analysis in the previous section, however, suggests that whilst the IR data do eliminate the closed-shell 1A1g state as a candidate for the ground state, they do not effectively discriminate between the 1A2u and 3Bg states, both of which are biradicals – certainly not to the extent that we could confidently eliminate the former. Meanwhile the XMCD data is clearly in favour of a singlet, so there appears to be no compelling evidence from experiment alone to support a triplet ground state. A closer analysis of the computational data shows that it is only the hybrid B3LYP functional that strongly favours the triplet (ΔE = 0.21 eV), and we must treat this number with caution as the tendency of hybrids to over-stabilise high spin states is well established. The gap is much smaller for the gradient-corrected functional BLYP (ΔE = 0.07 eV) and also at the CASPT2 level of theory (ΔE = 0.07–0.09 eV), values that lie below the threshold where we could confidently make a definitive assignment of the ground state. The case for a triplet from theory is therefore certainly not overwhelming. What is not in doubt, however, is that the ground state of [Mn@Si12]+ is fundamentally different from that of isoelectronic Cr@Si12: the former is a biradical (either triplet or singlet) while the latter has a closed-shell singlet ground state, and this difference manifests itself in characteristic shifts in the vibrational spectrum.
Acknowledgements
We thank the EPSRC for financial support (JEM, VA, EP/K021435/1) and also for the TMCS CDT programme (DF, EP/L015722/1). We also acknowledge the Spanish Administration (CdG, Project CTQ2014-51938-P), the Generalitat de Catalunya (CdG, Project 2014SGR199) and the European Union (JEM, CdG, COST Action ECOSTBio CM1305) for financial support. Finally, we thank Professor André Fielicke, Professor Tobias Lau and Dr Linn Leppert for helpful discussions and the former for providing the original spectroscopic data shown in Fig. 4.
Notes and references
- S. M. Beck, J. Chem. Phys., 1987, 87, 4233–4234 CrossRef CAS.
- S. M. Beck, J. Chem. Phys., 1989, 90, 6306–6312 CrossRef CAS.
- H. Hiura, T. Miyazaki and T. Kanayama, Phys. Rev. Lett., 2001, 86, 1733–1736 CrossRef CAS PubMed.
- S. N. Khanna, B. K. Rao and P. Jena, Phys. Rev. Lett., 2002, 89, 016803 CrossRef CAS PubMed.
- W. Zheng, J. M. Nilles, D. Radisic and K. H. Bowen, Jr., J. Chem. Phys., 2005, 122, 071101 CrossRef PubMed.
- N. Uchida, L. Bolotov, T. Miyazaki and T. Kanayama, J. Phys. D: Appl. Phys., 2003, 36, L43–L46 CrossRef CAS.
- Z. Sun, H. Oyanagi, N. Uchida, T. Miyazaki and T. Kanayama, J. Phys. D: Appl. Phys., 2009, 42, 015412 CrossRef.
- J. M. Goicoechea and J. E. McGrady, Dalton Trans., 2015, 44, 6755–6766 RSC.
- T. Krämer, J. C. Duckworth, M. D. Ingram, B. Zhou, J. E. McGrady and J. M. Goicoechea, Dalton Trans., 2013, 42, 12120–12129 RSC.
- B. B. Zhou, M. S. Denning, D. L. Kays and J. M. Goicoechea, J. Am. Chem. Soc., 2009, 131, 2802–2803 CrossRef CAS PubMed.
- S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS PubMed.
- J. Q. Wang, S. Stegmaier and T. F. Fässler, Angew. Chem., Int. Ed., 2009, 48, 1998–2002 CrossRef CAS PubMed.
- G. Espinoza-Quintero, J. C. Duckworth, W. K. Myers, J. E. McGrady and J. M. Goicoechea, J. Am. Chem. Soc., 2014, 136, 1210–1213 CrossRef CAS PubMed.
- R. B. King, Struct. Bonding, 2011, 140, 1–24 CrossRef CAS.
- R. B. King, I. Silaghi-Dumitrescu and M. M. Uta, Dalton Trans., 2007, 364–372 RSC.
- J. Ulises Reveles and S. N. Khanna, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 165413 CrossRef.
- J. Ulises Reveles and S. N. Khanna, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035435 CrossRef.
- M. B. Abreu, A. C. Reber and S. N. Khanna, J. Phys. Chem. Lett., 2014, 5, 3492–3496 CrossRef CAS PubMed.
- V. T. Ngan, E. Janssens, P. Claes, J. T. Lyon, A. Fielicke, M. T. Nguyen and P. Lievens, Chem. – Eur. J., 2012, 18, 15788–15793 CrossRef CAS PubMed.
- J. U. Reveles, P. A. Clayborne, A. C. Reber, S. N. Khanna, K. Pradhan, P. Sen and M. R. Pederson, Nat. Chem., 2009, 1, 310–315 CrossRef CAS PubMed.
- R. Robles and S. N. Khanna, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 115414 CrossRef.
- J. Zhao, X. Huang, P. Jin and Z. Chen, Coord. Chem. Rev., 2015, 289–290, 315–340 CrossRef CAS.
- V. Zamudio-Bayer, L. Leppert, K. Hirsch, A. Langenberg, J. Rittmann, M. Kossick, M. Vogel, R. Richter, A. Terasaki, T. Möller, B. v. Issendorff, S. Kümmel and J. T. Lau, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 115425 CrossRef.
-
L. Leppert and J. T. Lau, personal communication.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 CAS.
- E. J. Baerends, SCM, Theoretical chemistry, Vrije Universiteit, Amsterdam, The Netherlands, ADF2013, http://www.scm.com.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- L. Versluis and T. Ziegler, J. Chem. Phys., 1988, 88, 322–328 CrossRef CAS.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. F. Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. X. Ma, P. A. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. B. Pedersen, D. L. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Marti, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
- O. B. Roos, V. Veryazov and P. O. Widmark, Theor. Chem. Acc., 2003, 111, 345–351 Search PubMed.
- F. Aquilante, P. A. Malmqvist, T. B. Pedersen, A. Ghosh and B. O. Roos, J. Chem. Theory Comput., 2008, 4, 694–702 CrossRef CAS PubMed.
- N. Uchida, T. Miyazaki and T. Kanayama, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 205427 CrossRef.
- V. T. Ngan, K. Pierloot and M. T. Nguyen, Phys. Chem. Chem. Phys., 2013, 15, 5493–5498 RSC.
- K. Andersson and B. O. Roos, Chem. Phys. Lett., 1992, 191, 507–514 CrossRef CAS.
- K. Pierloot, Int. J. Quantum Chem., 2011, 111, 3291–3301 CrossRef CAS.
- B. Y. Liang and L. Andrews, J. Phys. Chem. A, 2002, 106, 6945–6951 CrossRef CAS.
- Y. I. Ishikawa and K. Kawakami, J. Phys. Chem. A, 2007, 111, 9940–9944 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1: Relative energies, structural data, Mulliken spin densities and values of 〈S2〉 for various states of Cr@Si12. Table S2: relative energies for various states of [Mn@Si12]+ obtained with CASPT2 (10,10) (no 4d orbitals) and (10,15) (with 4d orbitals) active spaces. Tables S3–S5: analysis of potential energy surfaces using CASPT2. Table S6: Cartesian coordinates and total energies of all stationary points for [Mn@Si12]+ and Cr@Si12. See DOI: 10.1039/c6cp03534e |
| ‡ The C2h-symmetric 3Ag and 3Bg states are conceptually related to the familiar tetragonal elongation and compression modes in octahedral complexes. |
| § The corresponding CASPT2 energies for Cr@Si12 are listed in Table S1: the closed-shell 1A1g state is clearly the most stable (by 0.72–0.78 eV) just as predicted by DFT. |
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 




