Decarbonylation of ethanol to methane, carbon monoxide and hydrogen by a [PNP]Ir complex†
Jonathan G.
Melnick
,
Alexander T.
Radosevich
,
Dino
Villagrán
and
Daniel G.
Nocera
*
Department of Chemistry, 6-335, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139-4307, USA. E-mail: nocera@mit.edu; Fax: +1 617 253 7670; Tel: +1 617 253 5537
First published on 23rd November 2009
Abstract
The putative three-coordinate Ir(I) PNPPri (PNPPri = [N{2-P(CHMe2)2-4-MeC6H3}2]–) pincer complex decarbonylates ethanol to yield methane, hydrogen and [PNPPri]Ir(CO). The mechanism involves the isolable trans-[PNPPri]Ir(H)(Me)(CO), which is susceptible to photochemical reductive elimination of methane.
The oxidative addition of hydroxyl functionalities is of importance for a variety of energy conversion reactions. For example, oxidative addition of water1–3 is a potential method for water oxidation,4 an important reaction for chemical energy storage.5–7 In addition, hydroxyl oxidative addition is pertinent to the increasing interest of using low-molecular weight alcohols, the most prominent of which is ethanol,8,9 as a primary fuel source. Consequently, a deeper understanding of alcohol synthesis and decomposition at catalytic centers may prove relevant to a range of energy conversion applications.
Whereas Ir(I) complexes promote alcohol dehydrogenation10–12 and aldehyde decarbonylation,13–16 the coupling of these two steps to drive alcohol decarbonylation is less explored.17,18 We have undertaken studies on ethanol decarbonylation, employing iridium(I) complexes of the PNPPri ligand (PNPPri = [N{2-P(CHMe2)2-4-MeC6H3}2]–).19 Alcohol decarbonylation has been promoted by Ir pincer complexes in only two cases: an Ir(I) PCP (PCP = [C6H3-2,6-(CH2PBut2)2]) pincer complex promotes methanol decarbonylation to generate hydrogen equivalents and the corresponding Ir–CO complex;20 and treatment of an Ir(I) PNP (PNP = [C5H3N-2,6-(CH2PBut2)2]) complex with methanol gives trans-Ir(H)2(CO), which is stable to dehydrogenation.21 We now report that [PNPPri]Ir(I) reacts with EtOH via initial O–H oxidative addition to afford the isolable trans-[PNPPri]Ir(H)(Me)(CO), which has been structurally characterized.‡ Photolysis of the hydrido-methyl complex subsequently drives the reductive elimination of methane to effect overall decarbonylation of ethanol.
Photolysis of [PNPPri]Ir(N2)§ (Fig. S3, ESI†) in EtOH yields the known compound [PNPPri]Ir(CO)22 as the only metal containing product and H2 and CH4 as the organic products in 79% and 94% yield, respectively, as determined by GC. To determine intermediates along the reaction pathway, possible EtOH addition products were targeted by non-photochemical means. Treatment of [PNPPri]Ir(H)2 and norbornylene with EtOH at room temperature yields trans-[PNPPri]Ir(H)(Me)(CO); the same product is obtained by the thermolysis of [PNPPri]Ir(N2) in EtOH at 80 °C:
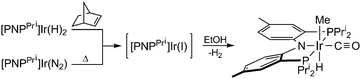 | (1) |
[PNPPri]Ir is capable of decarbonylating longer chain alcohols as well. Treatment of a mixture of [PNPPri]Ir(H)2 and NBE with n-PrOH gives trans-[PNPPri]Ir(H)(Et)(CO).¶ The trans configuration of alkyl and hydride ligands in the decarbonylation product is analogous to the reported trans-dihydride carbonyl obtained via decarbonylation of paraformaldehyde with Ir(I) pincer complexes, which has been attributed to stereoselective migratory deinsertion of the aldehyde following C–H activation.21
The products from the thermal decarbonylation reactions of EtOH and n-PrOH were isolated and their structures were characterized by X-ray diffraction, the results of which are shown in Fig. 1. The solid-state structure reveals iridium in an octahedral coordination environment with the carbonyl ligand trans to the amide nitrogen of the [PNPPri]. The decarbonylation product can be furnished independently. trans-[PNPPri]Ir(H)(Me)(CO) can also be prepared from the reaction of [PNPPri]Ir(H)223 with acetaldehyde.24 The observation that EtOH and acetaldehyde both furnish the same decarbonylation product suggests that the acetaldehyde is an intermediate along the alcohol decarbonylation pathway. Thermolysis or photolysis of [PNPPri]Ir(N2) is known to liberate N2 to generate the coordinatively unsaturated species, [PNPPri]Ir(I).25 EtOH addition to this coordinatively unsaturated intermediate to yield acetaldehyde and H2 followed by subsequent decarbonylation would result in trans-[PNPPri]Ir(H)(Me)(CO) as illustrated in Scheme 1.
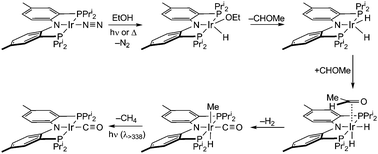 | ||
| Scheme 1 | ||
![Molecular structures of (top) trans-[PNPPri]Ir(H)(Me)(CO) and (bottom) trans-[PNPPri]Ir(H)(Et)(CO), which are the products obtained from the thermal decarbonylation of EtOH and n-PrOH, respectively. H-atoms (refined at calculated positions) are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level.](/image/article/2010/CC/b914083b/b914083b-f1.gif) | ||
| Fig. 1 Molecular structures of (top) trans-[PNPPri]Ir(H)(Me)(CO) and (bottom) trans-[PNPPri]Ir(H)(Et)(CO), which are the products obtained from the thermal decarbonylation of EtOH and n-PrOH, respectively. H-atoms (refined at calculated positions) are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. | ||
To determine whether trans-[PNPPri]Ir(H)(Me)(CO) is a viable intermediate for EtOH decarbonylation, we sought to effect reductive elimination of methane from the complex. Methane reductive elimination from trans-[PNPPri]Ir(H)(Me)(CO) is not observed to temperatures of 80 °C. However, methane elimination was observed under photolysis reaction conditions. As Fig. 2 displays, solutions of trans-[PNPPri]Ir(H)(Me)(CO) smoothly and quantitatively convert to [PNPPri]Ir(CO) under irradiation with λexc ≥ 338 nm; the product and its quantitative formation were corroborated by 1H NMR spectra of photolyzed solutions.26 The maintenance of isosbestic points in the absorption spectra of Fig. 2 indicates that no long-lived intermediates form during the photoreductive elimination reaction.
![Spectral changes of the absorption profile upon photolysis (λexc > 338 nm) of trans-[PNPPri]Ir(H)(Me)(CO) (). The final product spectrum corresponds to [PNPPri]Ir(CO) (—). Spectra were recorded over the span of 6 min every 30 s.](/image/article/2010/CC/b914083b/b914083b-f2.gif) | ||
Fig. 2 Spectral changes of the absorption profile upon photolysis (λexc > 338 nm) of trans-[PNPPri]Ir(H)(Me)(CO) (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ). The final product spectrum corresponds to [PNPPri]Ir(CO) (—). Spectra were recorded over the span of 6 min every 30 s. ). The final product spectrum corresponds to [PNPPri]Ir(CO) (—). Spectra were recorded over the span of 6 min every 30 s. | ||
Examples of photochemical trans reductive elimination from octahedral complexes are rare and typically involve homolytic bond cleavage.27,28 However, in our photolytic experiments with trans-[PNPPri]Ir(H)(Me)(CO) neither H2 nor C2H6 is observed, indicating that a homolytic bond cleavage mechanism is unlikely. Rather, ligand isomerization via photodissociation of CO could give cis-[PNPPri]Ir(H)(Me)(CO), permitting spontaneous unimolecular reductive elimination of CH4. This mechanism is consistent with the observation that treatment of [PNPPri]Ir(H)2 with CO furnishes [PNPPri]Ir(CO) along with putative elimination of H2.22
The initial dehydrogenation of EtOH can occur across the O(H)–C(H) bond to directly furnish the acetaldehyde or from across C(H)–C(H) to produce the enol, which then can isomerize to acetaldehyde. Iridium catalyzed alkane dehydrogenation is common,29–32 and fewer examples of diethyl ether dehydrogenation with iridium to give an Ir(η2-CH2CHOCH2CH3) species have been reported.33–35 In fact, the reaction of a mixture of [PNPPri]Ir(H)2 with NBE in Et2O yields [PNPPri]Ir(η2-CH2CHOCH2CH3).36 Its availability suggests the possibility that initial dehydrogenation proceeds from the ethoxy fragment to generate the enol. To address this question more incisively, [PNPPri]Ir(N2) was photolyzed in EtOD and the isotope distribution of methane, CHxD4–x, was analyzed. Mass spectra of the evolved methane show all possible isotopomers. The fact that higher order deuterium incorporation occurs requires that a mechanism for isotopic scrambling involving solvent is present. Indeed, D2O exchange with the hydride ligands of trans-Ir(H)2(CO)(Cl)(PR3)2 has been observed.37 The H–D exchange of Ir–H intermediates involving solvent necessarily obscures a detailed mechanistic description based on isotope experiments. Notwithstanding, an appreciable amount of CH4 (ca. 25%) is observed. This implies that dehydrogenation occurs across the O(H)–C(H) bond. If dehydrogenation were to exclusively occur from the C(H)–C(H) bond, the maximum proton composition of the liberated methane would be CH3D, since after H2 liberation, the generated acetaldehyde would only contain three protons. Although C(H)–C(H) dehydrogenation cannot be excluded as a competing mechanism, only dehydrogenation across the O(H)–C(H) can account for a 4H equivalency.
Of the two possible initial addition products involving the dehydrogenation of EtOH across the O(H)–C(H) bond, DFT computations support that the lowest energy addition product arises from addition across the O–H bond as opposed to the methylene C–H bond. The geometries of the respective transition states were calculated by scanning the reaction trajectories for O–H (Fig. S18–S20, ESI†) and C–H (Fig. S21–S23, ESI†) oxidative addition. The activation energy for O–H oxidative addition is 14.6 kcal mol–1, while the activation barrier for C–H is 18.9 kcal mol–1 (Fig. 3). In addition, the reverse reaction, reductive elimination to regenerate EtOH and [PNPPri]Ir(I), is more favorable for the C–H activation pathway by 7.4 kcal mol–1.
![DFT calculated structures and transition states of the oxidative addition of the O–H and methylene C–H of EtOH to [PNPPri]Ir(i). Only the EtOH and immediate coordination sphere are shown (Ir shown in green, P purple, N blue, O red, C gray, H white).](/image/article/2010/CC/b914083b/b914083b-f3.gif) | ||
| Fig. 3 DFT calculated structures and transition states of the oxidative addition of the O–H and methylene C–H of EtOH to [PNPPri]Ir(I). Only the EtOH and immediate coordination sphere are shown (Ir shown in green, P purple, N blue, O red, C gray, H white). | ||
The mechanism shown in Scheme 1 for the decarbonylation of ethanol to methane, dihydrogen and carbon monoxide is consistent with the foregoing observations.
In summary, we have observed room temperature decarbonylation of EtOH by [PNPPri]Ir(I) to generate H2 and trans-[PNPPri]Ir(H)(Me)(CO), which quantitatively undergoes photolytically induced reductive elimination of methane to generate [PNPPri]Ir(CO). Experiments and computations indicate the mechanism involves initial oxidative addition of the hydroxyl moiety of EtOH to the coordinatively unsaturated [PNPPri]Ir(I) center followed by dehydrogenation to produce the in situ generated acetaldehyde.
Research was supported by the National Science Foundation (Grant No. CHE-0750239). ATR and JGM acknowledge the NIH for NRSA postdoctoral fellowships. DV acknowledges the Camille and Henry Dreyfus Postdoctoral Program in Environmental Chemistry for a fellowship.
Notes and references
- O. V. Ozerov, Chem. Soc. Rev., 2009, 38, 83–88 RSC.
- D. Morales-Morales, D. W. Lee, Z. Wang and C. M. Jensen, Organometallics, 2001, 20, 1144–1147 CrossRef CAS.
- R. Dorta, H. Rozenberg, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2002, 124, 188–189 CrossRef CAS.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, B. D. Yehoshoa, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001–10017 CrossRef CAS.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697–1699 CrossRef CAS.
- A. E. Farrell, R. J. Plevin, B. T. Turner, A. D. Jones, M. O'Hare and D. M. Kammen, Science, 2006, 311, 506–508 CrossRef CAS.
- BP Statistical Review of World Energy June 2009, BP p.l.c., London, UK, 2009 Search PubMed.
- M. Königsmann, N. Donati, D. Stein, H. Schönberg, J. Harmer, A. Sreekanth and H. Grützmacher, Angew. Chem., Int. Ed., 2007, 46, 3567–3570 CrossRef.
- K. Fujita, N. Tanino and R. Yamaguchi, Org. Lett., 2007, 9, 109–111 CrossRef CAS.
- A. Gabrielsson, P. V. Leeuwen and W. Kaim, Chem. Commun., 2006, 4926–4927 RSC.
- T. Iwai, T. Fujihara and Y. Tuji, Chem. Commun., 2008, 6215–6217 RSC.
- P. Serp, M. Hernandez, B. Richard and P. Kalck, Eur. J. Inorg. Chem., 2001, 2327–2336 CrossRef CAS.
- P. J. Alaimo, B. A. Arndtsen and R. G. Bergman, Organometallics, 2000, 19, 2130–2143 CrossRef CAS.
- K. A. Bernard and J. D. Atwood, Organometallics, 1998, 7, 235–236.
- L. Vaska and J. W. DiLuzio, J. Am. Chem. Soc., 1961, 83, 2784–2785 CrossRef CAS.
- S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092–2093 CrossRef CAS.
- O. V. Ozerov, C. Guo, V. A. Papkov and B. A. Foxman, J. Am. Chem. Soc., 2004, 126, 4792–4793 CrossRef CAS.
- D. Morales-Morales, R. Redòn, Z. Wang, D. W. Lee, C. Yung, K. Manguson and C. M. Jensen, Can. J. Chem., 2001, 79, 823–829 CrossRef CAS.
- S. M. Kloek, D. M. Heinekey and K. I. Goldberg, Organometallics, 2006, 25, 3007–3011 CrossRef CAS.
- M. T. Whited and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 5874–5875 CrossRef CAS.
- L. Fan, S. Parkin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 16772–16773 CrossRef CAS.
- H2 is evolved from the treatment of [PNPPri]Ir(H)2 with acetaldehyde in 71% yield as determined by GC.
- M. T. Whited and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 16476–16477 CrossRef CAS.
- trans-[PNPPri]Ir(H)(Et)(CO) undergoes photolytic reductive elimination under irradiation with λexc ≥ 338 nm to yield C2H6 and [PNPPri]Ir(CO) quantitatively as determined by 1H NMR.
- A. Vogler, A. Kern and J. Hüttermann, Angew. Chem., Int. Ed. Engl., 1978, 17, 524–525 CrossRef.
- J. Sykora and J. Sima, Coord. Chem. Rev., 1990, 107, 1–212 CrossRef.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef CAS.
- I. Gttker-Schnetmann and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 9330–9338 CrossRef.
- K. Zhu, P. D. Achord, X. Zhang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13044–13053 CrossRef CAS.
- M. Gupta, C. Hagen, W. C. Kaska, R. E. Cramer and C. M. Jensen, J. Am. Chem. Soc., 1997, 119, 840–841 CrossRef CAS.
- H. F. Luecke, B. A. Arndtsen, P. Burger and R. G. Bergman, J. Am. Chem. Soc., 1996, 118, 2517–2518 CrossRef CAS.
- B. A. Arndtsen and R. G. Bergman, Science, 1995, 270, 1970–1973 CAS.
- P. Rodríguez, M. M. Díaz-Requejo, T. R. Belderrain, S. Trofimenko, M. C. Nicasio and P. J. Pérez, Organometallics, 2004, 23, 2162–2167 CrossRef CAS.
- M. T. Whited, Y. Zhu, S. D. Timpa, C. Chen, B. M. Foxman, O. V. Ozerov and R. H. Grubbs, Organometallics, 2009, 28, 4560–4570 CrossRef CAS.
- D. P. Paterniti, R. J. Roman and J. D. Atwood, Organometallics, 1997, 16, 3371–3376 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional spectra and charts. CCDC 740056–740058. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b914083b |
‡ Crystallographic data for trans-[PNPPri]Ir(H)(Me)(CO): C28H44IrNOP2, M = 664.78, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.3135(8), b = 9.8060(10), c = 17.8528(17), α = 84.829(2)°, β = 83.244(2)°, γ = 75.962(2)°, V = 1399.3(2), Z = 2, µ = 4.906 mm–1, T = 100 K, R1 = 0.0471, wR2 = 0.0732 (based on observed reflections), GooF = 1.024, reflections measured = 29 , a = 8.3135(8), b = 9.8060(10), c = 17.8528(17), α = 84.829(2)°, β = 83.244(2)°, γ = 75.962(2)°, V = 1399.3(2), Z = 2, µ = 4.906 mm–1, T = 100 K, R1 = 0.0471, wR2 = 0.0732 (based on observed reflections), GooF = 1.024, reflections measured = 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 281, Rint = 0.0855, CCDC 740057. 281, Rint = 0.0855, CCDC 740057. |
| § Crystallographic data for [PNPPri]IrN2·MesNH2: C35H53IrN4P2, M = 783.95, triclinic, space group P, a = 9.8992(12), b = 11.2251(14), c = 16.388(2), α = 101.780(2)°, β = 93.376(2)°, γ = 93.801(2)°, V = 1773.9(4), Z = 2, µ = 3.882 mm–1, T = 100 K, R1 = 0.0535, wR2 = 0.0944 (based on observed reflections), GooF = 1.005, reflections measured = 39573, Rint = 0.0882, CCDC 740058. |
| ¶ Crystallographic data for trans-[PNPPri]Ir(H)(Et)(CO): C29H46IrNOP2, M = 678.81, monoclinic, space group P2(1)/n, a = 14.7286(13), b = 12.3197(11), c = 16.0250(14), β = 90.754(2)°, V = 2907.5(4), Z = 4, µ = 4.724 mm–1, T = 100 K, R1 = 0.0226, wR2 = 0.0529 (based on observed reflections), GooF = 1.002, reflections measured = 67091, Rint = 0.0415, CCDC 740056. |
| This journal is © The Royal Society of Chemistry 2010 |