
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Re-engineering luminol: new frontiers in chemiluminescence chemistry
Amir M.
Alsharabasy

CÚRAM, Research Ireland Centre for Medical Devices, University of Galway, H91 W2TY, Ireland. E-mail: amir.abdo@universityofgalway.ie
First published on 20th June 2025
Abstract
Luminol and its derivatives have emerged as powerful chemiluminescent agents with broad applications in biomedical diagnostics, forensic science, and environmental monitoring. Despite their widespread use, luminol's limitations, including poor solubility, short luminescence duration, and sensitivity to environmental conditions, have driven extensive research into the synthesis of more efficient derivatives. This concise review presents recent advances in the molecular engineering of luminol derivatives, focusing on design strategies that employ electronic modulation (e.g., introduction of electron-donating or withdrawing substituents) and steric tuning (e.g., alkylation and ring substitutions) to optimize its chemiluminescence efficiency, kinetics, emission wavelength, solubility, stability, and applicability for specific environments (e.g., biological systems). The review also discusses how these structural modifications impact luminol's performance within integrated systems, including forensics, bioimaging platforms, immunoassay technologies and microfluidic sensors, thereby linking molecular-level design with macroscopic function. Emerging macromolecular and polymer-based luminol systems, such as those incorporating hydrophilic carriers, nanoparticles, enzyme-responsive linkers, surface-immobilized polymer brushes, and multi-functional hybrid platforms, are also highlighted for their potential to overcome solubility and biocompatibility barriers while enabling targeted delivery or signal amplification. Finally, key challenges and future perspectives are outlined, including the development of near-infrared-emitting derivatives, improved storage stability, and interdisciplinary strategies for translating luminol chemistry into next-generation diagnostics and environmental sensing platforms. By summarizing these advancements, this review underscores the evolving role of luminol chemistry in modern analytical science and its potential to revolutionize next-generation detection technologies.
 Amir Alsharabasy | Dr. Amir Alsharabasy is a postdoctoral fellow at CÚRAM, the SFI Research Centre for Medical Devices, University of Galway. His research spans biopolymer modification, hydrogel design, redox-active materials, and the synthesis of metal–organic compounds. His current work focuses on designing functional materials for the selective detection and modulation of reactive oxygen and nitrogen species (ROS/RNS), with particular emphasis on nitric oxide and its role in cancer and inflammation. Dr. Alsharabasy has published extensively on chemiluminescent sensors, gasotransmitter-responsive biomaterials, and redox-based therapeutics. His work integrates synthetic chemistry and materials science to advance next-generation diagnostic and therapeutic platforms for biomedical use. |
Design, System, ApplicationThis concise review distils recent progress in the molecular design of luminol derivatives, with an emphasis on structural modifications that enhance chemiluminescence performance. By organizing current synthetic strategies and mechanistic insights into a coherent framework, the review offers a system-level understanding of how electronic and steric modifications influence luminol's luminescence properties. It also explores emerging applications in diagnostics, sensing, and environmental monitoring, and highlights future directions such as near-infrared emitters and lab-on-a-chip integration. As such, the review serves as a valuable design resource for researchers developing soft molecular systems with tuneable optical responses for healthcare and environmental applications. |
1. Introduction
Chemiluminescence (CL) has recently emerged as an advanced strategy for detecting various analytes, including heavy metals, pollutants, metabolites, and pharmaceuticals. It has become a standard tool in environmental analysis,1 forensic science,2 clinical diagnostics and theranostics,3,4 as well as pharmaceutical and biomedical research.5,6 This widespread application is attributed to its simplicity, background-free nature, ultra-high sensitivity (down to attomolar concentrations), rapid detection, and flexible integration into diverse analytical strategies.5,7,8CL detection relies on a chemical reaction that generates electronically excited species, which emit light upon returning to their ground state. Luminol (3-amino-phthalhydrazide) is the most extensively studied chemiluminescent reagent due to its broad range of applications.4,9 Its oxidation is triggered by specific reactive oxygen and nitrogen species (ROS and RNS), such as hydrogen peroxide (H2O2), superoxide radical (O2˙−), hydroxyl radical (˙OH), nitric oxide (˙NO), and nitrogen dioxide radical (˙NO2), leading to the decomposition of luminol and the formation of 3-aminophthalate, which emits light.10–13 Additionally, certain catalysts, including peroxidase,14,15 hemoglobin,16 and hemin (Fe(III)-protoporphyrin),17,18 enhance these oxidation reactions, significantly increasing luminescence intensity. Moreover, the CL quantum yield of the different luminol derivatives depend on the solvent polarity, nature, position and number of substituents.19,20
To enhance the applicability of luminol, researchers have focused on improving its sensitivity, stability, and solubility through the synthesis of more potent chemiluminescent derivatives. This concise review is structured into three main sections (Fig. 1). The first section provides an overview of luminol synthesis, its structural features and functional groups, the underlying CL reaction mechanisms, and key factors influencing luminescence efficiency. The second section explores various modification strategies for luminol derivatives, including structural alterations to enhance sensitivity, stability, and solubility, functionalization with electron-donating (EDG) or withdrawing groups (EWG), and the development of hybrid luminol derivatives with superior properties. The final section examines challenges and future perspectives, focusing on stability and storage limitations, the need for more biocompatible and selective derivatives, emerging synthesis trends, and potential interdisciplinary applications.
 | ||
| Fig. 1 Overview of the review structure, including luminol synthesis, chemiluminescence reaction mechanism and light generation, strategies for luminol derivative design, and future challenges and applications. | ||
2. Chemistry of luminol and mechanism of luminescence generation
2.1. Luminol synthesis approaches
Luminol synthesis begins with 3-nitrophthalic acid, which can be obtained via the oxidation of α-nitronaphthalene or the nitration of phthalic acid or phthalic anhydride (Scheme 1). The synthesis starts with naphthalene nitration, which can be performed using: (1) nitric acid at room temperature for 5–6 days, followed by purification via methanol recrystallization.21 (2) Sulphonitric acid at 50–60 °C for 7 hours, followed by purification with xylene.22 (3) Sulphonitric acid at 45–60 °C for 90 minutes, followed by purification with diluted ethanol.23 (4) Sulphonitric acid at 95 °C, nitrating molten naphthalene for 30 minutes, followed by purification with methanol (step 1, Scheme 1).21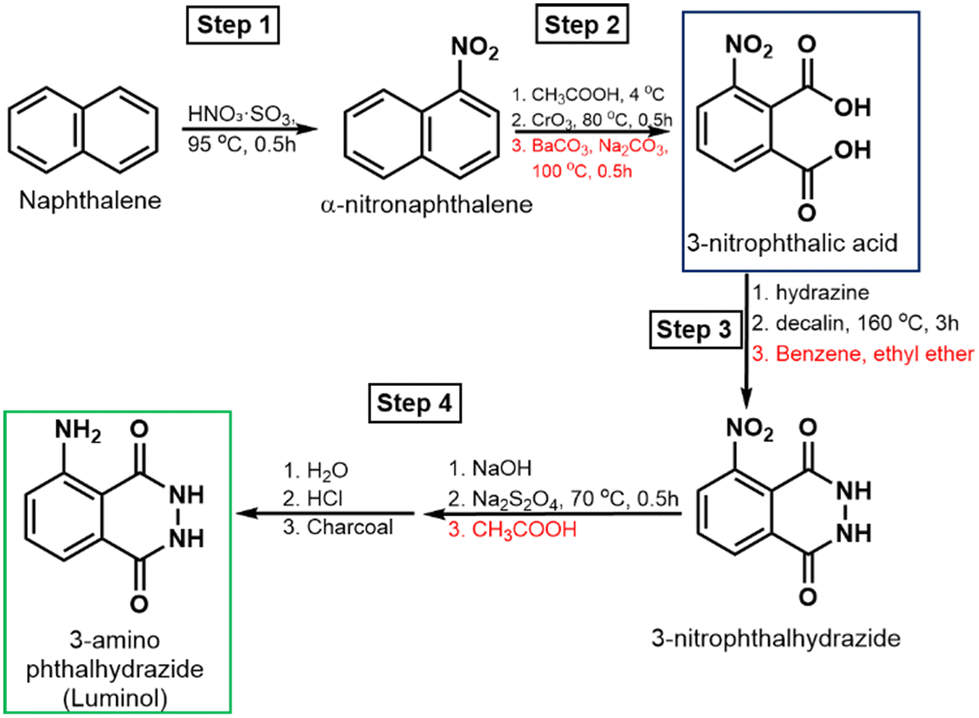 | ||
| Scheme 1 Luminol synthesis pathways, starting with naphthalene nitration (step 1), followed by oxidation to 3-nitro-phthalic acid (step 2), which, upon hydrazinolysis (step 3), and subsequential treatment with NaOH, sodium dithionite, and acidic precipitation yield luminol (step 4). | ||
The resulting α-nitronaphthalene is then oxidized by mixing with glacial acetic acid and chromic anhydride, heating at 70–80 °C for 90 minutes, followed by cooling, filtration of insoluble impurities, and washing.21 The solution is subsequently treated with barium carbonate and sodium carbonate, boiled, and then acidified with HCl at 50–60 °C to yield 3-nitrophthalic acid (step 2, Scheme 1). The nitration of phthalic acid or phthalic anhydride using Culhane's method produces a mixture of 3- and 4-nitrophthalic acids, which are difficult to separate.24
To obtain 3-nitrophthalhydrazide, 3-nitrophthalic acid is mixed with hydrazine sulphate and allowed to evaporate to dryness, followed by heating with decalin at 160 °C for 3 hours.25 The cooled product is then precipitated with benzene, filtered, and washed with ethyl ether (step 3, Scheme 1). Alternatively, 3-nitrophthalic acid can be mixed with a hydrazine solution in triethylene glycol, refluxed at 215 °C for 2 minutes, and cooled to yield 3-nitrophthalhydrazide.26 In the final stage, 3-nitrophthalhydrazide is mixed with sodium sulphate and NaOH or NH4OH, followed by the addition of sodium dithionite and heating at 70 °C for 30 minutes.27 The solution is then acidified with glacial acetic acid, and the precipitate is washed with water, dissolved in hot HCl, briefly decolorized with charcoal, filtered, washed, and dried (step 4, Scheme 1).
2.2. Mechanism of luminescence generation
Light emission in luminol solutions occurs under alkaline conditions in the presence of oxygen, following specific mechanisms in both protic and aprotic media.28Scheme 2 summarizes these CL reactions. In aprotic solutions, the luminol dianion forms and facilitates the spin inversion of molecular oxygen from its triplet state (3O2) to singlet oxygen (1O2).29 This leads to a concerted addition reaction, forming a bicyclic endoperoxide intermediate. The nitrogen atoms are then replaced by oxygen atoms through a peroxide bond, resulting in the formation of a chemiluminophore and the release of nitrogen gas. Electron transfer from the aniline ring to the O–O bond enables either high-energy or low-energy chemiexcitation. This process involves cleavage of the –O–O– bond, followed by either radiative decay (light emission) or radiationless decay. | ||
| Scheme 2 Chemiluminescence of luminol in aprotic solvents, starting with the oxidation to luminol dianion (step 1), followed by addition reaction and spin inversion of molecular oxygen (step 2). Upon nitrogen elimination and formation of chemiluminophore (step 3), 3-aminophthalate is formed via chemiexcitation, followed by radiative or non-radiative decay (step 4). | ||
The emission wavelength in aprotic solvents (e.g., DMSO) typically ranges from 485 to 510 nm. In protic solvents (e.g., water), the emission undergoes a blue shift to 420–430 nm due to intramolecular hydrogen transfer from the amino group to a nearby oxygen atom.30–32 The duration of light emission varies from a few seconds to several hours, depending on factors such as the medium composition,33 the reacting species,34 and the presence of additives or co-oxidants. These include peroxidases,35 various metalloporphyrins,36 ROS and RNS.13
3. Advances in the synthesis of luminol derivatives
Despite its wide range of applications, luminol has several limitations, including poor solubility in aqueous solutions, short luminescence duration, and weak intensity under certain conditions. To address these drawbacks, various chemical modification strategies have been developed, primarily focusing on introducing new functional groups to the C–NH2 position,37,38 and substitutions on the phenyl ring to modulate electron-donating effects.20,39 These substituent effects generally alter the electron density, highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) energy levels, pKα, and redox potentials. For instance, the conjugation of luminol to EDG is expected to stabilize the luminol dianion via resonance, reducing the energy penalty for deprotonation,19 which consequently make the CL reaction more feasible at physiological pH (7–8) instead of requiring strong alkali (pH ≥ 10). While these modifications generally aim to improve CL efficiency, some substitutions at the C–NH2 position have been found to reduce luminescence intensity by disrupting proton transfer between the amino and carbonyl groups, which is essential for light emission.29 Moreover, the luminol conjugation to EWG (e.g., chloro and nitro groups) reduced the excited-state stability, with a general decrease in luminescence intensity.40,41 Additionally, the chloride derivatives of mefenamic acid, ibuprofen, diclofenac sodium, and ampicillin were reacted with luminol in DMSO and triethylamine for the development of a new class of luminol derivatives with antibacterial activity.42 However, the luminescence activity of these derivatives was not investigated further. This section summarizes key advancements in the development of luminol derivatives with enhanced luminescence properties, highlighting the characteristics of promising candidate compounds.3.1. Substitutions on phthalimides
Mikroulis et al. synthesized a series of 6,8-disubstituted luminol derivatives using Suzuki coupling and hydrazinolysis.43 The synthesis began with the refluxing of 3-nitro-phthalic acid and acetic anhydride (Ac2O), followed by solvent evaporation in the presence of toluene and precipitation of the anhydride upon diethyl ether addition.38 The resulting anhydride was then refluxed with sec-butylamine and acetic acid, dried, dissolved in dichloromethane (DCM), and washed with an aqueous NaHCO3 solution. After drying with Na2SO4, this process yielded 3-nitro-phthalimide. The nitro group was subsequently reduced to an amino group by degassing the 3-nitro-phthalimide in methanol, adding Pd/C, and exposing the mixture to H2 gas, forming 3-amino-phthalimide. Bromination was carried out using either excess N-bromosuccinimide in DCM or bromine with acetic acid and sodium acetate, yielding 6-monobrominated and 4,6-dibrominated derivatives, respectively (Scheme 3A). Suzuki methylation with trimethylboroxine produced the corresponding 6-monosubstituted and 4,6-disubstituted 3-aminophthalimide derivatives. | ||
| Scheme 3 (A and B) Synthesis route of the 3-amino-phthalimide, followed by the synthetic pathways of 6-methyl-8(4-hydroxymethylphenyl)luminol and 6,8-dimethyl luminol, respectively. (C) Synthesis route of phosphonium phthalhydrazide beginning with the reaction between 3-aminophthalimide and bromoalkyl carboxylic acids for phthalimide acylation, followed by synthesis of phosphonium phthalimide, and final hydrazinolysis. Abbreviations: 4-HBPA: 4-(hydroxymethyl)phenylboronic acid; TMB: trimethylboroxine. | ||
To enhance luminescence intensity in the 6-monosubstituted derivative, an additional bromination step was performed, followed by reaction with 4-(hydroxymethyl)phenylboronic acid to introduce a hydroxyl-functionalized group (Scheme 3B). Hydrazinolysis of these products yielded luminol derivatives with enhanced CL efficiency under biologically relevant conditions. Notably, the CL quantum yields of 6,8-dimethyl luminol and 6-methyl-8-(4-hydroxymethylphenyl) luminol were 0.17 and 0.39, respectively, at pH 12, compared to just 0.012 for luminol (Table 1). Importantly, the enhanced luminescence observed with 8-methyl substitution compared to 8-phenyl is attributed to its ability to donate negative charge to the cyclic peroxide, facilitating O–O bond cleavage. Substitution at position 6 has a smaller impact on this effect. Similar enhancements in luminescence intensity were observed with luminol derivatives containing a 6-monomethyl substitution, which exhibited a twofold increase in CL intensity compared to luminol.44 However, no significant differences were noted for propyl- or tert-butyl-substituted derivatives, as the steric interactions of these alkyl groups with the carbonyl group hindered the transition to the excited state.
| Luminol derivative | Structural modification | Quantum yield | Emission wavelength (nm) | Ref. |
|---|---|---|---|---|
| Luminol | Parent compound | 0.012 | 425 | 44 |
| 6,8-Dimethyl luminol | Methyl substitutions at positions 6 and 8 on the benzene ring | 0.17 | 425 | 44 |
| 6-Methyl-8-(4-hydroxymethylphenyl) luminol | Methyl group at position 6 and 4-hydroxymethylphenyl at position 8 | 0.39 | 425 | 44 |
| Phosphonium phthalhydrazide | Phosphonium group substitution on the phthalhydrazide structure | < 0.001 | 425 | 38 |
| 6-Methyl luminol | Single methyl substitution at position 6 | 0.237 | 425 | 47 |
| 6-Isopropyl luminol | Isopropyl group at position 6 | 0.012 | 425 | 47 |
| Naphthalene-based analogues (GL-1 to GL-4) | Benzene ring replaced by naphthalene to extend conjugation | 0.3 (GL-1) | 520 | 50 |
| Luminol-fluorescein based activated cassettes | Luminol chemically linked to fluorescein via a linker | 0.007 | 524 | 52 |
| Luminol-Nile red based activated cassettes | Luminol chemically linked to Nile red via a linker | 0.012 | 634 | 52 |
| Isoluminol | Amino group at the meta-position | ≈0.0012 | 416 | 41 |
| O-Etherified luminol (Lum-glucose) | Enol group etherified with glucose via O-linkage | Quenched (restored after β-glu cleavage) | Turn-on signal after enzyme cleavage | 53 |
| m-Carboxyl luminol | Carboxyl group introduced at the meta-position | 0.06 | 39 | |
| Acylated luminol hydrazides | Succinic/glutaric acid linkers on the amino group | 0.004 | Minor shifts | 75 |
Interestingly, phosphonium cations were introduced into luminol derivatives to enable mitochondrial targeting for detecting intracellular ROS.38 Phosphonium-functionalized amino-acylated luminol derivatives were synthesized by stirring bromoalkyl carboxylic acids with oxalyl chloride (COCl)2 under an argon atmosphere, followed by evaporation of volatiles, redissolution in DCM, and reaction with 3-amino-phthalimide in the presence of pyridine (Scheme 3C). The resulting product was purified via aqueous two-phase extraction using water and DCM, followed by drying with Na2SO4. The acylated product was then reacted with phosphine in dry acetonitrile and refluxed under argon for three days, yielding the phosphonium phthalimide, which was subsequently refluxed with hydrazine in ethanol for hydrazinolysis. Although these final mitochondriotropic chemiluminescent probes successfully targeted mitochondria, they exhibited lower luminescence efficiency than luminol. Their poor CL performance and reduced intensity (Table 1) were attributed to fluorescence inhibition during the oxidation reaction. A similar reduction in luminescence intensity was observed after modifying isoluminol using the same approach.
Another approach for synthesizing luminol derivatives with substitutions at positions 6 and 8 was reported before,45,46 as shown in Fig. 2A. These studies aimed to modulate the steric interactions between the introduced alkyl groups and the carbonyl group of luminol. The synthesis began with the coupling of O-benzyl carbamate, N-methyl maleimide, and a series of unsaturated aldehydes, yielding a group of protected aminocyclohexenes. Subsequent deprotection using palladium-on-charcoal, followed by hydrazinolysis, produced a series of 6,8-dialkyl luminol derivatives. Among the synthesized derivatives, 6-methyl luminol exhibited a 20-fold increase in luminescence efficiency compared to luminol, accompanied by a twofold enhancement in chemiluminescence quantum yield and a slight blue shift (Table 1). In contrast, 6-isopropyl luminol displayed luminescence properties like those of luminol.47 These studies highlighted the role of steric effects introduced by alkyl substituents, which facilitate the transition to the excited state and enhance CL yields. This steric-based mechanism complements the previously discussed electronic modifications and suggests the potential of these luminol derivatives for bioluminescence imaging.
 | ||
| Fig. 2 (A) Schematic representation of the synthesis of alkyl luminol derivatives. (B) Synthetic routes of luminol derivatives with naphthalene, and the corresponding CL emission spectra in H2O/DMSO solution with potassium persulfate in the inset. Reprinted with permission.49 | ||
Notably, some derivatives demonstrated solubility in both aqueous and aprotic solutions, broadening their applicability. However, the synthesis of certain derivatives still requires long reaction times and palladium catalysis, which is both costly and highly sensitive to reaction conditions. Further optimization is needed to improve the efficiency and scalability of these synthetic routes.
Another strategy for enhancing luminol's luminescence involved extending its aromatic system by replacing the benzene ring with a naphthalene derivative functionalized with an –NH2 group or further modifying this exocyclic group. The synthesis began with the hydrazinolysis of 2,3-naphthalenedicarboxylic acid in acetic acid, followed by nitration using nitronium tetrafluoroborate (BF4NO2) in acetonitrile and subsequent mild reduction of the –NO2 group at position 5 (Fig. 2B).48,49 For derivatives with a protected –NH2 group, the reaction pathway started with esterification of 2,3-naphthalenedicarboxylic acid, followed by nitration and reduction. This was followed by mono- and di-benzylation of the amine group in the presence of KHCO3 or NaOH in DMF, with final hydrolysis and hydrazinolysis steps. Additional derivatives featuring amine substitution at position 6 were also synthesized, though they are not shown here. These naphthalene-based luminol derivatives exhibited green luminescence within the 490–590 nm range, with longer decay times and stronger CL signals than luminol (the inset in Fig. 2). As a result, they were proposed for the detection of low-concentration analytes, offering improved sensitivity compared to luminol.
Another strategy involved the synthesis of green-emitting luminol analogues through a three-step reaction sequence starting from 1-naphthylamine, including diazotization, nucleophilic substitution, and cyclization (Fig. 3A). This synthetic modification replaced luminol's benzene core with a naphthalene moiety, effectively extending the π-conjugation and red-shifting the emission wavelength to approximately 520 nm (Fig. 3B). In addition to the spectral shift, the structural changes enhanced the compound's stability under ambient conditions. Among the synthesized derivatives, the analogue GL-1 exhibited the highest CL yield compared to both luminol and other analogues (Fig. 3C), with luminescence intensity correlating linearly with concentration (Fig. 3D, Table 1).50
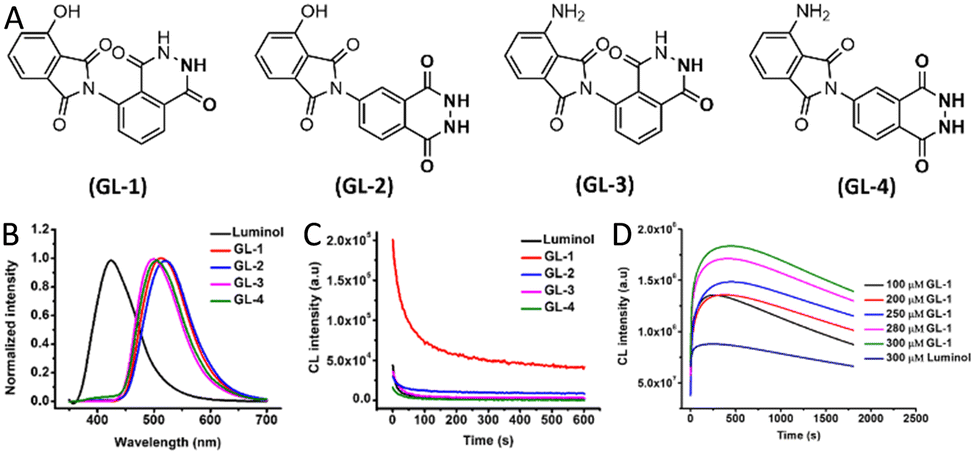 | ||
| Fig. 3 (A) Chemical structures of green-emitting luminol analogues (GL-1 to GL-4). (B) Luminescence emission spectra comparison between luminol and its analogues. (C) CL kinetic profiles of luminol and GL analogues. (D) CL intensity over time for varying concentrations of GL-1 and luminol in the presence of HRP, hydrogen peroxide H2O2, 4-morpholinopyridine, and 3-(10′-phenothiazinyl)-propane-1-sulfonate.53 | ||
3.2. Luminol modification via O-esterification and O-etherification
To enhance the CL intensity of luminol, an O-esterification reaction was performed by mixing it with triethylamine in DMF at room temperature or 50 °C (Fig. 4A).51 Among the synthesized derivatives, 5-amino-4-oxo-3,4-dihydrophthalazin-1-yl acetate (LC-1) and its deuterated counterpart LC-2 exhibited the highest luminescence efficiency, with significantly improved intensity and prolonged emission time compared to luminol and other derivatives within the pH range of 9 to 11. However, no details on the quantum yields were reported. Representative results from mixtures containing H2O2 and hemin at pH 9 are shown in Fig. 4B. Notably, reactions containing LC-2, which features a deuterated acetyl group, exhibited the highest reaction rate in the presence of blood (Fig. 4C). This enhancement is attributed to the esterification of the hydroxyl group, which stabilizes luminol in its enol form, thereby increasing luminescence intensity. Additionally, both luminol and LC-2 demonstrated comparable luminescence efficiency in western blot (WB) imaging for GAPDH detection, using goat anti-mouse IgG (H + L)-HRP as the secondary antibody (Fig. 4D). However, further esterification with larger substituents may negatively impact solubility and, due to steric hindrance, could reduce luminescence efficiency (Fig. 2B). These steric effects likely explain the lower performance observed in other synthesized derivatives. | ||
| Fig. 4 (A) Schematic representation of the synthesis of 5-amino-4-oxo-3,4-dihydrophthalazin-1-yl acetate (LC-1) and its deuterated counterpart (LC-2). (B) CL response curves, showing intensity versus time for luminol and its derivatives in the presence of H2O2 and hemin as a catalyst. (C) Gray value analysis of bloodstain images captured using a smartphone, following detection using a CL solution containing luminol, LC-1, or LC-2. (D) Western blot (WB) analysis of GAPDH protein expression, detected in mixtures containing luminol or LC-2 with H2O2. Reprinted with permission.51 | ||
Development of luminescence-based probes for biotechnological applications emitting at wavelengths above 450 nm has shown some success. For example, Han et al. studied chemiluminescent energy-transfer cassettes by coupling luminol derivatives with fluorescein and Nile red via Sonogashira reactions.52 The luminescence yield of luminol–fluorescein and luminol–Nile red was 61% and 100%, respectively, relative to luminol, producing yellow/green and red CL (Table 1). However, despite the efficient energy transfer, the synthesis involved multiple steps, lacked high convergence, and resulted in poor solubility in aqueous media, limiting biological applications. Additionally, these derivatives lack the amino group of luminol, which affect their CL efficiency and reactivity. Furthermore, switchable luminol-based electro-chemiluminescence (ECL) biosensors were developed by O-etherifying luminol at the enol group and conjugating it to glucose via an ether bond (Table 1).53 The resulting conjugate exhibited suppressed ECL emission, which was restored upon enzymatic cleavage by β-glucosidase, enabling sensitive detection of the enzyme with a linear range of 0.4–400 U L−1 and a detection limit of 0.1 U L−1.
3.3. Approaches for enhancing the solubility of luminol
Despite luminol's wide applicability in various analytical approaches, its poor solubility in aqueous solutions remains a challenge for specific assays. To address this, several studies have explored the synthesis of more hydrophilic luminol derivatives. For example, introducing a carboxyl (–COOH) group into luminol's benzene ring was achieved through the nitration of trimetallic anhydride at 120 °C, followed by treatment with dimethylurea, reduction of the exocyclic nitro group to an amine, and a final hydrazinolysis step (Fig. 5A).54,55 | ||
| Fig. 5 (A) Schematic representation of the m-carboxy luminol synthesis. (B) CL response curves for luminol and m-carboxy luminol with H2O2 in the presence of cobalt chloride (Co2+), hemin, and horseradish peroxidase (HRP) as catalysts. (C) CL response curves for varying concentrations of luminophores (luminol and m-carboxy luminol) mixed with H2O2 and hemin. (D) CL response curves corresponding to different lactate concentrations. Data are presented as mean ± SD (n = 3). Reprinted with permission.39 | ||
The resulting m-carboxyl luminol exhibited improved water solubility and, when loaded into liposomes, produced a fourfold increase in luminescence intensity with a 150-fold lower DNA detection limit compared to luminol.55 Moreover, in the presence of cobalt chloride, hemin, and HRP, m-carboxyl luminol exhibited a fivefold increase in quantum yield compared to luminol, along with ∼18-fold higher sensitivity for H2O2 detection (Fig. 5B, Table 1).39 Additionally, the lower detection limit for hemin was reduced fivefold with m-carboxyl luminol compared to luminol (Fig. 5C). Similarly, the luminescence signal in response to lactate was 2.6 times higher with m-carboxyl luminol, with generally lower detection limits than those obtained using colorimetric measurements (Fig. 5D). While this derivative shows potential for integration into microfluidic systems for point-of-care diagnostics, its low yield (15%) requires further optimization.55
3.4. Macromolecular strategies for luminol functionalization and signal amplification
Conjugating luminol to the backbones of various macromolecules has been employed to unlock a range of chemical, physical, and application-specific advantages. This strategy aims to achieve the following objectives: (1) enhanced stability of the chemiluminophore through covalent attachment, which helps prevent leaching and degradation, thereby increasing the overall longevity of the system.56 (2) Customizable chemical design that enables tuning of the chemical structure of redox-active materials by controlling the local environment around luminol to achieve specific functional properties. The main polymerization methods for luminol include oxidative chemical polycondensation, microwave-assisted oxidative polymerization, free-radical polymerization, and other miscellaneous approaches, which have been described in detail previously.57 (3) Reusability, where polymer-bound luminol allows for reagentless and repeatable detection, as demonstrated in various electropolymerized sensors incorporating luminol derivatives.58–64 (4) Signal amplification, as multiple luminol units can be attached to a single polymer chain, significantly boosting the chemiluminescent output.57 (5) Multifunctionality, enabling the incorporation of luminol alongside other functional groups (e.g., targeting moieties) on the same polymer for advanced biosensing applications.65,66 (6) Improved physical properties of the final luminol-based materials, where the inherent mechanical strength, processability, and thermal stability of polymers allow luminol to be applied in durable forms such as films, fibres, and nanoparticles.57 (7) Stimuli-responsive behaviour, where luminol–polymer conjugates act as self-reporting materials that emit light in response to specific environmental triggers such as oxidants, pH changes, or target analytes.58,67 (8) Biocompatibility, achieved by using suitable polymer carriers that enhance luminol's solubility and compatibility with biological fluids, enabling its use in biomedical applications.65,66 (9) Device integration, wherein luminol–polymer conjugates facilitate the incorporation of luminol into various electronic and optical systems, showing promise for embedded light-emitting functionalities.68,69 These advantages are summarized in Fig. 6. The recent advances in the development of luminol based polymers have been reviewed before.57 | ||
| Fig. 6 Main objectives for conjugating luminol to the backbones of macromolecules. | ||
Various strategies for loading of branched polymers with different luminophores towards achieving certain fluorescence properties and certain applications have been developed.70 Dendrimers, such as poly(amidoamine) (PAMAM) and poly(dithiodipropionic acid) (PDTPA), have been strategically integrated into luminol-based ECL biosensor platforms to enhance performance through multiple synergistic mechanisms. Their highly branched three-dimensional architecture provides abundant terminal groups, enabling dense luminol loading for multi-labelling and significant signal amplification. For instance, a luminol–PDTPA–Fe3O4 nanoparticle-based immunosensor was developed for the detection of carbohydrate antigen 125 (CA125) using a sandwich structure consisting of a primary antibody and a luminol-labeled secondary antibody via a PDTPA dendrimer. This complex was anchored to Fe3O4 magnetic nanoparticles and immobilized on a carbon paste electrode through magnetic force, yielding a detection range of 0.2–100 mU mL−1 and a detection limit of 0.032 mU mL−1.71 A similar strategy was applied for the detection of microalbuminuria—a key marker for early-stage renal complications in diabetes and hypertensive pregnancy disorders—using a microalbuminuria-specific antibody in a luminol–dendrimer–Fe3O4 system. This sensor achieved a detection range of 2–322 pg mL−1 and a detection limit of 0.084 pg mL−1.72
In addition to enabling high luminol loading, dendrimers act as efficient scaffolds for bioconjugation, facilitating the co-attachment of luminol and recognition biomolecules while enhancing stability by minimizing luminol leaching or degradation. For example, PAMAM-functionalized ZnO nanorods were employed for the simultaneous immobilization of luminol and a detection antibody specific to cancer antigen 15-3 (CA15-3). The ZnO nanorods catalyzed the decomposition of hydrogen peroxide, generating reactive oxygen species (ROS) that triggered and amplified the ECL signal. This system enabled CA15-3 quantification with a dynamic range of 0.1–120 U mL−1 and a detection limit of 0.033 U mL−1.73
For the development of advanced CL immunoassays, labelling functional enzymes (e.g., peroxidase, phosphatase) with a CL-labelling molecule would be advantageous. However, compared to fluorescence-labelling reagents, the introduction of CL molecules remains challenging, as many are chemically sensitive, and modifications can significantly affect luminescence intensity.30,74 For instance, acylation of the exocyclic amino group in luminol was achieved by overnight reaction with either succinic or glutaric anhydride in alkaline water, followed by hydrazinolysis in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (Scheme 4A).75
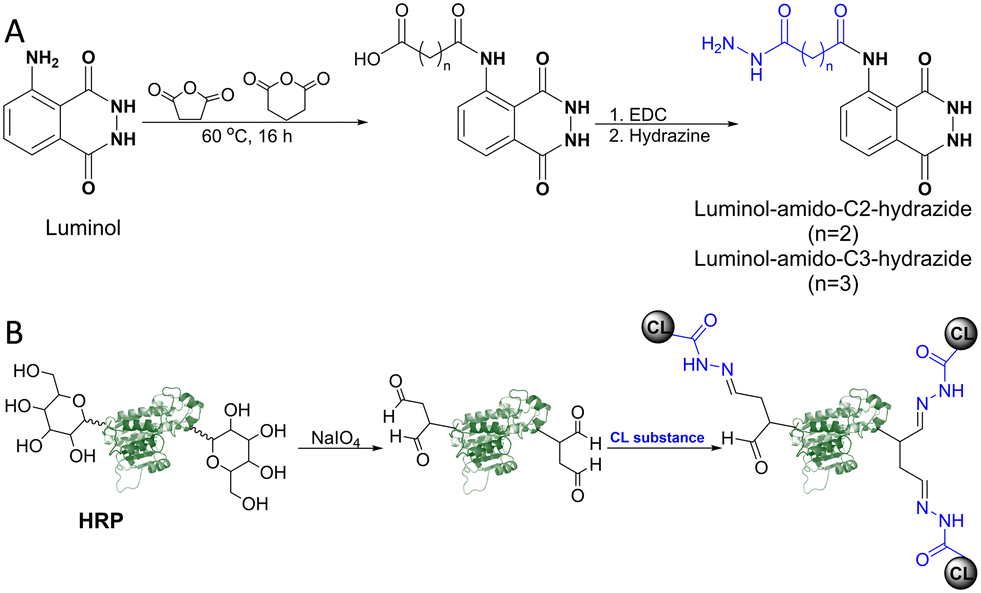 | ||
| Scheme 4 (A) Synthesis routes of luminol derivatives containing terminal hydrazide groups, using succinic acid and glutaric acid as linker moieties. (B) HRP labelling with the synthesized luminol-amido-C2-hydrazide, following enzyme oxidation with sodium periodate. | ||
4. Challenges and future perspectives
Stability, storage, and biocompatibility limitations
Despite the promise of luminol-based systems, key translational barriers remain. One of the most critical limitations is the instability of luminol derivatives under ambient conditions. Compounds such as hydrazides are prone to autoxidation and hydrolysis, while others suffer from photobleaching, collectively diminishing CL quantum yields within weeks.76 Current storage protocols, such as freezing at −20 °C under an inert atmosphere and light exclusion, are effective but impractical for field deployment or point-of-care applications.Equally important are biocompatibility and selectivity challenges, which limit in vivo utility. Unmodified luminol derivatives may cause dose-dependent cytotoxicity due to excessive ROS production and mitochondrial disruption.77,78 Additionally, nonspecific interactions with serum proteins or metal ions can hinder signal specificity and reduce assay accuracy.79 These limitations are summarized in Fig. 7.
 | ||
| Fig. 7 Challenges and emerging strategies in luminol–polymer chemiluminescent systems. | ||
Advanced synthetic chemistry approaches
To overcome these challenges, future innovations should prioritize chemical strategies that enhance molecular robustness and predictive stability. These include: (1) structural fortification through sterically hindered N-alkyl groups (e.g., tert-butyl) and oxidation-resistant motifs (e.g., thiadiazole analogues) to prevent degradation.80,81 (2) Machine learning-guided design to develop predictive models capable of identifying luminol derivatives with long-term shelf stability (Fig. 7).Development of advanced luminol–polymer systems
Polymer-based platforms provide a promising avenue to address the inherent weaknesses of free luminol, including short emission duration, pH dependency, and lack of targeting. These strategies not only amplify the signal but also introduce spatial control, enhanced stability, and multifunctionality (Fig. 7).The first strategy depends on covalent functionalization of polymers, where grafting luminol onto polymers (e.g., those with carboxylate groups) helps mitigate pH sensitivity, improve aqueous solubility, and support physiological performance. Branched architectures, such as dendritic polymers, offer steric shielding,82 which, upon conjugation to luminol, can reduce quenching, increase its local density, and enhance CL intensity in biosensing platforms. Redox-active polymers (e.g., poly(vinylferrocene)) offer alternative luminol activation pathways for self-powered sensing. Bioconjugation with PEG, peptides, or antibodies/aptamers enhances biocompatibility, reduces immune clearance, and enables target-specific recognition.83 Additionally, using biodegradable polymers (e.g., PLGA) allows for safe degradation and reduces cytotoxicity.84
The second strategy spans a number of encapsulation approaches, where hydrogel-based luminol entrapment provides structural protection and spatial control. Co-encapsulation with catalytic cofactors such as hemin, horseradish peroxidase (HRP), or heme proteins enables reagent-free, enzyme-driven luminescence suitable for point-of-care detection.85–88 Our recent work with hemin-loaded HA hydrogels shows promise for NO detection, and ongoing efforts aim to incorporate luminol directly into the matrix for simplified fabrication and enhanced reagent stability.89 Additionally, stimuli-responsive systems activated by disease-related enzymes (e.g., MMPs or MPO) enable site-specific CL for real-time biomarker monitoring. Moreover, encapsulation within protective nanomaterials can improve luminol's stability and quantum yield. Silica nanoparticles doped with luminol have demonstrated improved performance,90–92 though further development is needed. Similarly, luminol-loaded metal–organic frameworks (MOFs) have been employed as chemiluminescent scaffolds in biosensing.93–95
The third strategy is the development of hybrid and surface-immobilized systems. Multifunctional materials such as surface-immobilized polymer brushes and luminol–quantum dot micelles combine CL with resonance energy transfer (CRET) for tuneable biosensing.96,97 Conductive polymer conjugates also allow for low-voltage ECL by serving as both the catalyst and the electrode. These platforms offer highly modular, reagent-free solutions to conventional luminol limitations.
Advances in synthesis and interdisciplinary applications
Scalability and environmental sustainability are critical in luminol derivative synthesis. Recent trends emphasize green chemistry, such as microwave-assisted polymerization, which reduces reaction time and energy consumption.98,99 Aqueous-phase cross-coupling reactions (e.g., Suzuki–Miyaura) offer solvent-free alternatives for luminol functionalization.100 Eco-friendly catalysts and catalyst recycling further support sustainable practices.In parallel, computational chemistry, including density functional theory (DFT) and AI-based molecular design, can accelerate the discovery of new luminol derivatives by predicting quantum yields and optimizing chemiluminescence profiles.101,102
Luminol derivatives are also extending into interdisciplinary domains such as bioimaging, point-of-care diagnostics, and environmental monitoring. For instance, near-infrared (NIR) luminol analogues are being investigated for deep-tissue imaging, and multi-layer films generating long-lasting NIR emission via peroxyoxalate reactions may be adapted for these systems.103 Integration with microfluidic and lab-on-a-chip platforms further enhances luminol's diagnostic utility.104–106
Together, these emerging strategies position luminol–polymer systems as a next-generation platform for biosensing and diagnostic technologies. Future research should focus on scalable synthesis, AI-guided molecular engineering, and integration with biocompatible, degradable materials to enable real-world applications in clinical, environmental, and industrial settings.
Conclusions
Over the years, significant progress has been made in the synthesis of luminol derivatives, resulting in improved CL efficiency, enhanced solubility, and greater stability. Structural modifications such as alkylation, esterification, and heterocyclic substitution have allowed precise tuning of luminol's photophysical properties, supporting its successful integration into diverse analytical platforms. Additionally, the incorporation of luminol into advanced hybrid materials, such as nanocomposites, polymer matrices, and metal–organic frameworks, has significantly expanded its functional range for biomedical diagnostics, forensic analysis, and environmental sensing.Despite these advancements, critical challenges remain in translating luminol-based systems into real-world applications. Instability under ambient conditions, biocompatibility concerns, and lack of target specificity continue to limit in vivo and point-of-care deployment. To overcome these barriers, recent research has prioritized robust synthetic strategies, including sterically protected molecular designs and machine learning-guided optimization, to enhance stability and predictive performance. Polymer conjugation and nanoscale encapsulation approaches are also being leveraged to amplify signal output, extend luminescence duration, and introduce stimuli-responsive behaviour for dynamic sensing environments.
Looking forward, the development of scalable, green synthesis methods, such as microwave-assisted polymerization and aqueous-phase cross-coupling reactions, will be critical for sustainable manufacturing. Simultaneously, computational chemistry and artificial intelligence offer powerful tools for predicting and tailoring luminol's chemiluminescence behaviour. The continued convergence of synthetic innovation, materials science, and bioengineering will drive the evolution of luminol-based systems into next-generation diagnostic and sensing platforms. With interdisciplinary collaboration and a focus on clinical and environmental translation, luminol derivatives are poised to remain indispensable tools in analytical science and beyond.
Data availability
No new datasets were generated in this study. All data discussed are available in the published literature and cited accordingly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a research grant from Taighde Éireann – Research Ireland (formerly Science Foundation Ireland), co-funded by the European Regional Development Fund (Grant No. 13/RC/2073_P2) and by Grant No. GOIPD/2023/1640.Notes and references
- X. Wang, J. M. Lin, M. L. Liu and X. L. Cheng, TrAC, Trends Anal. Chem., 2009, 28, 75–87 CrossRef CAS.
- F. Barni, S. W. Lewis, A. Berti, G. M. Miskelly and G. Lago, Talanta, 2007, 72, 896–913 CrossRef CAS PubMed.
- D. Mao, W. Wu, S. Ji, C. Chen, F. Hu, D. Kong, D. Ding and B. Liu, Chem, 2017, 3, 991–1007 CAS.
- M. Yang, J. Huang, J. Fan, J. Du, K. Pu and X. Peng, Chem. Soc. Rev., 2020, 49, 6800–6815 RSC.
- C. Dodeigne, L. Thunus and R. Lejeune, Talanta, 2000, 51, 415–439 CrossRef CAS PubMed.
- A. J. Steckl and P. Ray, ACS Sens., 2018, 3, 2025–2044 CrossRef CAS PubMed.
- D. Hong, H.-A. Joung, D. Y. Lee, S. Kim and M.-G. Kim, Sens. Actuators, B, 2015, 221, 1248–1255 CrossRef CAS.
- A. Roda, M. Mirasoli, E. Michelini, M. Di Fusco, M. Zangheri, L. Cevenini, B. Roda and P. Simoni, Biosens. Bioelectron., 2016, 76, 164–179 CrossRef CAS PubMed.
- P. Khan, D. Idrees, M. A. Moxley, J. A. Corbett, F. Ahmad, G. Von Figura, W. S. Sly, A. Waheed and M. I. Hassan, Appl. Biochem. Biotechnol., 2014, 173, 333–355 CrossRef CAS PubMed.
- M. S. Kwon, G. Jang, D. Bilby, B. Milián-Medina, J. Gierschner, T. S. Lee and J. Kim, RSC Adv., 2014, 4, 46488–46493 RSC.
- M. Vacher, I. F. Galván, B.-W. Ding, S. Schramm, R. Berraud-Pache, P. Naumov, N. Ferré, Y.-J. Liu, I. Navizet, D. Roca-Sanjuán, W. J. Baader and R. Lindh, Chem. Rev., 2018, 118, 6927–6974 CrossRef CAS PubMed.
- Z. Wang, J. Huang, J. Huang, B. Yu, K. Pu and F. Xu, Aggregate, 2021, 2(6), 2692–4560 CrossRef.
- A. M. Alsharabasy, P. Farràs and A. Pandit, Anal. Chem., 2024, 96, 7763–7771 CrossRef CAS PubMed.
- K. Akimoto, Y. Shinmen, M. Sumida, S. Asami, T. Amachi, H. Yoshizumi, Y. Saeki, S. Shimizu and H. Yamada, Anal. Biochem., 1990, 189, 182–185 CrossRef CAS PubMed.
- H. Karatani, Anal. Sci., 2022, 38, 613–621 CrossRef CAS PubMed.
- T. Olsson, K. Bergström and A. Thore, Clin. Chim. Acta, 1982, 122, 125–133 CrossRef CAS PubMed.
- C. Plieth, ACS Omega, 2019, 4, 3268–3279 CrossRef CAS PubMed.
- Y. B. Tsaplev, R. F. Vasil'ev, V. D. Kancheva and A. V. Trofimov, Russ. J. Phys. Chem. B, 2020, 14, 431–435 CrossRef CAS.
- W. J. Baader, C. V. Stevani and E. L. Bastos, Chem. Peroxides, 2006, 1211–1278 CAS.
- K. O. Sulaiman, A. T. Onawole, D. T. Shuaib and T. A. Saleh, J. Mol. Liq., 2019, 279, 146–153 CrossRef CAS.
- P. Răzvan and H. Gheorghe, UPB Sci. Bull., Ser. B, 2013, 75, 23–34 Search PubMed.
- H. F. David and L. Blangey, Grundlegende Operationen der Farbenchemie, 1952 Search PubMed.
- I. Vogel, Practical organic chemistry, 1974 Search PubMed.
- R. R. Odle, European Pat., EP0165300B1, 1985 Search PubMed.
- C. Redemann and C. Diazomethane, Org. Synth., Coll., 1955, 244–247 Search PubMed.
- K. Williamson and K. Masters, Macroscale and Microscale Organic Experiments, 2017, pp. 355–357 Search PubMed.
- C. Redemann and C. Diazomethane, Org. Synth., Coll., 1945, 28 CAS.
- C. Dodeigne, L. Thunus and R. Lejeune, Talanta, 2000, 51, 415–439 CrossRef CAS PubMed.
- A. Giussani, P. Farahani, D. Martínez-Muñoz, M. Lundberg, R. Lindh and D. Roca-Sanjuán, Chem. – Eur. J., 2019, 25, 5202–5213 CrossRef CAS PubMed.
- E. H. White and M. M. Bursey, J. Am. Chem. Soc., 1964, 86, 941–942 CrossRef CAS.
- P. D. Wildes and E. H. White, J. Am. Chem. Soc., 1973, 95, 2610–2617 CrossRef CAS.
- Y. Omote, T. Miyake, S. Ohmori and N. Sugiyama, Bull. Chem. Soc. Jpn., 1967, 40, 899–903 CrossRef CAS.
- A. M. Alsharabasy, S. Glynn, P. Farràs and A. Pandit, Nitric Oxide, 2022, 124, 49–67 CrossRef CAS PubMed.
- L. S. Cui, A. J. Gillett, S. F. Zhang, H. Ye, Y. Liu, X. K. Chen, Z. Sen Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J. L. Bredas, C. Adachi and R. H. Friend, Nat. Photonics, 2020, 14, 636–642 CrossRef CAS.
- M. Nakamura and S. Nakamura, Free Radical Biol. Med., 1998, 24, 537–544 CrossRef CAS PubMed.
- A. M. Alsharabasy, D. Cherukaraveedu, J. Warneke, Z. Warneke, J. R. Galán-Mascarós, S. A. Glynn, P. Farràs and A. Pandit, Small Sci., 2024, 2400237 CrossRef CAS PubMed.
- S. Bag, J. C. Tseng and J. Rochford, Org. Biomol. Chem., 2015, 13, 1763–1767 RSC.
- A. Pantelia, I. Daskalaki, M. C. Cuquerella, G. Rotas, M. A. Miranda and G. C. Vougioukalakis, Molecules, 2019, 24, 3957 CrossRef CAS PubMed.
- S. Rink, A. Duerkop, A. Jacobi von Wangelin, M. Seidel and A. J. Baeumner, Anal. Chim. Acta, 2021, 1188, 339161 CrossRef CAS PubMed.
- Y. Omote, T. Miyake and N. Sugiyama, Bull. Chem. Soc. Jpn., 1967, 40, 2446–2448 CrossRef CAS.
- K. Todoroki, Y. Ohba and K. Zaitsu, Chem. Pharm. Bull., 2000, 48, 2011–2013 CrossRef CAS PubMed.
- T. S. Kadhim, M. M. Kareem and A. J. Atiyah, Bull. Chem. Soc. Ethiop., 2023, 37, 159–169 CrossRef CAS.
- T. Mikroulis, M. C. Cuquerella, A. Giussani, A. Pantelia, G. M. Rodríguez-Muñiz, G. Rotas, D. Roca-Sanjuán, M. A. Miranda and G. C. Vougioukalakis, J. Org. Chem., 2021, 86, 11388–11398 CrossRef CAS PubMed.
- R. B. Brundrett and E. H. White, J. Am. Chem. Soc., 1974, 96, 7497–7502 CrossRef CAS.
- H. Neumann, S. Klaus, M. Klawonn, D. Strübing, S. Hübner, D. Gördes, A. J. Von Wangelin, M. Lalk and M. Beller, Z. Naturforsch. B: Chem. Sci., 2004, 59, 431–438 CrossRef CAS.
- R. Pérez-Ruiz, R. Fichtler, Y. Diaz Miara, M. Nicoul, D. Schaniel, H. Neumann, M. Beller, D. Blunk, A. G. Griesbeck and A. Jacobi Von Wangelin, J. Fluoresc., 2010, 20, 657–664 CrossRef PubMed.
- A. G. Griesbeck, Y. Díaz-Miara, R. Fichtler, A. Jacobi Von Wangelin, R. Pérez-Ruiz and D. Sampedro, Chem. – Eur. J., 2015, 21, 9975–9979 CrossRef CAS PubMed.
- M. N. Cortona, N. Vettorazzi, J. J. Silber and L. Sereno, J. Electroanal. Chem., 1995, 394, 245–251 CrossRef.
- G. Periyasami, L. Martelo, C. Baleizão and M. N. Berberan-Santos, New J. Chem., 2014, 38, 2258 RSC.
- F. Chen, X. Xia, D. Ye, T. Li, X. Huang, C. Cai, C. Zhu, C. Lin, T. Deng and F. Liu, Anal. Chem., 2023, 95, 5773–5779 CrossRef CAS PubMed.
- F. Chen, Y. Zhang, T. Li, D. Peng, Z. Qi, J. Song, T. Deng and F. Liu, Chin. Chem. Lett., 2023, 34, 107496 CrossRef CAS.
- J. Han, J. Jose, E. Mei and K. Burgess, Angew. Chem., 2007, 119, 1714–1717 CrossRef.
- H. Ma, Q. Wang, L. Huang, Y. Wang, F. Chen, F. Liu and Y. Ma, Anal. Chem., 2025, 97, 594–601 CrossRef CAS PubMed.
- M. A. Ribi, C. C. Wei and E. H. White, Tetrahedron, 1972, 28, 481–492 CrossRef CAS.
- M. Mayer, S. Takegami, M. Neumeier, S. Rink, A. Jacobi von Wangelin, S. Schulte, M. Vollmer, A. G. Griesbeck, A. Duerkop and A. J. Baeumner, Angew. Chem., Int. Ed., 2018, 57, 408–411 CrossRef CAS PubMed.
- G. Li, X. Zheng and L. Song, Electroanalysis, 2009, 21, 845–852 CrossRef CAS.
- C. M. Geiselhart, C. Barner-Kowollik and H. Mutlu, Polym. Chem., 2021, 12, 1732–1748 RSC.
- Y. W. Liou and C. M. Wang, J. Electroanal. Chem., 2001, 495, 126 CrossRef CAS.
- K. Chiang Lin, S. Yu Lai and S. Ming Chen, Analyst, 2014, 139, 3991 RSC.
- G. F. Zhang and H. Y. Chen, Anal. Chim. Acta, 2000, 419, 25 CrossRef CAS.
- S. M. Chen and K. C. Lin, J. Electroanal. Chem., 2002, 523, 93 CrossRef CAS.
- Y. T. Chang, K. C. Lin and S. M. Chen, Electrochim. Acta, 2005, 51, 450 CrossRef CAS.
- K. C. Lin and S. M. Chen, J. Electroanal. Chem., 2006, 589, 52 CrossRef CAS.
- J. Ballesta-Claver, J. Ametis-Cabello, J. Morales-Sanfrutos, A. Megía-Fernández, M. C. Valencia-Mirón, F. Santoyo-González and L. F. Capitán-Vallvey, Anal. Chim. Acta, 2012, 754, 91 CrossRef CAS PubMed.
- H. Zhang, C. Smanmoo, T. Kabashima, J. Lu and M. Kai, Angew. Chem., Int. Ed., 2007, 46, 8226–8229 CrossRef CAS PubMed.
- H. Zhang, T. Shibata, T. Krawczyk, T. Kabashima, J. Lu, M. K. Lee and M. Kai, Talanta, 2009, 79, 700–705 CrossRef CAS PubMed.
- C. M. Geiselhart, H. Mutlu and C. Barner-Kowollik, Angew. Chem., 2021, 60(32), 17290–17313 CrossRef CAS PubMed.
- H. H. Chu, J. L. Yan and Y. F. Tu, Sensors, 2010, 10, 9481–9492 CrossRef CAS PubMed.
- Y. Lin, Y. Dai, Y. Sun, C. Ding, W. Sun, X. Zhu, H. Liu and C. Luo, Talanta, 2018, 182, 116–124 CrossRef CAS PubMed.
- R. Wu, Y. He, Z. Li, Z. Yu, X. Guo and H. Yan, Chem. – Eur. J., 2025, e202501133 CrossRef PubMed.
- J. Li, Q. Xu, C. Fu and Y. Zhang, Sens. Actuators, B, 2013, 185, 146–153 CrossRef CAS.
- C. Fu, Q. Xu, X. Wei and J. Li, RSC Adv., 2014, 4, 26102 RSC.
- X. Jiang, H. Wang, R. Yuan and Y. Chai, Biosens. Bioelectron., 2015, 63, 33–38 CrossRef CAS PubMed.
- Y. Omote, T. Miyake, S. Ohmori and N. Sugiyama, Bull. Chem. Soc. Jpn., 1966, 39, 932–935 CrossRef CAS.
- T. Shibata, H. Yoshimura, A. Yamayoshi, N. Tsuda and S. Dragusha, Chem. Pharm. Bull., 2019, 67, 772–774 CrossRef CAS PubMed.
- S. Depienne, D. Alvarez-Dorta, M. Croyal, R. C. T. Temgoua, C. Charlier, D. Deniaud, M. Mével, M. Boujtita and S. G. Gouin, Chem. Sci., 2021, 12, 15374–15381 RSC.
- O. K. Glebov, D. S. Abramian, S. R. Romanov and L. V. Smagina, Tsitologiya, 1994, 36, 441–452 CAS.
- G. Briheim, O. Stendahl and C. Dahlgren, Infect. Immun., 1984, 45, 1–5 CrossRef CAS PubMed.
- N. S. Moyon and S. Mitra, Chem. Phys. Lett., 2010, 498, 178–183 CrossRef CAS.
- S. Chan, A. Palone, M. Bietti and M. Costas, Angew. Chem., Int. Ed., 2024, 63, e202402858 CrossRef CAS PubMed.
- F. Liu and L. Chen, J. Mol. Liq., 2023, 390, 122904 CrossRef CAS.
- V. I. Gomzyak, N. G. Sedush, A. A. Puchkov, D. K. Polyakov and S. N. Chvalun, Polym. Sci., Ser. B, 2021, 63, 257–271 CrossRef CAS.
- D. M. Valcourt, J. Harris, R. S. Riley, M. Dang, J. Wang and E. S. Day, Nano Res., 2018, 11, 4999–5016 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- Q. Wang, L. Li and B. Xu, Chem. – Eur. J., 2009, 15, 3168–3172 CrossRef CAS PubMed.
- J. Ye, L. Zhu, M. Yan, T. Xiao, L. Fan, Y. Xue, J. Huang and X. Yang, Talanta, 2021, 230, 122351 CrossRef CAS PubMed.
- Y. Ning, F. Lu, Y. Liu, S. Yang, F. Wang, X. Ji and Z. He, Analyst, 2021, 146, 4775–4780 RSC.
- D. Calabria, A. Pace, E. Lazzarini, I. Trozzi, M. Zangheri, M. Guardigli, S. Pieraccini, S. Masiero and M. Mirasoli, Biosensors, 2023, 13, 650 CrossRef CAS PubMed.
- A. M. Alsharabasy, M. Sankar, M. Biggs, P. Farràs and A. Pandit, Talanta, 2024, 126522 CrossRef CAS PubMed.
- K. J. Qian, L. Zhang, M. L. Yang, P. G. He and Y. Z. Fang, Chin. J. Chem., 2004, 22, 702–707 CrossRef CAS.
- L. Zhang and X. Zheng, Anal. Chim. Acta, 2006, 570, 207–213 CrossRef CAS PubMed.
- M. Zhao, W. Qi, Y. Fu, H. He, D. Wu, L. Qi and R. Li, Microchim. Acta, 2019, 186, 409 CrossRef PubMed.
- H. Chen, D. Zou, W. Cao, Y. Cui, G. Qian and Z. Liao, J. Mater. Chem. C, 2024, 12, 11012–11019 RSC.
- J. Hassanzadeh, H. A. J. Al Lawati and N. Bagheri, Talanta, 2024, 276, 126219 CrossRef CAS PubMed.
- A. Hussain, F. A. Bushira, Z. Dong, A. M. A. Alboull, S. S. Tessema, M. Y. Suleiman and G. Xu, Anal. Chem., 2024, 96, 13504–13511 CrossRef CAS PubMed.
- X. Huang, L. Li, H. Qian, C. Dong and J. Ren, Angew. Chem., Int. Ed., 2006, 45, 5140–5143 CrossRef CAS PubMed.
- E. S. Lee, V. G. Deepagan, D. G. You, J. Jeon, G.-R. Yi, J. Y. Lee, D. S. Lee, Y. D. Suh and J. H. Park, Chem. Commun., 2016, 52, 4132–4135 RSC.
- S. Jadoun, S. M. Ashraf and U. Riaz, Polym. Adv. Technol., 2018, 29, 1007–1017 CrossRef CAS.
- U. Riaz, S. Jadoun, P. Kumar, R. Kumar and N. Yadav, RSC Adv., 2018, 8, 37165–37175 RSC.
- H. Türkmen, R. Can and B. Çetinkaya, Dalton Trans., 2009, 7039–7044 RSC.
- A. Borrego-Sánchez, A. Giussani, M. Rubio and D. Roca-Sanjuán, Phys. Chem. Chem. Phys., 2020, 22, 27617–27625 RSC.
- L. Türker, Earthline J. Chem. Sci., 2024, 515–537 Search PubMed.
- W. Qiao, X. Zheng and Z. Y. Wang, J. Lumin., 2016, 180, 88–94 CrossRef CAS.
- K. S. Lok, Y. C. Kwok and N.-T. Nguyen, Sens. Actuators, B, 2012, 169, 144–150 CrossRef CAS.
- B. Al Mughairy, H. A. J. Al-Lawati and F. O. Suliman, Spectrochim. Acta, Part A, 2019, 221, 117182 CrossRef CAS PubMed.
- B. Yang, L. Shi, Q. Tang, W. Liu, B. Li, C. Yang and Y. Jin, Lab Chip, 2023, 23, 785–792 RSC.
| This journal is © The Royal Society of Chemistry 2025 |