
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric intramolecular propargylation of cyclopropanols to access the cuparane core†
Yankun
Zhao
,
Hongya
Yan
,
Yulian
Zhang
,
Tao
Zhou
,
Mengxing
Tian
,
Chongzhou
Zhang
,
Shan
Yuan
,
Hanyue
Qiu
*,
Ling
He
 and
Min
Zhang
*
and
Min
Zhang
*
Chongqing Key Laboratory of Natural Product Synthesis and Drug Research, Innovative Drug Research Center, School of Pharmaceutical Sciences, Chongqing University, Chongqing 401331, China. E-mail: hanyue.qiu@cqu.edu.cn; minzhang@cqu.edu.cn
First published on 13th June 2024
Abstract
The catalytic asymmetric propargylation of enol(ate) intermediates is a well-established method for the synthesis of α-propargyl-substituted carbonyl compounds. However, the propargylation of homo-enol(ate) or its equivalents for the synthesis of β-propargyl-substituted carbonyl compounds remains underdeveloped. A catalytic enantioselective decarboxylative intramolecular propargylation of cyclopropanols has been developed using a PyBox-complexed copper catalyst. This reaction offers an effective approach to assemble a cyclopentanone skeleton bearing an all-carbon quaternary stereogenic center and an adjacent quaternary gem-dimethyl carbon center, which is the core scaffold of the naturally occurring cuparenoids. Key to the success of this protocol is the use of a new structurally optimized PyBox ligand. This study represents the first example of catalytic asymmetric intramolecular propargylation of cyclopropanols.
Introduction
The catalytic asymmetric allylation and propargylation of enol(ate) intermediates are well-established methods to synthesize α-allyl- or α-propargyl-substituted carbonyl compounds.1–8 In contrast, the analogous reactions of homo-enol(ate) or its equivalents for the synthesis of β-allyl- or β-propargyl-substituted carbonyl compounds have been lagging far behind (Fig. 1A).9–14 As one of the most prominent homo-enol equivalents, cyclopropanol has been demonstrated to undergo facile ring-opening coupling reactions with various electrophiles.9–14 Its catalytic asymmetric reaction with allyl electrophiles, however, remains underexplored until recently. Yin15 and Trost16 reported the catalytic asymmetric allylation of cyclopropanols with allyl phosphates.15–22 Despite that, the catalytic asymmetric ring-opening propargylation of cyclopropanols, to the best of our knowledge, has not been reported yet (Fig. 1A).23 In this context, establishment of a catalytic system ensuring an enantioselective propargylic substitution with cyclopropanols is highly desirable, not only because it expands the scope of the applicable electrophiles for the cyclopropanol, but also because it generates synthetically valuable β-propargyl ketones.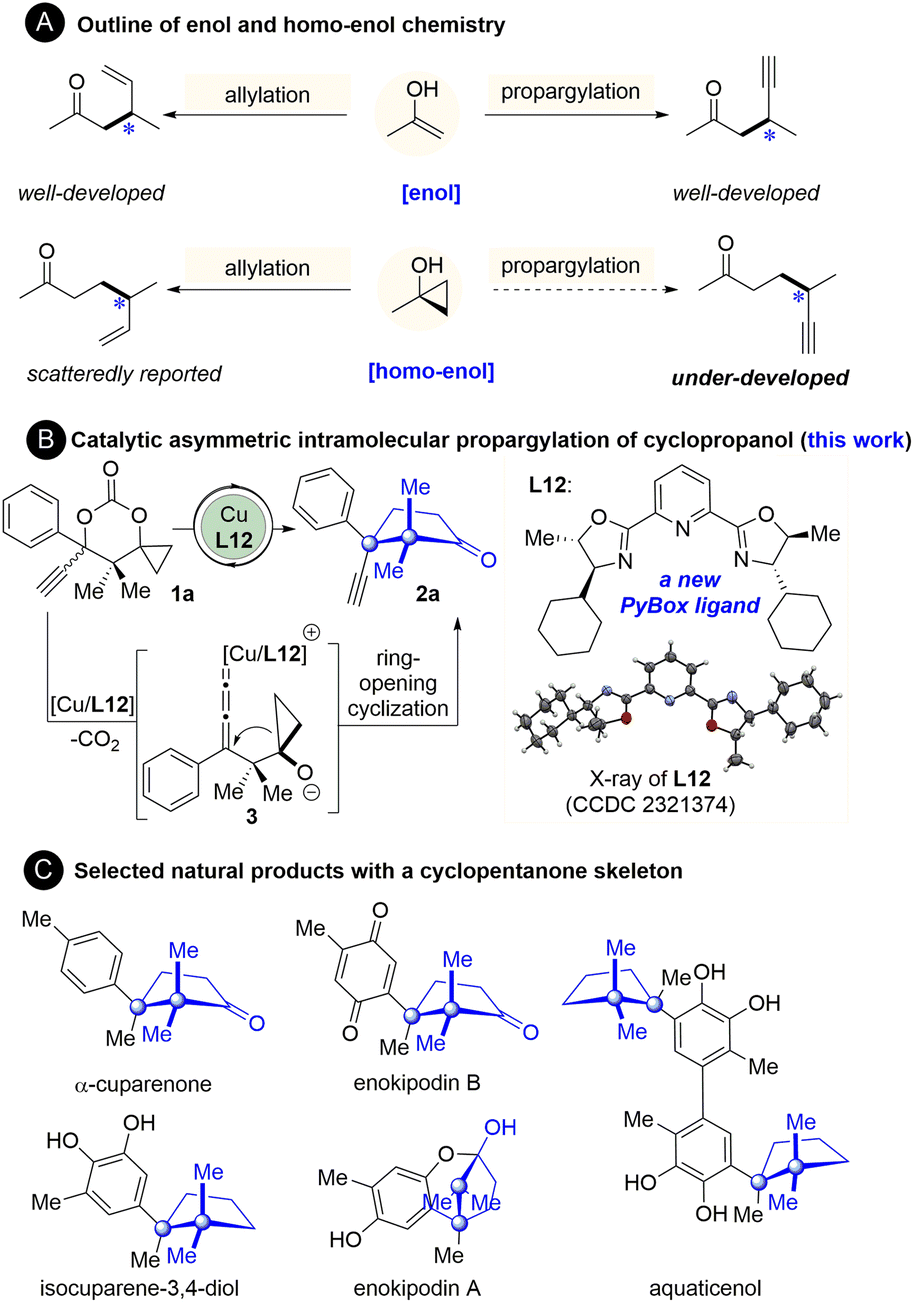 | ||
| Fig. 1 Background and study synopsis. (A) Outline of the enol and homo-enol chemistry. (B) Catalytic asymmetric intramolecular propargylation of cyclopropanol (this work). (C) Selected natural products with a cyclopentanone skeleton. | ||
At the outset of the present study, the feasibility and challenges of propargylation of cyclopropanol were first considered. The great advances made in the area of catalytic asymmetric propargylic substitutions of propargylic electrophiles with an array of nucleophiles in the past few years indicate the feasibility of this reaction.2–8,24 Despite that, there are inherent issues that need to be addressed. In Cha,18 Yoshikai,19 Yin,15 and Trost's16 ring-opening allylation of cyclopropanol, preactivation of the cyclopropanol unit as a zinc or potassium alkoxide salt is essential prior to the transmetallation with copper or nickel for the subsequent allylic substitution.15–22 These studies together with our experience25–30 and others' reports31–36 on ring-opening reactions of cyclopropanols indicate that how to activate the cyclopropanol unit is key to the success of the ring-opening reactions of cyclopropanols and thus should be identified primarily. Furthermore, an asymmetric catalytic system applicable to the reaction of cyclopropanol remains underdeveloped,9–14,37–43 which highlights the need to establish an effective asymmetric catalytic system for this new reaction.
To address the aforementioned issues, we have developed the first and highly enantioselective intramolecular propargylation of cyclopropanol, which was enabled by a new structurally optimized pybox ligand-complexed copper catalyst.44 The cyclopropanol and propargyl units were simultaneously activated as cyclopropoxide and copper allenylidene respectively by decarboxylation. This protocol furnishes a series of cyclopentanones bearing an all-carbon quaternary stereogenic center adjacent to a quaternary gem-dimethyl carbon center (Fig. 1B). This sterically congested motif is the cuparane core shared by a number of naturally occurring cuparenoids, an eminent family of sesquiterpenes which were isolated from a culture broth of a mushroom (Flammulina velutipes) and other natural sources and exhibited a series of antimicrobial activities (Fig. 1C).45–52
Results and discussion
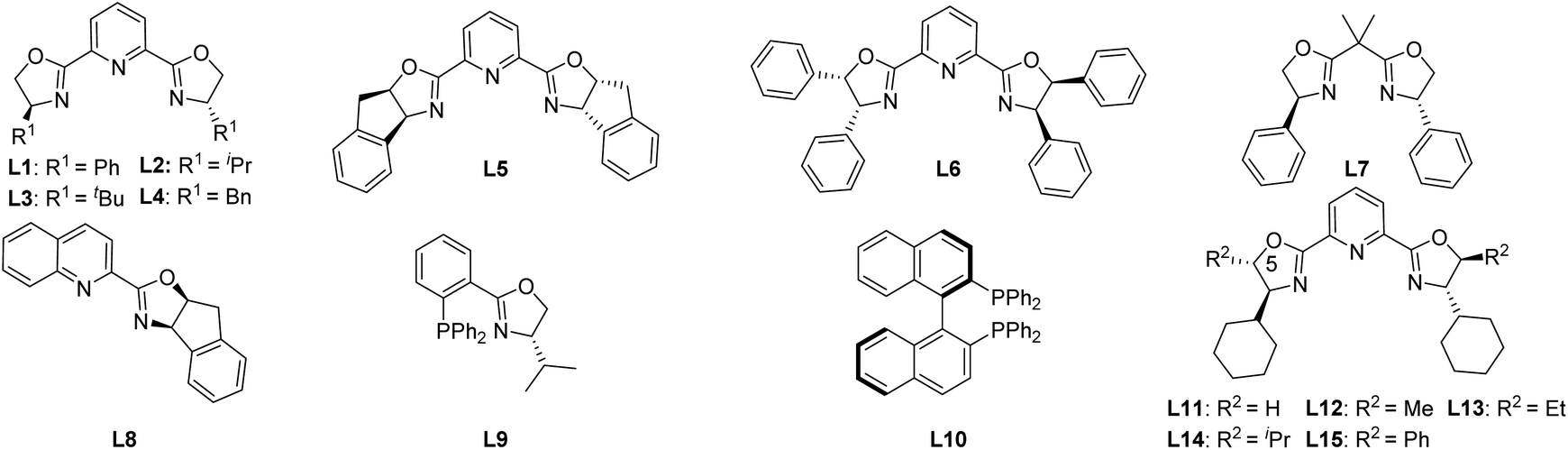
We commenced the study by using carbonate 1a as the model substrate and by screening an array of chiral ligands (Table 1) (see the ESI† for details). Encouragingly, an initial attempt with Cu(OTf)2 and Ph-PyBox ligand L1 produced the desired propargylation product 2a albeit in 35% yield and with 20% ee value (Table 1, entry 1). With this promising result, other copper salts were then screened. After evaluation of several copper(I) and (II) salts (see the ESI† for details), Cu(MeCN)4PF6 was identified as the best one, which improved both the yield and ee value significantly (Table 1, entry 2). After evaluation of other bases (see the ESI† for details), NaHCO3 proved to be the best choice, improving the yield to 86% and ee value to 68% (Table 1, entry 3). Since the subtle structural changes of the ligands would have a significant impact on the reaction outcome, an array of known bidentate and tridentate ligands including the PyBox ligands (L2–L6, L11), Box ligand L7, QuinOx ligand L8, PhOx ligand L9, and Binap L10 were then evaluated to improve the enantioselectivity (Table 1, entries 4–9). All these ligands resulted in a substantial decrease of not only the yield but also the enantioselectivity except the Cy-PyBox ligand L11 which provided an improved level of enantiocontrol (90% ee, Table 1, entry 10). On the basis of these results, we resorted to the design and synthesis of new PyBox ligands to further improve the reaction efficiency. Gratifyingly, introducing a methyl group at C5 of each oxazoline motif of the Cy-PyBox ligand L11 delivered an excellent yield and enantioselectivity (90% yield, 94% ee, Table 1, entry 11), which was slightly improved by reducing the reaction temperature from 80 to 60 °C (Table 1, entry 12), while introducing more hindered groups such as Et-, iPr-, and Ph- at C5 was fruitless (L13–L15, Table 1, entries 13–15).|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | Ligand | Base | Yieldb (%) | ee (%) |
| a Typical reaction conditions unless otherwise noted: 1a (0.1 mmol), metal salt (10 mol%), ligand (12 mol%), base (1 equiv.), DCE (4 mL), 80 °C, 48 h. b Isolated yield of 2a. c Determined by chiral HPLC analysis. d Reaction was conducted at 60 °C. | |||||
| 1 | Cu(OTf)2 | L1 | DIPEA | 35 | 20 |
| 2 | Cu(MeCN)4PF6 | L1 | DIPEA | 75 | 66 |
| 3 | Cu(MeCN)4PF6 | L1 | NaHCO3 | 86 | 68 |
| 4 | Cu(MeCN)4PF6 | L2 | NaHCO3 | 86 | 64 |
| 5 | Cu(MeCN)4PF6 | L3 | NaHCO3 | 19 | 40 |
| 6 | Cu(MeCN)4PF6 | L4 | NaHCO3 | 67 | 15 |
| 7 | Cu(MeCN)4PF6 | L5 | NaHCO3 | 57 | −22 |
| 8 | Cu(MeCN)4PF6 | L6 | NaHCO3 | 48 | 6 |
| 9 | Cu(MeCN)4PF6 | L7–L10 | NaHCO3 | 0 | — |
| 10 | Cu(MeCN)4PF6 | L11 | NaHCO3 | 85 | 90 |
| 11 | Cu(MeCN)4PF6 | L12 | NaHCO3 | 90 | 94 |
| 12 | Cu(MeCN) 4 PF 6 | L12 | NaHCO 3 | 95 | 95 |
| 13d | Cu(MeCN)4PF6 | L13 | NaHCO3 | 85 | 95 |
| 14d | Cu(MeCN)4PF6 | L14 | NaHCO3 | 85 | 86 |
| 15d | Cu(MeCN)4PF6 | L15 | NaHCO3 | 20 | 70 |
|
|||||

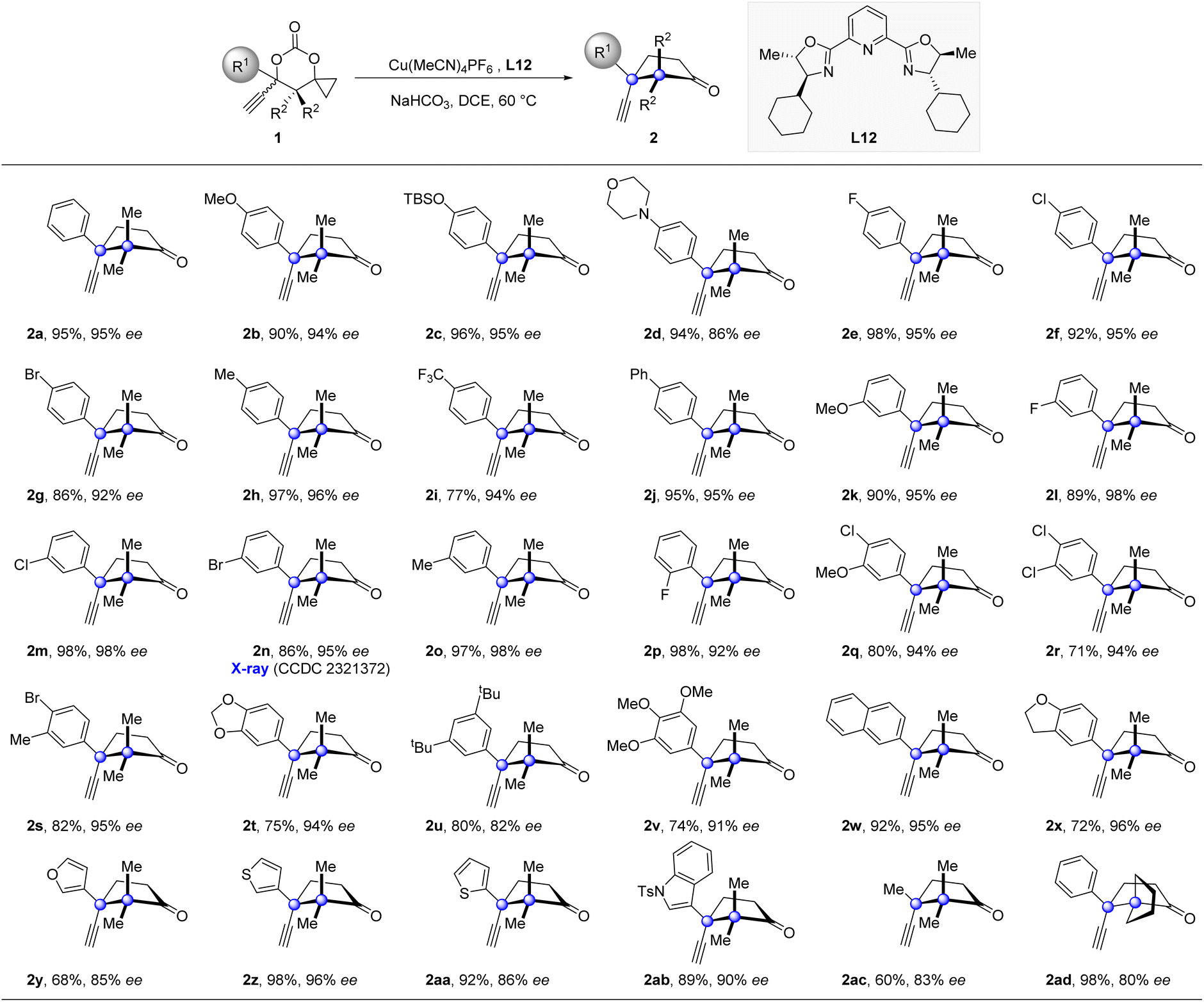
Having established the optimized catalytic system, we then explored the generality of this enantioselective propargylation of cyclopropanols (Table 2). A broad range of substrates, possessing both electro-donating and -withdrawing groups at the para-position of the phenyl ring, were well tolerated to furnish the desired products in high yields with excellent enantioselectivies (2b–2j). The variation of the substituent pattern has a subtle impact on the reaction outcome. For instance, the substrates 1k, 1l, 1m, 1n, 1o, and 1p, which possess MeO-, F-, Cl-, Br-, and Me- substituents at the meta- or ortho-position of the phenyl ring, all participated in the transformations and thus provided cyclopentanones (2k–2p) in high enantioselectivities with handles for further derivations. Furthermore, substrates with two or three substituents of different nature at the phenyl ring were tolerated as well (2q–2v). 1w and 1x bearing a bicyclic aromatic substituent also proved to be viable substrates. The substrate scope was further expanded to include substrates substituted with heteroaryl groups, including furyl, thienyl, and indolyl groups (2y–2ab). Notably, replacing the phenyl group with a methyl group also produced product 2ac with good enantioselectivity in synthetically useful yield. Moreover, 1ad with a spiropentyl group was also a suitable substrate. The relative and absolute configurations of 2n were determined by single-crystal X-ray crystallographic analysis (see the ESI† for details), and the same configurations were analogously assigned to the other products.
| a Reaction conditions: 1 (0.1 mmol), Cu(MeCN)4PF6 (10 mol%), L12 (12 mol%), NaHCO3 (1 equiv.), DCE (4 mL), 60 °C, 24–48 h; isolated yields were reported and ee values were determined by chiral HPLC analysis. |
|---|
|
Since the products generated by this new protocol bear a naturally occurring cuparane core, introduction of such a fragment into therapeutic molecules would be of pharmaceutical significance.53 To demonstrate the potential synthetic utility of this protocol in drug development, derivations of drugs were carried out (Fig. 2). The fructose with two acetal groups, core scaffold of the anticonvulsant drug topiramate, was introduced with cuparane cores having different absolute configurations in high diastereoselectivities under the typical reaction conditions with L12 or ent-L12 (2ae and 2ae′) as the ligand. The same manipulation was also applied to the anti-inflammatory drugs naproxen and indomethacin (2af, 2af′, and 2ag).
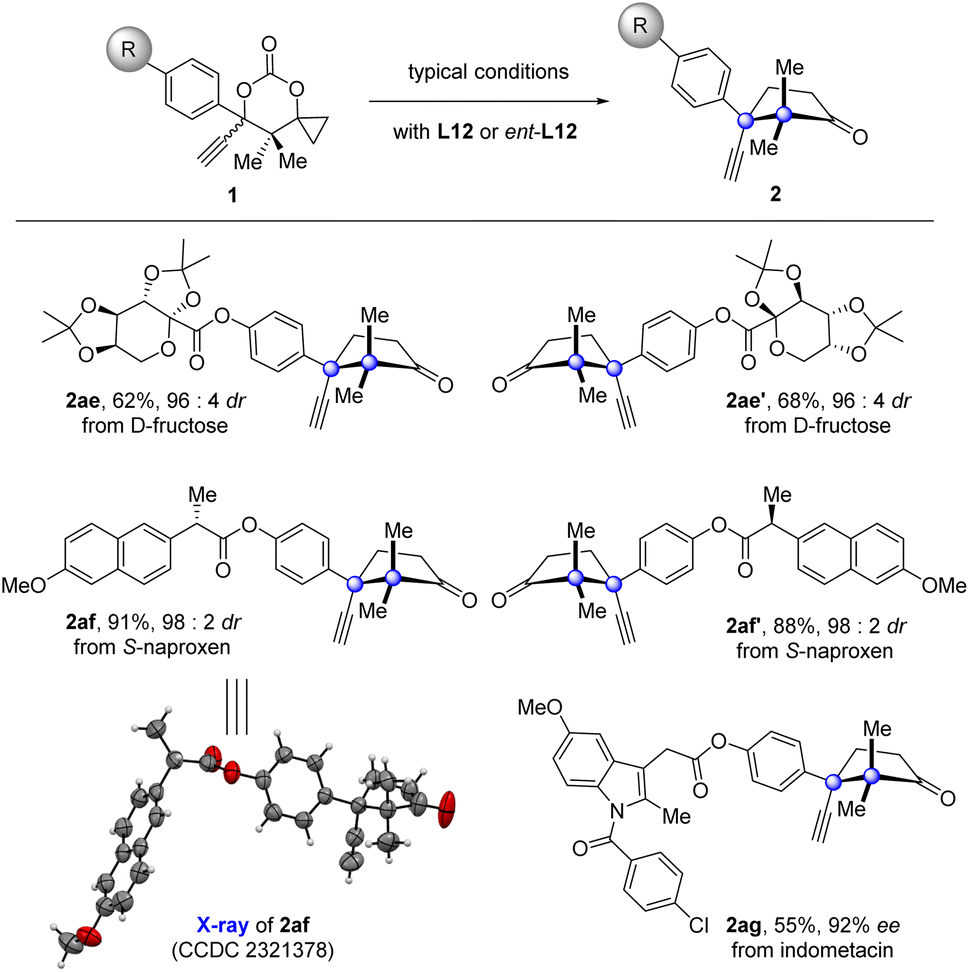 | ||
| Fig. 2 Drug derivations. L12 was used for 2ae, 2af, and 2ag; ent-L12 was used for 2ae′ and 2af′. | ||
Besides the high yields and enantioselectivity, this key cyclization reaction boasts good scalability. Grams of 2h were conveniently accumulated by the reaction conducted at the gram scale without affecting the reaction efficiency and enantioselectivity (Fig. 3). Further elaborations at the alkynyl-substituted cuparane core of 2h were carried out. Saturation of the alkynyl group of 2h delivered the natural product analogue 6-methyl-α-cuparenone (4) in 95% yield. The click reaction of 2h and azide 5 possessing a biotin segment provided 6, which could serve as a chemical probe to investigate the biological properties of the cuparenoids. Taking advantage of the alkynyl group, a reaction of Larock indole synthesis delivered indolyl-substituted product 7. Furthermore, a sequence of partial saturation of the alkyne, allyl addition to the ketone group, and ring closing metathesis with the Grubbs II catalyst furnished the bridged product 8 in a synthetically useful yield.
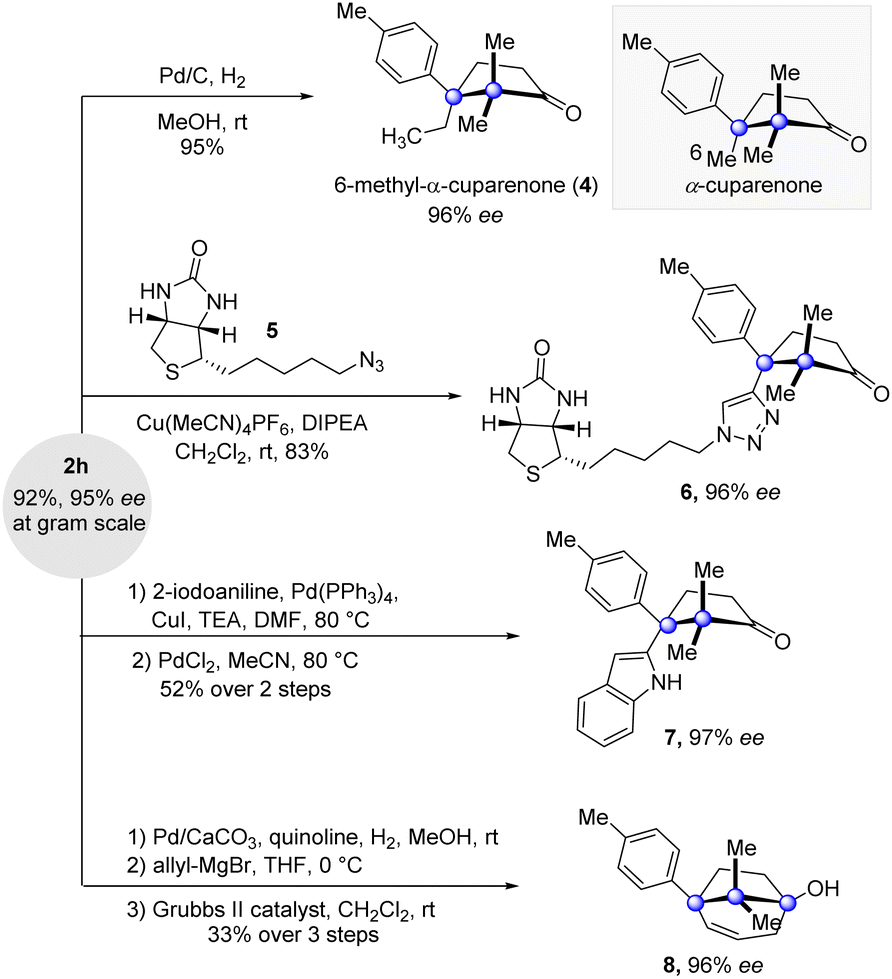 | ||
| Fig. 3 Product derivations. | ||
To gain more mechanistic insights into the current reaction, several experiments were conducted (Fig. 4). Substrate 1ah with an additional PMB group at the cyclopropane moiety generated product 2ah with the cleavage of the less substituted bond (Fig. 4A). This result indicates that an ionic cyclopropanol ring-opening mechanism might operate since it is generally accepted that the radical cyclopropanol ring-opening process favors breaking the more substituted to produce a more stable radical intermediate, while the ionic ring-opening process tends to break the less substituted one.9–14 The reaction of 1a proceeded well in the presence of the radical scavenger TEMPO or BHT, further confirming that this is a non-radical process (Fig. 4B). Non-linear effect experiments were carried out with ee-varied chiral L12 (Fig. 4C). The results clearly showed the linear relationship between the ee value of L12 and the ee value of product 2a, indicating that a monomeric complex may work as a catalyst in the reaction.8 On the basis of our mechanistic experiments and the literature precedents,2–8,24 a plausible catalytic cycle and stereoinduction model were proposed (Fig. 4D). First, deprotonation of 1a generated the copper acetylide intermediate 9. The monocopper allenylidene complex 3 with the cyclopropanol segment activated as a cyclopropoxide is then formed by decarboxylation. Ring-opening cyclization of the cyclopropoxide unit to the copper allenylidene affords intermediate 11. The asymmetric induction may be rationalized by a spatial repulsion exerted by the cyclohexyl group, in which re-face attack on the copper allenylidene occurs preferentially. Finally, protodemetalation occurs to furnish product 2a and to release the L12-complexed copper species, which re-enters the catalytic cycle.
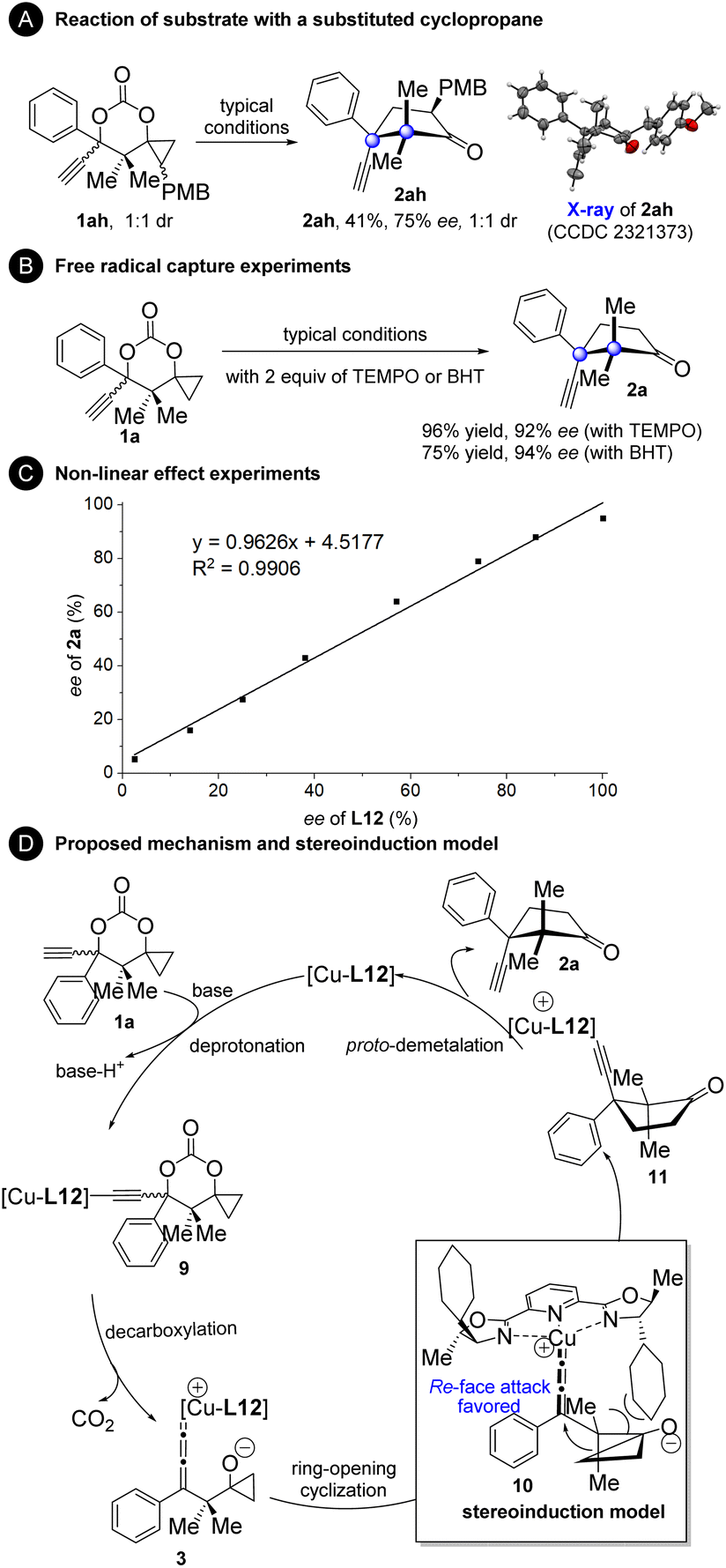 | ||
| Fig. 4 Mechanism studies. (A) Reaction of substrate with a substituted cyclopropane. (B) Free radical capture experiments. (C) Non-linear effect experiments. (D) Proposed mechanism and stereoinduction model. | ||
Conclusion
In summary, we have developed a highly enantioselective copper-catalyzed intramolecular propargylation of cyclopropanols. Key to the success of this method is the meticulous structural optimization of the PyBox ligand, which ensures good yields and excellent enantioselectivities for this reaction. This novel protocol presents a reasonably broad substrate scope and excellent functional group tolerance and provides straightforward access to a diverse range of difficult to access cyclopentanones bearing an all-carbon quaternary stereogenic center and an adjacent quaternary gem-dimethyl carbon center. This study represents the first catalytic asymmetric intramolecular propargylation of cyclopropanols and thus enriches the homoenol(ate) chemistry.Data availability
All data associated with this article are available in the ESI.†Author contributions
Conceptualization: M. Z.; investigation: Y. Z., H. Y., Y. Z., T. Z., M. T., C. Z. and S. Y.; formal analysis: Y. Z., H. Y., Y. Z., T. Z., M. T., C. Z. and S. Y. writing – original draft: M. Z., Y. Z. and H. Q.; writing – review and editing: Y. Z., H. Q. and L. H. All authors discussed the results and gave their approval of the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21922102 and 22271033), Graduate Scientific Research and Innovation Foundation of Chongqing, China (CYB22075), Chongqing Science and Technology Commission (CSTB2022NSCQLZX0036), Luzhou Science and Technology Program (2022-XDY-191), and the Fundamental Research Funds for the Central Universities (2022CDJQY-001). We are grateful to Mr Xiangnan Gong (CQU) for X-ray crystallographic analysis.Notes and references
- T. B. Wright and P. A. Evans, Chem. Rev., 2021, 121, 9196–9242 CrossRef CAS PubMed.
- S. W. Roh, K. Choi and C. Lee, Chem. Rev., 2019, 119, 4293–4356 CrossRef CAS PubMed.
- F.-L. Zhu, Y. Zou, D.-Y. Zhang, Y.-H. Wang, X.-H. Hu, S. Chen, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 1410–1414 CrossRef CAS PubMed.
- Q. Wang, T.-R. Li, L.-Q. Lu, M.-M. Li, K. Zhang and W.-J. Xiao, J. Am. Chem. Soc., 2016, 138, 8360–8363 CrossRef CAS PubMed.
- J. Song, Z.-J. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 5212–5216 CrossRef CAS PubMed.
- X. Gao, R. Cheng, Y.-L. Xiao, X.-L. Wan and X. Zhang, Chem, 2019, 5, 2987–2999 CAS.
- B. G. Das, S. Shah, A. Das and V. K. Singh, Org. Lett., 2021, 23, 6262–6266 CrossRef CAS PubMed.
- H.-H. Kong, C.-J. Zhu, S. Deng, G. Xu, R.-N. Zhao, C.-C. Yao, H.-M. Xiang, C.-H. Zhao, X.-T. Qi and H. Xu, J. Am. Chem. Soc., 2022, 144, 21347–21355 CrossRef CAS PubMed.
- O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597–2632 CrossRef CAS PubMed.
- A. Nikolaev and A. Orellana, Synthesis, 2016, 48, 1741–1768 CrossRef CAS.
- T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Chem. Rev., 2021, 121, 3–79 CrossRef CAS PubMed.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS PubMed.
- X. Cai, W. Liang and M. Dai, Tetrahedron, 2019, 75, 193–208 CrossRef CAS.
- H. Yan, G. S. Smith and F.-E. Chen, Green Synth. Catal., 2022, 3, 219–226 CrossRef CAS.
- Q. Zhang, S.-W. Zhou, C.-Y. Shi and L. Yin, Angew. Chem., Int. Ed., 2021, 60, 26351–26356 CrossRef CAS PubMed.
- B. M. Trost, G. Zhang, M. Xu and X. Qi, Chem.–Eur. J., 2022, 28, e202104268 CrossRef CAS PubMed.
- E. Nakamura, S. Aoki, K. Sekiya, H. Oshino and I. Kuwajima, J. Am. Chem. Soc., 1987, 109, 8056–8066 CrossRef CAS.
- P. P. Das, K. Belmore and J. K. Cha, Angew. Chem., Int. Ed., 2012, 51, 9517–9520 CrossRef CAS PubMed.
- Y. Sekiguchi, Y. Y. Lee and N. Yoshika, Org. Lett., 2021, 23, 5993–5997 CrossRef CAS PubMed.
- J. Lin, T. Zhu, M. Jia and S. Ma, Chem. Commun., 2019, 55, 4523–4526 RSC.
- A. Kitabayashi, S. Mizushima, K. Higashida, Y. Yasuda, Y. Shimizu and M. Sawamura, Adv. Synth. Catal., 2022, 364, 1855–1862 CrossRef CAS.
- J. Hou, X. Li, K. Yan, L. Zhang, T. P. Loh and P. Xie, Chem. Sci., 2024, 15, 1143–1149 RSC.
- P. Wu, M. Jia, W. Lin and S. Ma, Org. Lett., 2018, 20, 554–557 CrossRef CAS PubMed.
- Z. Fu, N. Deng, S.-N. Su, H. Li, R.-Z. Li, X. Zhang, J. Liu and D.-W. Niu, Angew. Chem., Int. Ed., 2018, 57, 15217–15221 CrossRef CAS PubMed.
- Q. Tan, Z. Yang, D. Jiang, Y. Cheng, J. Yang, S. Xi and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 6420–6424 CrossRef CAS PubMed.
- Z. Yang, Q. Tan, Y. Jiang, J. Yang, X. Su, Z. Qiao, W. Zhou, L. He, H. Qiu and M. Zhang, Angew. Chem., Int. Ed., 2021, 60, 13105–13111 CrossRef CAS PubMed.
- W. Zhou, T. Zhou, M. Tian, Y. Jiang, J. Yang, S. Lei, Q. Wang, C. Zhang, H. Qiu, L. He, Z. Wang, J. Deng and M. Zhang, J. Am. Chem. Soc., 2021, 143, 19975–19982 CrossRef CAS PubMed.
- D. Jiang, P. Tang, H. Xiong, S. Lei, Y. Zhang, C. Zhang, L. He, H. Qiu and M. Zhang, Angew. Chem., Int. Ed., 2023, 62, 202307286 CrossRef PubMed.
- H. Qiu, X. Fei, J. Yang, Z. Qiao, S. Yuan, H. Zhang, L. He and M. Zhang, Nat. Commun., 2023, 14, 5560–5567 CrossRef CAS PubMed.
- W. Zhou, S. Xi, H. Chen, D. Jiang, J. Yang, S. Liu, L. He, H. Qiu, Y. Lan and M. Zhang, Nat. Chem., 2023, 15, 1074–1082 CrossRef CAS PubMed.
- S. Diethelm and E. M. Carreira, J. Am. Chem. Soc., 2015, 137, 6084–6096 CrossRef CAS PubMed.
- N. N. Rao and J. K. Cha, J. Am. Chem. Soc., 2015, 137, 2243–2246 CrossRef CAS PubMed.
- D. C. Davis, K. L. Walker, C. Hu, R. N. Zare, R. M. Waymouth and M. Dai, J. Am. Chem. Soc., 2016, 138, 10693–10699 CrossRef CAS PubMed.
- K. Ma, X. Yin and M. Dai, Angew. Chem., Int. Ed., 2018, 57, 15209–15212 CrossRef CAS PubMed.
- K. Yu, Z.-N. Yang, C.-H. Liu, S.-Q. Wu, X. Hong, X.-L. Zhao and H. Ding, Angew. Chem., Int. Ed., 2019, 58, 8556–8560 CrossRef CAS PubMed.
- L. Wu, L. Wang, P. Chen, Y.-L. Guo and G. Liu, Adv. Synth. Catal., 2020, 362, 2189–2194 CrossRef.
- X. Cai, W. Liang, M. Liu, X. Li and M. Dai, J. Am. Chem. Soc., 2020, 142, 13677–13682 CrossRef CAS PubMed.
- Ł. Woźniak, G. Magagnano and P. Melchiorre, Angew. Chem., Int. Ed., 2018, 57, 1068–1072 CrossRef PubMed.
- C. Jiang, L. Wang, H. Zhang, P. Chen, Y.-L. Guo and G. Liu, Chem, 2020, 6, 2407–2419 CAS.
- Y. Sekiguchi and N. Yoshikai, J. Am. Chem. Soc., 2021, 143, 4775–4781 CrossRef CAS PubMed.
- W. Huang and F. Meng, Angew. Chem., Int. Ed., 2021, 60, 2694–2698 CrossRef CAS PubMed.
- J. Wang and X. Li, Chem. Sci., 2022, 13, 3020–3026 RSC.
- M. A. S. Blackburn, C. C. Wagen, M. Raul Bodrogean, P. M. Tadross, A. J. Bendelsmith, D. A. Kutateladze and E. N. Jacobsen, J. Am. Chem. Soc., 2023, 145, 15036–15042 CrossRef CAS PubMed.
- R. Connon, B. Roche, B. V. Rokade and P. J. Guiry, Chem. Rev., 2021, 121, 6373–6521 CrossRef CAS PubMed.
- G. L. Chetty and S. Dev, Tetrahedron Lett., 1964, 5, 73–77 CrossRef.
- N. K Ishikawa, K. Yamaji, S. Tahara, Y. Fukushi and K. Takahashi, Phytochemistry, 2000, 54, 777–782 CrossRef PubMed.
- Y. Fukuyama and Y. Asakawa, J. Chem. Soc., Perkin Trans., 1991, 1, 2737–2741 RSC.
- N. K. Ishikawa, Y. Fukushi, K. Yamaji, S. Tahara and K. Takahashi, J. Nat. Prod., 2001, 64, 932–934 CrossRef CAS PubMed.
- D. M. Hodgson, Y. K. Chung, I. Nuzzo, G. Freixas, K. K. Kulikiewicz, E. Cleator and J. M. Paris, J. Am. Chem. Soc., 2007, 129, 4456–4462 CrossRef CAS PubMed.
- A. Natarajan, D. Ng, Z. Yang and M. A. Garcia-Garibay, Angew. Chem., Int. Ed., 2007, 46, 6485–6487 CrossRef CAS PubMed.
- P. Zhang, H. Le, R. E. Kyne and J. P. Morken, J. Am. Chem. Soc., 2011, 133, 9716–9719 CrossRef CAS PubMed.
- W. Zhou and A. Voituriez, J. Am. Chem. Soc., 2021, 143, 17348–17353 CrossRef CAS PubMed.
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2321372, 2321373, 2321374 and 2321378. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02504k |
| This journal is © The Royal Society of Chemistry 2024 |