
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomically dispersed cobalt catalysts for tandem synthesis of primary benzylamines from oxidized β-O-4 segments†
Sen
Luan‡
ab,
Wei
Wu‡
c,
Bingxiao
Zheng‡
cf,
Yuxuan
Wu
ab,
Minghua
Dong
ab,
Xiaojun
Shen
 d,
Tianjiao
Wang
ab,
Zijie
Deng
ab,
Bin
Zhang
a,
Bingfeng
Chen
a,
Xueqing
Xing
e,
Haihong
Wu
*c,
Huizhen
Liu
*a and
Buxing
Han
*abc
d,
Tianjiao
Wang
ab,
Zijie
Deng
ab,
Bin
Zhang
a,
Bingfeng
Chen
a,
Xueqing
Xing
e,
Haihong
Wu
*c,
Huizhen
Liu
*a and
Buxing
Han
*abc
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid and Interface and Thermodynamics, Center for Carbon Neutral Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liuhz@iccas.ac.cn; hanbx@iccas.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
cShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: hhwu@chem.ecnu.edu.cn
dBeijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing 100083, China
eBeijing Synchrotron Radiation Facility (BSRF), Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
fFunctional Polymer Materials R&D and Engineering Application Technology Innovation Center of Hebei, XingTai University, Xingtai, Hebei 050041, China
First published on 18th June 2024
Abstract
This work presents an innovative approach focusing on fine-tuning the coordination environment of atomically dispersed cobalt catalysts for tandem synthesis of primary benzylamines from oxidized lignin model compounds. By meticulously regulating the Co–N coordination environment, the activity of these catalysts in the hydrogenolysis and reductive amination reactions was effectively controlled. Notably, our study demonstrates that, in contrast to cobalt nanoparticle catalysts, atomically dispersed cobalt catalysts exhibit precise control of the sequence of hydrogenolysis and reductive amination reactions. Particularly, the CoN3 catalyst with a triple Co–N coordination number achieved a remarkable 94% yield in the synthesis of primary benzylamine. To our knowledge, there is no previous documentation of the synthesis of primary benzylamines from lignin dimer model compounds. Our study highlights a promising one-pot route for sustainable production of nitrogen-containing aromatic chemicals from lignin.
Introduction
Nitrogen-containing aromatic chemicals find extensive applications in fields such as medicine, agriculture, and materials research.1–8 Currently, the synthesis of these chemicals heavily relies on petroleum-derived feedstocks. However, in the context of a circular economy, utilizing biomass as a carbon source to produce N-containing compounds presents an attractive alternative.9–14 Lignin, a renewable resource, has emerged as a promising precursor for generating aromatic building blocks.15 The synthesis of heteroatom-containing aromatics from lignin is crucial for meeting the value-added requirements of bio-refining. While considerable research has focused on manipulating the cleavage of C–O or C–C bonds in lignin to obtain oxygen-containing aromatic compounds,16–20 exploration of lignin as a source for nitrogen-containing aromatic chemicals remains relatively limited.Current strategies for obtaining N-containing aromatics from lignin primarily focus on reactions between lignin-derived monomer or dimer model compounds and N-containing precursors. Anilines could be synthesized by cross-coupling of lignin-derived phenols with organic amines, hydrazine hydrate, ammonium acetate or ammonia (NH3), over palladium-based catalysts.21–27 In addition, some other N-containing compounds can be generated by reacting organic amines or NH3 with lignin monomers such as 4-propanolguaiacol and p-coumaric acid.28–32 The diaryl ether model compounds have also been explored to synthesize high value-added anilines.33 Since the most abundant linkage in lignin is the β-O-4 structure, the conversion of lignin often begins with the study of dimer models containing similar structures. The β-O-4 model compounds have shown potential in synthesizing valuable N-containing aromatic chemicals, as shown in Scheme 1.34–42 Recently, β-O-4 model compounds with the α-OH group were successfully transformed into secondary or tertiary benzylamines in the presence of organic amines.35 In this process, five equivalents of organic amine are required, as it serves not only as the source of the amine but also plays a crucial role as a reducing agent by providing molecular hydrogen (H2) through in situ dehydrogenation. However, this strategy cannot be used to efficiently synthesize primary benzylamines from industrial inorganic ammonia due to the inability of ammonia to supply H2. The generation of oxidized β-O-4 structures through the selective oxidation of the α-OH group has been vigorously developed.36–38 To the best of our knowledge, for oxidized β-O-4 models, although aromatic amides, isoxazoles and nitriles could be generated,39–42 no literature reports the direct conversion strategy for transforming these models into primary amines.
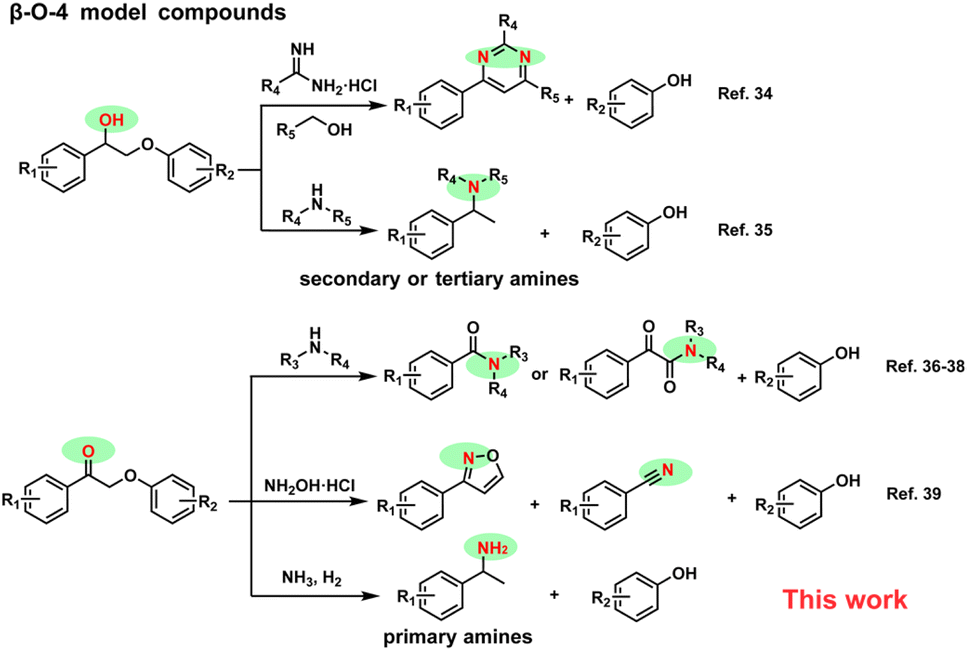 | ||
| Scheme 1 Strategies for the synthesis of N-containing aromatic chemicals from β-O-4 model compounds. | ||
Moreover, the catalytic conversion of lignin or its model compounds into N-containing compounds typically relies on noble-metal-based catalysts. There is growing interest in developing non-noble-metal-based catalysts for these significant catalytic processes due to their cost-effectiveness and widespread availability. Nevertheless, fabricating an efficient catalytic system for producing N-containing compounds poses a significant challenge. This is because the coordination between the nitrogen atom and the metal can impact both the catalytic performance and the lifetime of the catalyst.43,44 One promising avenue for addressing this challenge is the use of single-atom catalysts.45–54 These catalysts possess well-defined active sites and adjustable electronic structures, offering the potential to develop efficient catalytic systems. Additionally, they provide an opportunity to study the intrinsic properties of high-performance catalysts by modifying the coordination environment of the metal active sites.
Herein, we present a new route for the direct conversion of oxidized β-O-4 models to primary benzylamines and phenols using NH3 and H2 in a tandem reaction which involves C–O hydrogenolysis and reductive amination. In this route, it is challenging to avoid the hydrogenation of β-O-4 ketones to β-O-4 alcohols and the hydrogenation of aromatic rings to enhance selective hydrogenolysis.55 Simultaneously, it is also very important to avoid excessive alkylation of primary amines, which results in the undesired formation of secondary and tertiary amines.56,57 Compared with the cobalt (Co) nanoparticle catalyst, atomically dispersed Co catalysts perform hydrogenolysis selectively due to the weakening of hydrogenation activity. Regulating the coordination number of Co–N only affects the hydrogenation ability of the Co atomically dispersed catalysts during the reduction amination process rather than hydrogenolysis. Notably, this is the first reported application of a single atom in the cascade reaction of hydrolysis and reductive amination. The CoN3 catalyst with the Co–N triple coordination number showed the best performance, with a yield of 94% for primary benzylamine from a series of oxidative β-O-4 model substrates. We believe that this work offers a potential one-pot route for the sustainable production of primary benzylamines and phenols from lignin.
Results and discussion
A series of atomically dispersed Co–N–C (CoSA) catalysts with different Co–N coordination numbers (CoN4, CoN3, and CoN2) were synthesized by adjusting the temperature during the carbonization of bimetallic Co/Zn zeolitic imidazole frameworks (Co/Zn-ZIFs), as depicted in Fig. 1a.58–60 As previously reported, the pyrolysis temperature exhibits an inverse correlation with the N coordination number around the single-atom metal sites.59,61,62 Three catalysts with different Co–N coordination numbers (CoN4, CoN3 and CoN2) were obtained.59 This approach enabled the investigation of catalytic performance at the single-atom level, highlighting the impact of the coordination environment on the active sites. A comparison catalyst, named CoNP, was prepared by directly pyrolyzing monometallic Co-ZIFs at 900 °C. The Co loadings in the CoSA catalysts were below 2% (Table S1†), as determined by inductively coupled plasma atomic emission spectroscopy (ICP-OES). Transmission electron microscopy (TEM) images revealed the presence of Co particles in CoNP but not in the CoSA catalysts (Fig. S1†). X-ray diffraction (XRD) patterns confirmed the characteristic peaks of Co crystals in CoNP but not in the CoSA catalysts (Fig. S2†). Co, N, and C species exhibited uniform distribution in the CoSA catalysts, as observed in the energy dispersive X-ray spectral (EDS) element mappings (Fig. 1b and S3†). Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images demonstrated that the Co atoms in the three catalysts were atomically dispersed (Fig. 1c and S4†). X-ray absorption near-edge structure (XANES) spectra indicated that the oxidation state of the Co species in the CoSA catalysts closely resembled Co(II) (Fig. 1d), which is correlated with the X-ray photoelectron spectroscopy (XPS) results of Co 2p (Fig. S5†). The oxidation state of Co decreased for the CoN4, CoN3, and CoN2 catalysts, as evidenced by a shift to lower energy in the Co K-edge XANES spectra. The extended X-ray absorption fine structure (EXAFS) spectra showed a primary peak attributed to the Co–N shell in all catalysts (Fig. 1e). The Co–N coordination numbers were determined through EXAFS fitting as 4.1, 3.3, and 2.3 for the CoN4, CoN3, and CoN2 catalysts, respectively (Fig. S6 and Table S2†). The XPS N 1s spectra of the prepared cobalt single-atom catalysts reveal the presence of abundant pyridinic nitrogen structures on the carbon support (Fig. S7†). Higher pyrolysis temperature leads to reduced pyridinic N and increased graphitic N, suggesting a decrease in Co–N coordination.62 Based on this, a structural model depicting the coordination between cobalt and pyridinic nitrogen was adopted (Fig. S8†), which is consistent with previous reports.58,59,63 The XANES spectra of the CoN3 catalyst were calculated based on the Co1–N3C1 model and compared with the experimental spectra of the catalysts (Fig. 1f), confirming the well-defined structures of the CoN3 catalyst.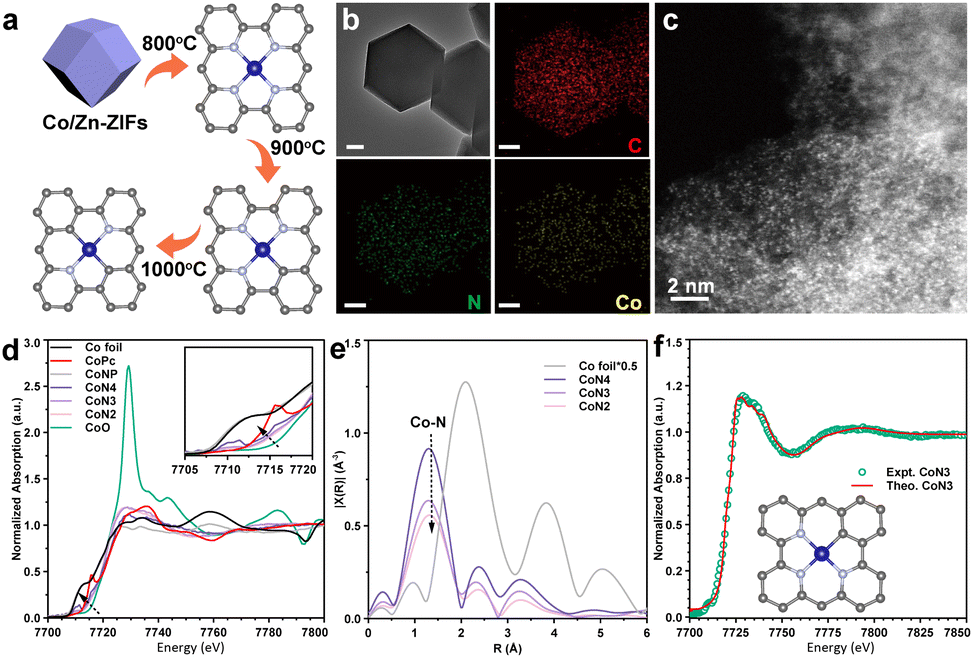 | ||
| Fig. 1 (a) Schematic diagram of coordination number regulation of a single Co site. (b) STEM and corresponding EDS mappings of the CoN3 catalyst. (c) HAADF-STEM images of the CoN3 catalyst. (d) Co K-edge XANES analysis of Co foil, CoPc, CoNP, CoN4, CoN3, CoN2 and CoO. (e) Co K-edge EXAFS analysis of Co foil, CoN4, CoN3 and CoN2, shown in R-space (FT magnitude). (f) Comparison between the experimental K-edge XANES spectra and the theoretical spectra of the CoN3 catalyst. | ||
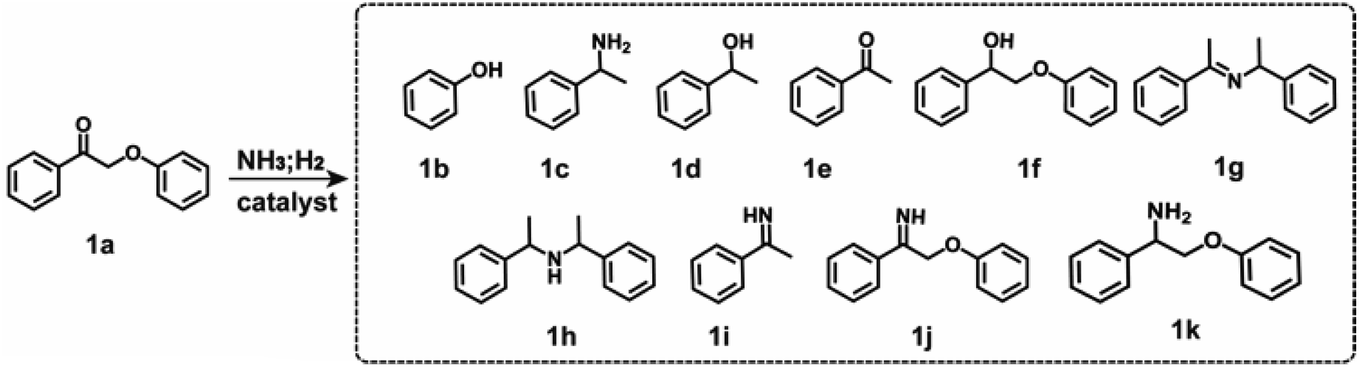
In the initial stage of our study, we employed 2-phenoxy-1-phenylethanone (PP-one, 1a) as the model substrate to evaluate the performance of the prepared catalysts (Table 1). Without a catalyst, 96% conversion of 1a resulted in the exclusive formation of 2-phenoxy-1-phenylethanimine (PP-imine, 1j) as the product (Table 1, entry 1). Nitrogen-containing carbon analogues of Co-based catalysts, named NC, were obtained by pyrolysis of Zn-ZIFs. In the presence of NC, 1a was fully converted to imine 1j, similar to the reaction without a catalyst (Table 1, entry 2). The CoNP catalyst exhibited complete conversion of 1a into phenol (1b) and primary amine 1c with yields of 64% and 62%, respectively (Table 1, entry 3). Remarkably, the CoSA catalysts exclusively facilitated the cleavage of Cβ–O bonds, resulting in a higher yield of phenol (1b). The byproducts detected were phenylethanol (1d), acetophenone (1e), 2-phenoxy-1-phenylethanol (PP-ol, 1f), secondary amine (1h), Schiff bases (1g, 1i), imines (1j) and 2-phenoxy-1-phenylethylamine (PP-amine, 1k). Based on the product distribution over different catalytic systems and some reported knowledge, the possible reaction paths are shown in Fig. 2. Two reaction pathways were involved in the transformation of PP-one to primary benzylamine using NH3 and H2. The first pathway involves the formation of PP-imine through the reaction between PP-one and NH3. Subsequently, the Cβ–O bond in PP-imine is cleaved, leading to the production of imines and phenols. The imines are then hydrogenated, resulting in the desired primary benzylamines. In the second pathway, the Cβ–O bond in 2-phenoxyacetophenone is initially cleaved, generating acetophenone and phenol. Next, imines are rapidly formed from the reaction between acetophenones and NH3. These imines, originating from both pathway-1 and pathway-2, undergo hydrogenation to yield primary benzylamine.
|
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Conv. (%) | Yields (%) | |||||||||
| 1b | 1c | 1d | 1e | 1f | 1g | 1h | 1i | 1j | 1k | |||
| a Reaction conditions: 2-phenoxy-1-phenylethanone, 0.2 mmol; 7 M ammonia solution in methanol, 2 mL; H2, 1 MPa; catalyst, 15 mg; 140 °C; 6 h. Yields were determined by GC with n-dodecane as an internal standard. | ||||||||||||
| 1 | Blank | 96 | — | — | — | — | — | — | — | — | 96 | — |
| 2 | NC | >99 | — | — | — | — | — | — | — | — | >99 | — |
| 3 | CoNP | >99 | 64 | 62 | — | — | 4 | — | — | — | — | 30 |
| 4 | CoN4 | >99 | >99 | 6 | 1 | 26 | — | 27 | — | 37 | — | — |
| 5 | CoN3 | >99 | >99 | 91 | 2 | 1 | — | 1 | 3 | — | — | — |
| 6 | CoN2 | >99 | >99 | 88 | — | — | — | — | 5 | — | — | — |
| 7 | CoNP–HCl | >99 | >99 | 76 | — | 6 | — | — | 5 | 9 | — | — |
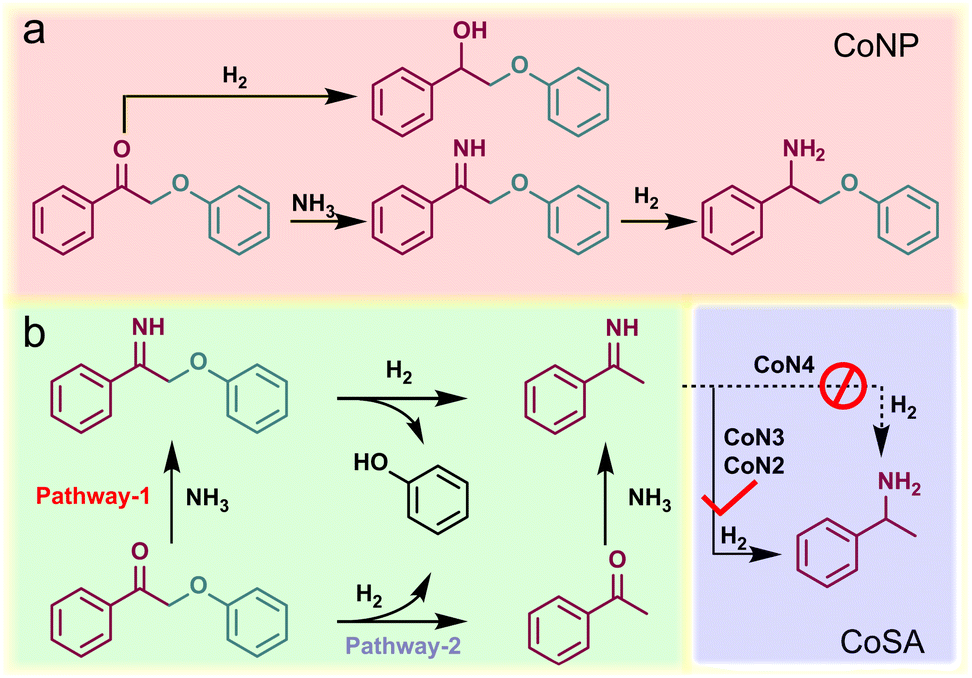 | ||
| Fig. 2 Proposed mechanism for the reductive amination of PP-one to primary benzylamine using NH3 and H2 over (a) CoNP and (b) CoSA catalysts. | ||
Comparison of CoNP and CoSA catalysts revealed that CoNP exhibited a higher yield of 2-phenoxy-1-phenylethanol (PP-ol, 1f) and 2-phenoxy-1-phenylethylamine (PP-amine, 1k), suggesting a preference for catalyzing the hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CN bonds over the hydrogenolysis of Cβ–O bonds. Interestingly, the yield of 2-phenoxy-1-phenylethylamine (PP-amine, 1k) was significantly higher than that of 2-phenoxy-1-phenylethanol (PP-ol, 1f), indicating rapid and efficient formation of 2-phenoxy-1-phenylethanimine (PP-imine, 1j), subsequently hydrogenating to 2-phenoxy-1-phenylethylamine (1k). Both CoNP and CoSA catalysts showed no activity in converting the byproducts 1f and 1k, likely due to the higher Cβ–O bond dissociation energy in 1f and 1k compared to 1a and 1j (Table S4†). The CoN4 catalyst achieved a phenol yield exceeding 99%, with only a 6% yield of the desired product 1c and a 27% yield of Schiff base 1g obtained by the reaction of 1c with acetophenone. Additionally, the yield of 1-phenylethanimine (1i), an intermediate to form 1c, was 37%. In contrast, CoN3 and CoN2 catalysts exhibited significantly enhanced yields of 1c at 91% and 88%, respectively. These findings suggest that CoN4 efficiently catalyzes the hydrogenolysis of Cβ–O bonds, while its activity for the hydrogenation of CN is low, hindering the efficient transformation of 1e and 1j into primary amine (1c). Thus, it is possible to tailor the chemical environment surrounding the Co site to regulate the selectivity of the reductive amination of acetophenone. We treated CoNP with 2 M hydrochloric acid solution to remove surface exposed Co nanoparticles and named it CoNP–HCl (Fig. S9†). The Co content decreased from 41.5 wt% to 3.7 wt% (Table S1†), indicating that most of the Co nanoparticles on the surface were removed. Removing surface-exposed Co nanoparticles from CoNP resulted in increased yield of the target product 1c to 76%, demonstrating selective reductive cleavage of the Cβ–O bond, with the phenol yield exceeding 99% (Table 1, entry 7).
O/CN bonds over the hydrogenolysis of Cβ–O bonds. Interestingly, the yield of 2-phenoxy-1-phenylethylamine (PP-amine, 1k) was significantly higher than that of 2-phenoxy-1-phenylethanol (PP-ol, 1f), indicating rapid and efficient formation of 2-phenoxy-1-phenylethanimine (PP-imine, 1j), subsequently hydrogenating to 2-phenoxy-1-phenylethylamine (1k). Both CoNP and CoSA catalysts showed no activity in converting the byproducts 1f and 1k, likely due to the higher Cβ–O bond dissociation energy in 1f and 1k compared to 1a and 1j (Table S4†). The CoN4 catalyst achieved a phenol yield exceeding 99%, with only a 6% yield of the desired product 1c and a 27% yield of Schiff base 1g obtained by the reaction of 1c with acetophenone. Additionally, the yield of 1-phenylethanimine (1i), an intermediate to form 1c, was 37%. In contrast, CoN3 and CoN2 catalysts exhibited significantly enhanced yields of 1c at 91% and 88%, respectively. These findings suggest that CoN4 efficiently catalyzes the hydrogenolysis of Cβ–O bonds, while its activity for the hydrogenation of CN is low, hindering the efficient transformation of 1e and 1j into primary amine (1c). Thus, it is possible to tailor the chemical environment surrounding the Co site to regulate the selectivity of the reductive amination of acetophenone. We treated CoNP with 2 M hydrochloric acid solution to remove surface exposed Co nanoparticles and named it CoNP–HCl (Fig. S9†). The Co content decreased from 41.5 wt% to 3.7 wt% (Table S1†), indicating that most of the Co nanoparticles on the surface were removed. Removing surface-exposed Co nanoparticles from CoNP resulted in increased yield of the target product 1c to 76%, demonstrating selective reductive cleavage of the Cβ–O bond, with the phenol yield exceeding 99% (Table 1, entry 7).
The evolution of product distribution over CoN3 with time provided insight into the reaction pathway. As depicted in Fig. 3a, a significant portion of 1a was swiftly hydrogenolyzed within 1 hour, reaching >99% conversion. The yield of phenol reached 98% at 1 h. Although no 1j was detected within 1 h, a high primary imine 1i yield of up to 48% was observed, indicating that most of 1a has been converted to imine 1j at the initial stage and then transformed into 1i. The primary amine 1c was only 4% at 1 h since it was rapidly condensed with acetophenone or primary imine to form a Schiff base 1g. The Schiff base reacted with ammonia to form an equilibrium with amine 1c and imine 1i, rather than being reduced to form the secondary amine 1h. As the reaction proceeded, the primary imine and acetophenone were converted to the target product by reductive amination, and the Schiff base was also consumed. The yield of 1c reached 91% at 6 h, with a negligible increase in yield thereafter. Based on kinetic analysis, we propose possible reaction pathways of one-step synthesis of the primary benzylamines from β-O-4 model compounds over CoN3 as shown in Fig. 3c. The recyclability of CoN3 was evaluated by washing and drying the used catalyst for reuse. As shown in Fig. 3b, the CoN3 catalyst could be recycled at least 4 times without obvious changes in both the substrate conversion and target product yield and the yield of 1c was maintained at 87%, demonstrating that it was stable under the reaction conditions. No diffraction peaks attributed to cobalt particles were identified after four cycles (Fig. S10†). Furthermore, the catalyst remains atomically dispersed after four cycles, as evidenced by HAADF-STEM analysis (Fig. S11†).
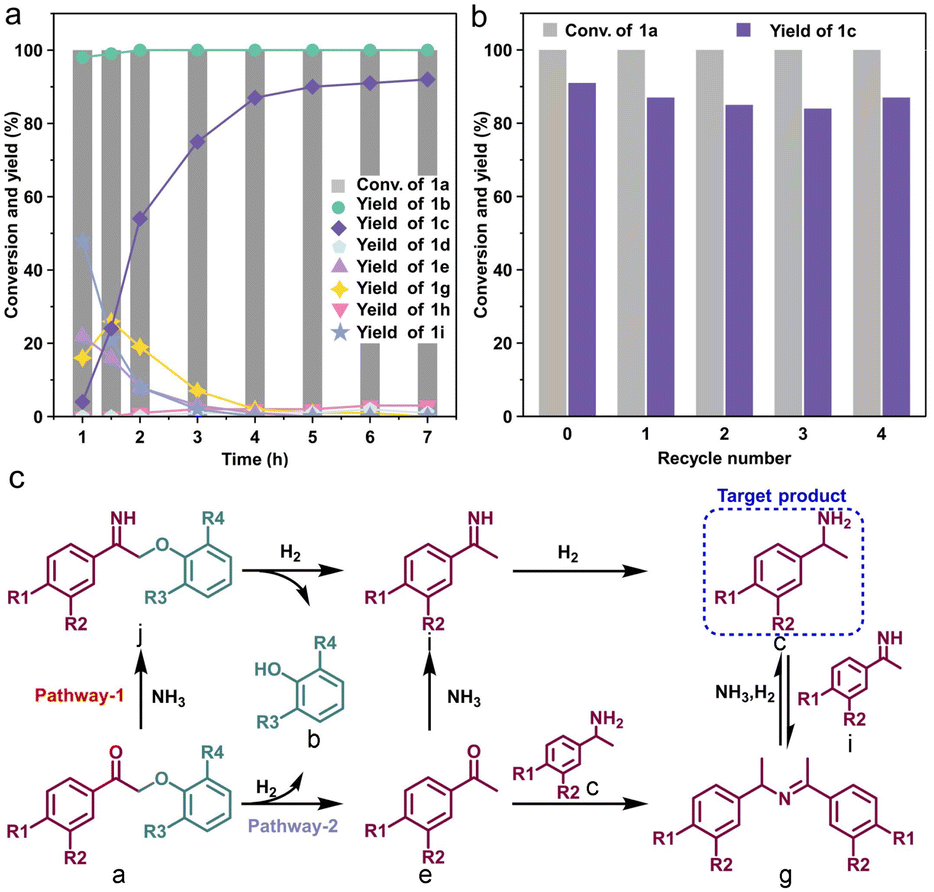 | ||
| Fig. 3 (a) Kinetic profile for tandem synthesis of primary benzylamine 1c from oxidized β-O-4 model compound 1a. Reaction conditions: 2-phenoxy-1-phenylethanone, 0.2 mmol; 7 M ammonia solution in methanol, 2 mL; H2, 1 MPa; catalyst, 15 mg; 140 °C. (b) Reusability of the CoN3 catalyst. (c) Proposed reaction pathways of one-step synthesis of the primary benzylamines from β-O-4 model compounds over CoN3. Reaction conditions: 2-phenoxy-1-phenylethanone, 0.2 mmol; 7 M ammonia solution in methanol, 2 mL; H2, 1 MPa; catalyst, 15 mg; 140 °C; 6 h. Yields were determined by GC with n-dodecane as an internal standard. (c) Proposed reaction pathways of one-step synthesis of the primary benzylamines from β-O-4 model compounds over CoN3. | ||
To investigate the underlying factors contributing to these distinct properties, several control experiments were conducted. The catalytic performance of different catalysts was evaluated for the hydrogenolysis of 2-phenoxy-1-phenylethanone (1a) in the absence of NH3. Fig. 4a illustrates that the CoNP catalyst displayed only a 68% conversion for 1a, yielding 2-phenoxy-1-phenylethanol (1f) primarily through CαO bond hydrogenation at a 45% yield and 66% selectivity. The Cβ–O bond hydrogenolysis to produce phenol (1b) yielded merely 23%, along with notably low yields of acetophenone (1e) and phenylethanol (1d). The ineffectiveness of CoNP in Cβ–O bond hydrogenolysis aligns with previous findings in the presence of ammonia (Table 1, entry 3). Acid treatment of the CoNP catalyst led to an increased phenol yield of 47% due to the removal of surface Co nanoparticles. In comparison, CoN4, CoN3, and CoN2 exhibited higher hydrogenolysis activity for the Cβ–O bond and the selectivity of phenol all could reach 84%. Moreover, acetophenone serves as an intermediate that can be further transformed into primary amine (1c). Evaluating the catalytic performance of CoN4, CoN3, and CoN2 revealed that CoN4 exhibited the lowest activity, with a conversion of 87% for 1a. Significantly, under ammonia (Table 1, entry 4), 1a was fully converted, likely due to imine 1g formation. In addition, the yield of alcohol 1f was higher over CoN3 and CoN2 compared to CoN4, implying the enhanced reductive hydrogenation ability. Comparing the catalytic performance of CoNP before and after acid treatment, the ratio of hydrogenolysis and hydrogenation of 1a improved from 34% to 81% under molecular hydrogen (Fig. 4a). This indicates that the key to enhancing the selectivity of Cβ–O bond cleavage lies in the removal of Co particles, thereby providing evidence for the atomic dispersion of the prepared CoSA catalysts.
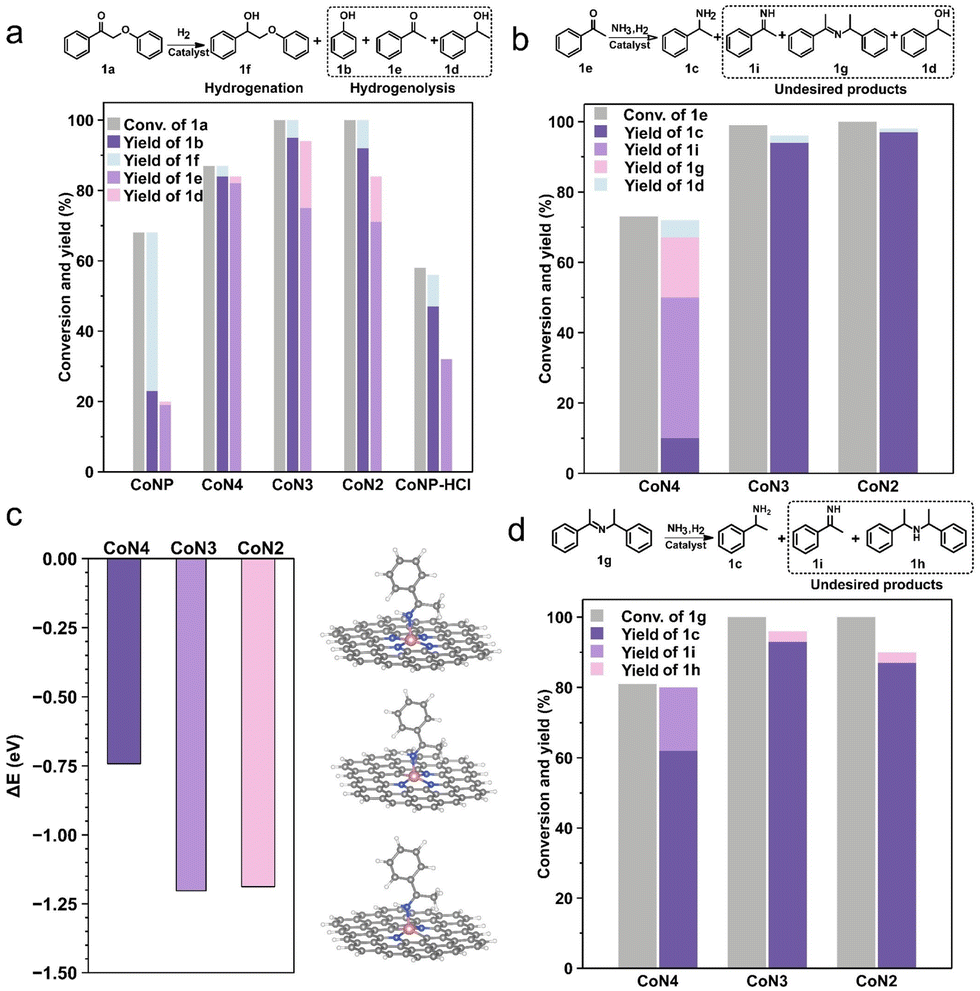 | ||
| Fig. 4 (a) Hydrogenolysis performance of Co-based catalysts for oxidized lignin model compound 1a. Reaction conditions: 2-phenoxy-1-phenylethanone, 0.2 mmol; methanol, 2 mL; H2, 1 MPa; catalyst, 15 mg; 140 °C; 6 h. (b) Reductive amination performance of the CoSA catalysts for acetophenone. Reaction conditions: acetophenone, 0.2 mmol; 7 M ammonia solution in methanol, 2 mL; H2, 1 MPa; catalyst, 15 mg; 140 °C; 6 h. (c) DFT calculated adsorption energy of imine 1i on Co sites with different N coordination numbers. Co pink, N blue, C gray, Co pink. (d) Catalytic performance of Schiff base 1g to 1-phenylethanamine over the CoSA catalysts. Reaction conditions: 1-phenyl-N-(1-phenylethyl)ethan-1-imine, 0.2 mmol; 7 M ammonia solution in methanol, 2 mL; H2, 1 MPa; catalyst, 15 mg; 140 °C; 6 h. Yields were determined by GC with n-dodecane as an internal standard. | ||
As shown in Table 1, the acetophenone (1e) was detected in 26% yield over the CoN4 catalyst, indicating that a significant portion of the substrate 1a was hydrogenolyzed before the formation of PP-imine over the Co single-atom catalysts. Thus, it is reasonable to use 1e to analyze the reductive amination process. The results showed 73% conversion of 1e and only 10% yield of 1c over the CoN4 catalyst, while the yields of primary imine 1i and Schiff base 1g were 17% and 40%, respectively (Fig. 4b). Reductive amination proceeded incompletely, retaining intermediates 1i and 1e due to the weak hydrogenation ability of the CoN4 catalyst. Conversely, over the CoN3 and CoN2 catalysts with less N coordination around the Co sites, 1e was entirely converted, with a yield of primary amine 1c exceeding 94%. The reduced production of the secondary product alcohol 1d during the conversion of 1a under reductive amination conditions could stem from the limited hydrogenation capability of single-atom catalysts for 1e. The hydrogenation ability of single-atom catalysts to 1e was investigated under molecular hydrogen. The results showed that the CoN4 catalyst has the weakest hydrogenation ability among these three Co single-atomic catalysts (Table S5†). The CoN3 and CoN2 catalysts could completely convert 1e during the reductive amination process but partially convert 1e in the absence of ammonia, probably due to the preferential hydrogenation of the primary imine 1i. DFT calculations were performed to understand the reduction of primary imine 1i (Fig. 4c and S12†). The adsorption energy of the 1i molecule on CoN4 is −0.74 eV, whereas the adsorption energies on CoN3 and CoN2 are −1.20 and −1.19 eV, respectively. This indicates that the intermediate imine 1i is favored for activation by CoN3 and CoN2, resulting in its further conversion to benzylamine 1c.
During the reductive amination of ketones, the abundant presence of secondary imines (Schiff bases) is a well-documented characteristic, as depicted in Fig. 3a. Upon exposure to molecular hydrogen, the yield of primary amine 1c decreases due to the formation of secondary amine 1h from Schiff base 1g. The yield of secondary amine 1h was below 6% when 1g was tested under reductive amination conditions over Co single-atomic catalysts (Fig. 4d). The yield of 1c from the intermediate 1g was 56% over the CoN4 catalyst. However, over the CoN3 and CoN2 catalysts, the yield of 1c is notably increased, achieving 93% and 89% respectively (Fig. 4d). These results demonstrated the efficient tandem hydrogenolysis and reductive amination reactions using 1a as the substrate over the CoN3 and CoN2 catalysts. The yield of 1c on the CoN2 catalyst exhibited a slight reduction compared to that over the CoN3 catalyst, probably due to the formation of undetectable products, as evidenced by a slightly lower carbon balance.
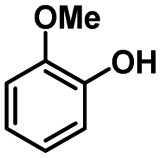
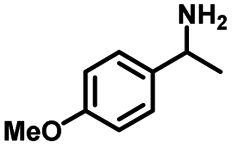
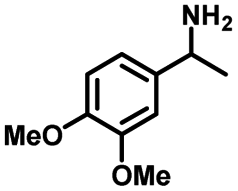
We also investigated the scopes of the substrates for the synthesis of primary anilines and phenols from oxidized β-O-4 model compounds (Table 2, 2a–8a). It was found that the catalytic system was effective for various β-O-4 model compounds. The yield of the target product methylbenzylamines was above 88%, and the yield of 3a reached up to 94%.
| Substrate | Products | |
|---|---|---|
| a Reaction conditions: acetophenone, 0.2 mmol; 7 M ammonia solution in methanol, 2 mL; H2, 1 MPa; catalyst, 15 mg; 140 °C; 6 h. Yields were determined by GC with n-dodecane as an internal standard. N.D. represents the substrate that cannot be detected by gas chromatography. | ||
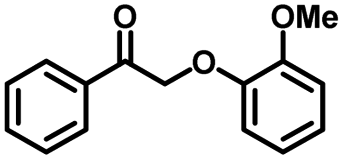
|
|
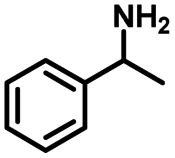
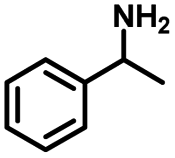
|
| 2a | 2b | 1c |
| Conversion: >99% | Yield: >99% | Yield: 91% |

|
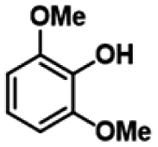
|
|
| 3a | 3b | 1c |
| >99% | >99% | 89% |
|
|
|
| 4a | 1b | 2c |
| >99% | >99% | 92% |
|
|
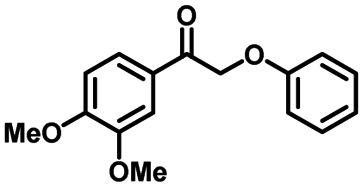
|
| 5a | 3b | 2c |
| N.D. | 98% | 94% |
|
|
|
| 6a | 1b | 3c |
| >99% | >99% | 88% |
|
|
|
| 7a | 2b | 3c |
| N.D. | 92% | 90% |
|
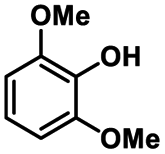
|
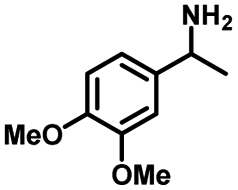
|
| 8a | 3b | 3c |
| N.D. | >99% | 92% |
Conclusions
In summary, tandem hydrogenolysis and reductive amination under NH3 and H2 led to the successful synthesis of various primary benzylamines from lignin β-O-4 model compounds. Both PP-one and PP-imine can function as substrates for the reaction, given that PP-one readily condenses with ammonia to form PP-imine. During hydrogenolysis, it is essential to prevent concurrent hydrogenation reactions. The use of the CoNP catalyst exhibited a noticeable competing reduction reaction alongside hydrogenolysis, leading to undesired selectivity towards primary benzylamines. By eliminating clusters and particles of exposed Co species, the hydrogenation activity of the catalyst could be reduced. Consequently, Co single-atom catalysts selectively hydrogenolyzed the Cβ–O bond of the substrates under molecular hydrogen. The reductive amination activity of single-atom catalysts can be enhanced with minimal impact on the initial hydrogenolysis by controlling the coordination of N atoms around Co sites. In other words, Co single-atom catalysts can precisely control the sequence of hydrogenolysis and reductive amination reactions, thereby avoiding the competitive effect. Across various model compounds, the yield of the target primary amine could reach 94% over the CoN3 catalyst. We believe this catalytic system holds potential for the sustainable production of high value-added N-containing products through biomass conversion.Author contributions
Huizhen Liu, Sen Luan, Wei Wu and Bingxiao Zheng: conceptualization, validation, investigation, and writing the manuscript. Yuxuan Wu, Minghua Dong, Zijie Deng and Xueqing Xing provided assistance in collecting, analyzing and processing XAFS data. Xiaojun Shen, Tianjiao Wang, Bin Zhang and Bingfeng Chen contributed to the experimental work and formal analysis. Huizhen Liu, Haihong Wu and Buxing Han: writing – review & editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key Research and Development Program of China (2022YFA1504901) and the National Natural Science Foundation of China (22293012, 22179132, 22072157, 22121002, and 22308029). The XAFS experiment was conducted at Beijing Synchrotron Radiation Facility 1W2B. We thank Shanghai Synchrotron Radiation Facility beamlines BL14W1 and BL11B for beamtime. We are grateful to Prof. Jing Xia (Technical Institute of Physics and Chemistry, Chinese Academy of Sciences) for AC HAADF-STEM acquisition.Notes and references
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications, Molecules, 2020, 25(8), 1909 CrossRef CAS PubMed.
- D. Yang, B. An, W. Wei, L. Tian, B. Huang and H. Wang, Copper-Catalyzed Domino Synthesis of Nitrogen Heterocycle-Fused Benzoimidazole and 1,2,4-Benzothiadiazine 1,1-Dioxide Derivatives, ACS Comb. Sci., 2015, 17, 113–119 CrossRef CAS PubMed.
- G. Pai and A. P. Chattopadhyay, N-Arylation of nitrogen containing heterocycles with aryl halides using copper nanoparticle catalytic system, Tetrahedron Lett., 2016, 57, 3140–3145 CrossRef CAS.
- A. Tirsoaga, B. Cojocaru, C. Teodorescu, F. Vasiliu, M. N. Grecu, D. Ghica, V. I. Parvulescu and H. Garcia, C–N cross-coupling on supported copper catalysts: The effect of the support, oxidation state, base and solvent, J. Catal., 2016, 341, 205–220 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- L. Xu, C. Shi, Z. He, H. Zhang, M. Chen, Z. Fang and Y. Zhang, Recent Advances of Producing Biobased N-Containing Compounds via Thermo-Chemical Conversion with Ammonia Process, Energy Fuels, 2020, 34, 10441–10458 CrossRef CAS.
- P. Ruiz-Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- K. N. Wood, R. O'Hayre and S. Pylypenko, Recent progress on nitrogen/carbon structures designed for use in energy and sustainability applications, Energy Environ. Sci., 2014, 7, 1212–1249 RSC.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Biobased Amines: From Synthesis to Polymers; Present and Future, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- S. Song, V. Fung Kin Yuen, L. Di, Q. Sun, K. Zhou and N. Yan, Integrating Biomass into the Organonitrogen Chemical Supply Chain: Production of Pyrrole and d-Proline from Furfural, Angew. Chem., Int. Ed., 2020, 59, 19846–19850 CrossRef CAS PubMed.
- X. Ma, G. Gözaydın, H. Yang, W. Ning, X. Han, N. Y. Poon, H. Liang, N. Yan and K. Zhou, Upcycling chitin-containing waste into organonitrogen chemicals via an integrated process, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7719–7728 CrossRef CAS PubMed.
- T. T. Pham, X. Chen, T. Söhnel, N. Yan and J. Sperry, Haber-independent, diversity-oriented synthesis of nitrogen compounds from biorenewable chitin, Green Chem., 2020, 22, 1978–1984 RSC.
- X. Chen, Y. Liu and J. Wang, Lignocellulosic Biomass Upgrading into Valuable Nitrogen-Containing Compounds by Heterogeneous Catalysts, Ind. Eng. Chem. Res., 2020, 59, 17008–17025 CrossRef CAS.
- J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Sustainable access to renewable N-containing chemicals from reductive amination of biomass-derived platform compounds, Green Chem., 2020, 22, 6714–6747 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, The Catalytic Valorization of Lignin for the Production of Renewable Chemicals, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Catalytic Transformation of Lignin for the Production of Chemicals and Fuels, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Z. Zhang, J. Song and B. Han, Catalytic Transformation of Lignocellulose into Chemicals and Fuel Products in Ionic Liquids, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- C. Zhang, X. Shen, Y. Jin, J. Cheng, C. Cai and F. Wang, Catalytic Strategies and Mechanism Analysis Orbiting the Center of Critical Intermediates in Lignin Depolymerization, Chem. Rev., 2023, 123, 4510–4601 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Downstream processing of lignin derived feedstock into end products, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- Z. Chen, H. Zeng, H. Gong, H. Wang and C.-J. Li, Palladium-catalyzed reductive coupling of phenols with anilines and amines: efficient conversion of phenolic lignin model monomers and analogues to cyclohexylamines, Chem. Sci., 2015, 6, 4174–4178 RSC.
- Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C.-J. Li, Formal Direct Cross-Coupling of Phenols with Amines, Angew. Chem., Int. Ed., 2015, 54, 14487–14491 CrossRef CAS PubMed.
- Z. Qiu, L. Lv, J. Li, C.-C. Li and C.-J. Li, Direct conversion of phenols into primary anilines with hydrazine catalyzed by palladium, Chem. Sci., 2019, 10, 4775–4781 RSC.
- J.-S. Li, Z. Qiu and C.-J. Li, Palladium-Catalyzed Synthesis of N-Cyclohexyl Anilines from Phenols with Hydrazine or Hydroxylamine via N-N/O Cleavage, Adv. Synth. Catal., 2017, 359, 3648–3653 CrossRef.
- T. Ichitsuka, I. Takahashi, N. Koumura, K. Sato and S. Kobayashi, Continuous Synthesis of Aryl Amines from Phenols Utilizing Integrated Packed-Bed Flow Systems, Angew. Chem., Int. Ed., 2020, 59, 15891–15896 CrossRef CAS PubMed.
- A. Dominguez-Huerta, I. Perepichka and C.-J. Li, Direct Synthesis of Diphenylamines from Phenols and Ammonium Formate Catalyzed by Palladium, ChemSusChem, 2019, 12, 2999–3002 CrossRef CAS PubMed.
- T. Cuypers, P. Tomkins and D. E. De Vos, Direct liquid-phase phenol-to-aniline amination using Pd/C, Catal. Sci. Technol., 2018, 8, 2519–2523 RSC.
- S. Elangovan, A. Afanasenko, J. Haupenthal, Z. Sun, Y. Liu, A. K. H. Hirsch and K. Barta, From Wood to Tetrahydro-2-benzazepines in Three Waste-Free Steps: Modular Synthesis of Biologically Active Lignin-Derived Scaffolds, ACS Cent. Sci., 2019, 5, 1707–1716 CrossRef CAS PubMed.
- X. Wu, M. De Bruyn and K. Barta, Primary amines from lignocellulose by direct amination of alcohol intermediates, catalyzed by RANEY® Ni, Catal. Sci. Technol., 2022, 12, 5908–5916 RSC.
- J. Ma, D. Le and N. Yan, Single-step conversion of wood lignin into phenolic amines, Chem, 2023, 9, 2869–2880 CAS.
- W. Deng, Y. Wang, S. Zhang, K. M. Gupta, M. J. Hülsey, H. Asakura, L. Liu, Y. Han, E. M. Karp, G. T. Beckham, P. J. Dyson, J. Jiang, T. Tanaka, Y. Wang and N. Yan, Catalytic amino acid production from biomass-derived intermediates, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5093–5098 CrossRef CAS PubMed.
- S. Song, J. Qu, P. Han, M. J. Hülsey, G. Zhang, Y. Wang, S. Wang, D. Chen, J. Lu and N. Yan, Visible-light-driven amino acids production from biomass-based feedstocks over ultrathin CdS nanosheets, Nat. Commun., 2020, 11, 4899 CrossRef CAS PubMed.
- D. Cao, H. Zeng and C.-J. Li, Formal Cross-Coupling of Diaryl Ethers with Ammonia by Dual C(Ar)–O Bond Cleavages, ACS Catal., 2018, 8, 8873–8878 CrossRef CAS.
- B. Zhang, T. Guo, Z. Li, F. E. Kühn, M. Lei, Z. K. Zhao, J. Xiao, J. Zhang, D. Xu, T. Zhang and C. Li, Transition-metal-free synthesis of pyrimidines from lignin β-O-4 segments via a one-pot multi-component reaction, Nat. Commun., 2022, 13, 3365 CrossRef CAS PubMed.
- B. Zhang, T. Guo, Y. Liu, F. E. Kühn, C. Wang, Z. K. Zhao, J. Xiao, C. Li and T. Zhang, Sustainable Production of Benzylamines from Lignin, Angew. Chem., Int. Ed., 2021, 60, 20666–20671 CrossRef CAS PubMed.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, Chemoselective Metal-Free Aerobic Alcohol Oxidation in Lignin, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- W. Lan, J. B. de Bueren and J. S. Luterbacher, Highly Selective Oxidation and Depolymerization of α,γ-Diol-Protected Lignin, Angew. Chem., Int. Ed., 2019, 58, 2649–2654 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Isolation of Functionalized Phenolic Monomers through Selective Oxidation and CO Bond Cleavage of the β-O-4 Linkages in Lignin, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- X. Liu, L. Wang, L. Zhai, C. Wu and H. Xu, H2O2 -promoted C–C bond oxidative cleavage of β-O-4 lignin models to benzanilides using water as a solvent under metal-free conditions, Green Chem., 2022, 24, 4395–4398 RSC.
- H. Li, M. Liu, H. Liu, N. Luo, C. Zhang and F. Wang, Amine-Mediated Bond Cleavage in Oxidized Lignin Models, ChemSusChem, 2020, 13, 4660–4665 CrossRef CAS PubMed.
- J. Zhang, Y. Liu, S. Chiba and T.-P. Loh, Chemical conversion of β-O-4 lignin linkage models through Cu-catalyzed aerobic amide bond formation, Chem. Commun., 2013, 49, 11439–11441 RSC.
- H. Li, M. Wang, H. Liu, N. Luo, J. Lu, C. Zhang and F. Wang, NH2OH–Mediated Lignin Conversion to Isoxazole and Nitrile, ACS Sustain. Chem. Eng., 2018, 6, 3748–3753 CrossRef CAS.
- A. A. Philippov and O. N. Martyanov, Poisoning effect of N-containing compounds on performance of Raney® nickel in transfer hydrogenation, Catal. Commun., 2021, 161, 106361 CrossRef CAS.
- Y. Du, H. Chen, R. Chen and N. Xu, Poisoning effect of some nitrogen compounds on nano-sized nickel catalysts in p-nitrophenol hydrogenation, Chem. Eng. J., 2006, 125, 9–14 CrossRef CAS.
- M. Dan, X. Zhang, Y. Yang, J. Yang, F. Wu, S. Zhao and Z.-Q. Liu, Dual-axial engineering on atomically dispersed catalysts for ultrastable oxygen reduction in acidic and alkaline solutions, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2318174121 CrossRef CAS PubMed.
- W. Xue, Z. Zhu, S. Chen, B. You and C. Tang, Atomically Dispersed Co-N/C Catalyst for Divergent Synthesis of Nitrogen-Containing Compounds from Alkenes, J. Am. Chem. Soc., 2023, 145, 4142–4149 CrossRef CAS PubMed.
- P.-C. Poon, Y. Wang, W. Li, D. W.-S. Suen, W. W. Y. Lam, D. Z. J. Yap, B. L. Mehdi, J. Qi, X.-Y. Lu, E. Y. C. Wong, C. Yang and C.-W. Tsang, Synergistic effect of Co catalysts with atomically dispersed CoNx active sites on ammonia borane hydrolysis for hydrogen generation, J. Mater. Chem. A, 2022, 10, 5580–5592 RSC.
- V. B. Saptal, V. Ruta, M. A. Bajada and G. Vilé, Single-Atom Catalysis in Organic Synthesis, Angew. Chem., Int. Ed., 2023, 62, e202219306 CrossRef CAS PubMed.
- J. Du, Y. Peng, X. Guo, G. Zhang, F. Zhang, X. Fan, W. Peng and Y. Li, Atomically Dispersed Pd Sites on ZrO2 Hybridized N-Doped Carbon for Efficient Suzuki–Miyaura Reaction, Catalysts, 2023, 13, 651 CrossRef CAS.
- Y. Wang, X. Cui, J. Zhang, J. Qiao, H. Huang, J. Shi and G. Wang, Advances of atomically dispersed catalysts from single-atom to clusters in energy storage and conversion applications, Prog. Mater. Sci., 2022, 128, 100964 CrossRef CAS.
- Z. Chen, G. Zhang, Y. Wen, N. Chen, W. Chen, T. Regier, J. Dynes, Y. Zheng and S. Sun, Atomically Dispersed Fe-Co Bimetallic Catalysts for the Promoted Electroreduction of Carbon Dioxide, Nano-Micro Lett., 2021, 14, 25 CrossRef PubMed.
- G. Ding, L. Hao, H. Xu, L. Wang, J. Chen, T. Li, X. Tu and Q. Zhang, Atomically dispersed palladium catalyses Suzuki–Miyaura reactions under phosphine-free conditions, Commun. Chem., 2020, 3, 43 CrossRef CAS PubMed.
- M. Liu, L. Wang, K. Zhao, S. Shi, Q. Shao, L. Zhang, X. Sun, Y. Zhao and J. Zhang, Atomically dispersed metal catalysts for the oxygen reduction reaction: synthesis, characterization, reaction mechanisms and electrochemical energy applications, Energy Environ. Sci., 2019, 12, 2890–2923 RSC.
- B. C. Gates, M. Flytzani-Stephanopoulos, D. A. Dixon and A. Katz, Atomically dispersed supported metal catalysts: perspectives and suggestions for future research, Catal. Sci. Technol., 2017, 7, 4259–4275 RSC.
- M. Wang, X. Zhang, H. Li, J. Lu, M. Liu and F. Wang, Carbon Modification of Nickel Catalyst for Depolymerization of Oxidized Lignin to Aromatics, ACS Catal., 2018, 8, 1614–1620 CrossRef CAS.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, MOF-derived cobalt nanoparticles catalyze a general synthesis of amines, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- H. Qi, J. Yang, F. Liu, L. Zhang, J. Yang, X. Liu, L. Li, Y. Su, Y. Liu, R. Hao, A. Wang and T. Zhang, Highly selective and robust single-atom catalyst Ru1/NC for reductive amination of aldehydes/ketones, Nat. Commun., 2021, 12, 3295 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Single Cobalt Atoms with Precise N-Coordination as Superior Oxygen Reduction Reaction Catalysts, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Regulation of Coordination Number over Single Co Sites: Triggering the Efficient Electroreduction of CO2, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- X. Han, X. Ling, Y. Wang, T. Ma, C. Zhong, W. Hu and Y. Deng, Generation of Nanoparticle, Atomic-Cluster, and Single-Atom Cobalt Catalysts from Zeolitic Imidazole Frameworks by Spatial Isolation and Their Use in Zinc–Air Batteries, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed.
- X. Hai, X. Zhao, N. Guo, C. Yao, C. Chen, W. Liu, Y. Du, H. Yan, J. Li, Z. Chen, X. Li, Z. Li, H. Xu, P. Lyu, J. Zhang, M. Lin, C. Su, S. J. Pennycook, C. Zhang, S. Xi and J. Lu, Engineering Local and Global Structures of Single Co Atoms for a Superior Oxygen Reduction Reaction, ACS Catal., 2020, 10, 5862–5870 CrossRef CAS.
- Y. Pan, Y. Chen, K. Wu, Z. Chen, S. Liu, X. Cao, W.-C. Cheong, T. Meng, J. Luo, L. Zheng, C. Liu, D. Wang, Q. Peng, J. Li and C. Chen, Regulating the coordination structure of single-atom Fe-NxCy catalytic sites for benzene oxidation, Nat. Commun., 2019, 10, 4290 CrossRef PubMed.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang, J. Gao, J. Wang, Y. Huang, T. Zhang and B. Liu, Single Cobalt Atoms Anchored on Porous N-Doped Graphene with Dual Reaction Sites for Efficient Fenton-like Catalysis, J. Am. Chem. Soc., 2018, 140, 12469–12475 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01813c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |