
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A catch-and-release nano-based gene delivery system†
Christoph O.
Franck‡
a,
Andrea
Bistrovic Popov‡
a,
Ishtiaq
Ahmed
a,
Rachel E.
Hewitt
b,
Luise
Franslau
c,
Puneet
Tyagi
d and
Ljiljana
Fruk
 *a
*a
aBioNano Engineering Lab, Department of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK. E-mail: lf389@cam.ac.uk
bDepartment of Veterinary Medicine, University of Cambridge, Madingley Rd, Cambridge CB3 0ES, UK
cInstitut für Physikalische Chemie, Georg-August-Universität Göttingen, Tammanstraße 6, Göttingen 37077, Germany
dAstraZeneca, One MedImmune Way, Gaithersburg, MD 20878, USA
First published on 6th September 2023
Abstract
The design of nanomaterial-based nucleic acid formulations is one of the biggest endeavours in the search for clinically applicable gene delivery systems. Biopolymers represent a promising subclass of gene carriers due to their physicochemical properties, biodegradability and biocompatibility. By modifying melanin-like polydopamine nanoparticles with poly-L-arginine and poly-L-histidine blends, we obtained a novel catch-and-release gene delivery system for efficient trafficking of pDNA to human cells. A synergistic interplay of nanoparticle-bound poly-L-arginine and poly-L-histidine was observed and evaluated for pDNA binding affinity, cell viability, gene release and transfection. Although the functionalisation with poly-L-arginine was crucial for pDNA binding, the resulting nanocarriers failed to release pDNA intracellularly, resulting in limited protein expression. However, optimal pDNA release was achieved through the co-formulation with poly-L-histidine, essential for pDNA release. This effect enabled the design of gene delivery systems, which were comparable to Lipofectamine in terms of transfection efficacy and the catch-and-release surface modification strategy can be translated to other nanocarriers and surfaces.
New conceptsDevelopments in gene therapy, vaccine design and engineering biology require controlled delivery of nucleic acid cargo. Nanocarriers are excellent alternatives to viral vector, polymers and strategies such as electroporation and nanoneedles. Required nanocarriers need to be biocompatible and cost effective, easy to manufacture and modify, all while enabling binding, delivery and release of nucleic acids. To fulfil these requirements, we have developed biopolymer-based nanocarriers characterised by high stability and cell uptake ability, which enable delivery and release of active DNA cargo. Our system is inspired by histone proteins and employs surface modification with poly-arginine and poly-histidine elements. Once attached, DNA cargo remains bound under physiological conditions, successfully crosses cell membrane, and is then released in the cell, which is confirmed by successful expression of fluorescent protein. Our system combines easy-to-manufacture biopolymeric core with facile surface modification, resulting in a dynamic system capable of catching and releasing DNA cargo. Simplicity of the surface tuning opens the route to modification of other core materials (both nanocarriers and planar surfaces for implantable continuous release chips) broadening the application to a range of delivery modes and cargo sizes. |
In the early 1990s innovative vaccination strategies based on the administration of genetic material,1,2 deoxyribonucleic acid (DNA) or ribonucleic acid (RNA), emerged as a promising therapeutic strategy for the treatment of allergies,3,4 infectious5,6 and autoimmune diseases,7,8 as well as cancer.9 In contrast to conventional vaccines, which were based on the use of live or inactivated pathogens or their proteins, genetic vaccines rely on the expression of antigen-encoding nucleic acids. However, early formulations were often characterised by insufficient transfection efficacies and low stability under physiological conditions.10,11 In order to stabilise the delicate genes and boost their therapeutical value, a broad variety of nanomaterials, including liposomes, micelles, virosomes, peptides, dendrimers and polymers, were investigated as gene carrier materials.12–15 Liposome-mediated gene delivery was particularly successful resulting in the rapid production of the first globally employed mRNA-vaccine used during the recent SARS-CoV-2 caused pandemic.16
Despite the outstanding potential of liposomal gene delivery systems, safety concerns and the incessant drive to improve therapeutical efficacy have prompted the exploration of other delivery systems.17 Among nanocarriers, polymeric nanoparticles (NPs), in particular those made of biopolymers, are characterised by favourable physicochemical properties, biocompatibility and degradability, as well as cost-effective manufacturing.18,19 In addition, their size, charge and surface functional groups can be tuned to modulate circulation time, cell uptake and nucleic acid attachment.20
Biopolymeric polydopamine nanoparticles (PDA NPs) recently emerged as a particularly interesting drug delivery platform due to their size tunability,21,22 chemical versatility23,24 and ease of preparation. However, PDA NPs are inherently negatively charged and therefore unsuitable for efficient nucleic acid immobilisation. A strong, yet reversible interaction of nanocarriers and nucleic acids is crucial, as it protects the nucleic acid from nuclease degradation while preventing its premature release.25 Previously, the only reported PDA-based gene delivery system was reliant on the modification of the nanocarriers with the efficient but cytotoxic transfection agent polyethyleneimine (PEI).26–28
In nature, packing and protection of DNA is commonly achieved by protamines, which are exceptionally rich in cationic arginine residues.29 Taking this inspiration into account, the modification of PDA NPs with arginine-rich compounds, such as poly-L-arginine (pArg) blends, represented a promising approach to broaden the scope of PDA-based nucleic acid delivery. Although pArg were shown to facilitate cellular uptake,30 low buffering capacity of pArg may hinder intracellular gene release and its endosomal escape. To overcome this challenge, a proton-buffering compound could be co-formulated with pArg to aid nucleic acid release and the rupture of endosomal compartments. Poly-L-histidine (pHis) was previously reported as an endosomal escape agent in various drug and gene delivery formulations, including silica and polymeric NPs.31,32 Due to the abundant presence of ionisable imidazole moieties, pHis significantly contributes to proton buffering inside the endosomes, which in turn may cause their rupture through the hypothesised proton-sponge effect.33
Herein, we report the design of an efficient non-viral gene delivery system based on PDA nanocarriers functionalised with cationic pArg and pHis blends. The resulting nanomaterials were characterised by strong pDNA binding affinities and facile syntheses. Furthermore, we explored the synergistic effect of pArg and pHis on gene delivery and transfection efficacy (Scheme 1).
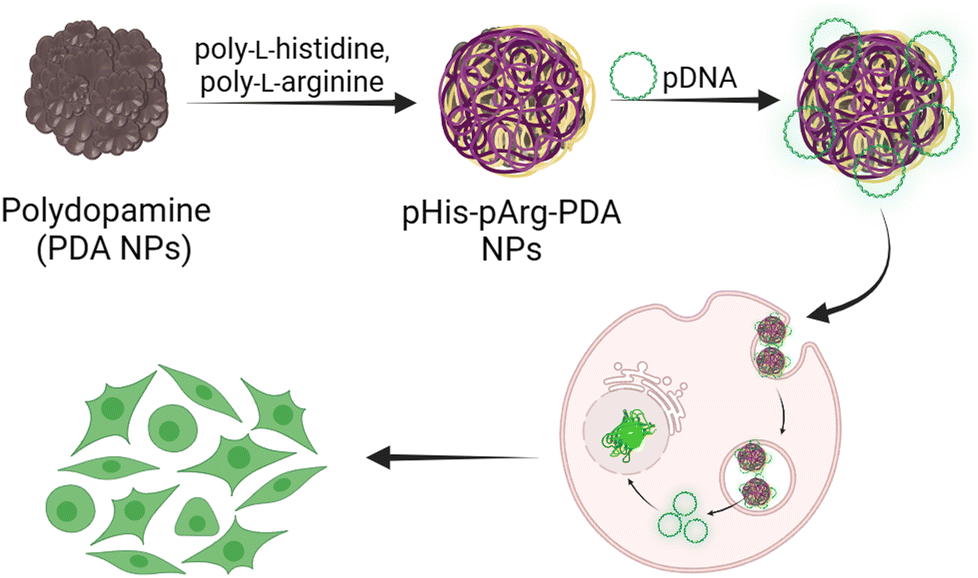 | ||
| Scheme 1 Schematic representation of the synthesis, pDNA binding and gene delivery using pHis-pArg-PDA NPs. | ||
PDA NPs were prepared through self-polymerization of dopamine in Tris-buffer (pH 8.5).34 The obtained spherical NPs had an average hydrodynamic radius of 158.4 ± 39.2 nm and the characteristic negative surface potential (−29.2 ± 1.9 mV), owing to the abundant presence of imine and quinone moieties within the PDA backbone.35,36 To obtain positively charged carriers and facilitate nucleic acid binding, PDA NPs were incubated with poly-L-arginine (pArg, Mw = 5–15 kDa) under mild basic conditions (10 mM NH4OH, aq.) for 24 h, resulting in the formation of pArg-PDA NPs. In addition, to explore the impact of pHis on the endosomal escape and intracellular release of the genetic cargo,37 PDA NPs were modified with a mixture of pArg and poly-L-histidine (pHis, Mw = 5–25 kDa), (Fig. 1A). Different ratios of pHis and pArg (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1 and 5:1 (w/w)) were added to PDA NPs, resulting in pHis-pArg-PDA_1, pHis-pArg-PDA_2 and pHis-pArg-PDA_5 NPs, respectively.
:1, 2:1 and 5:1 (w/w)) were added to PDA NPs, resulting in pHis-pArg-PDA_1, pHis-pArg-PDA_2 and pHis-pArg-PDA_5 NPs, respectively.
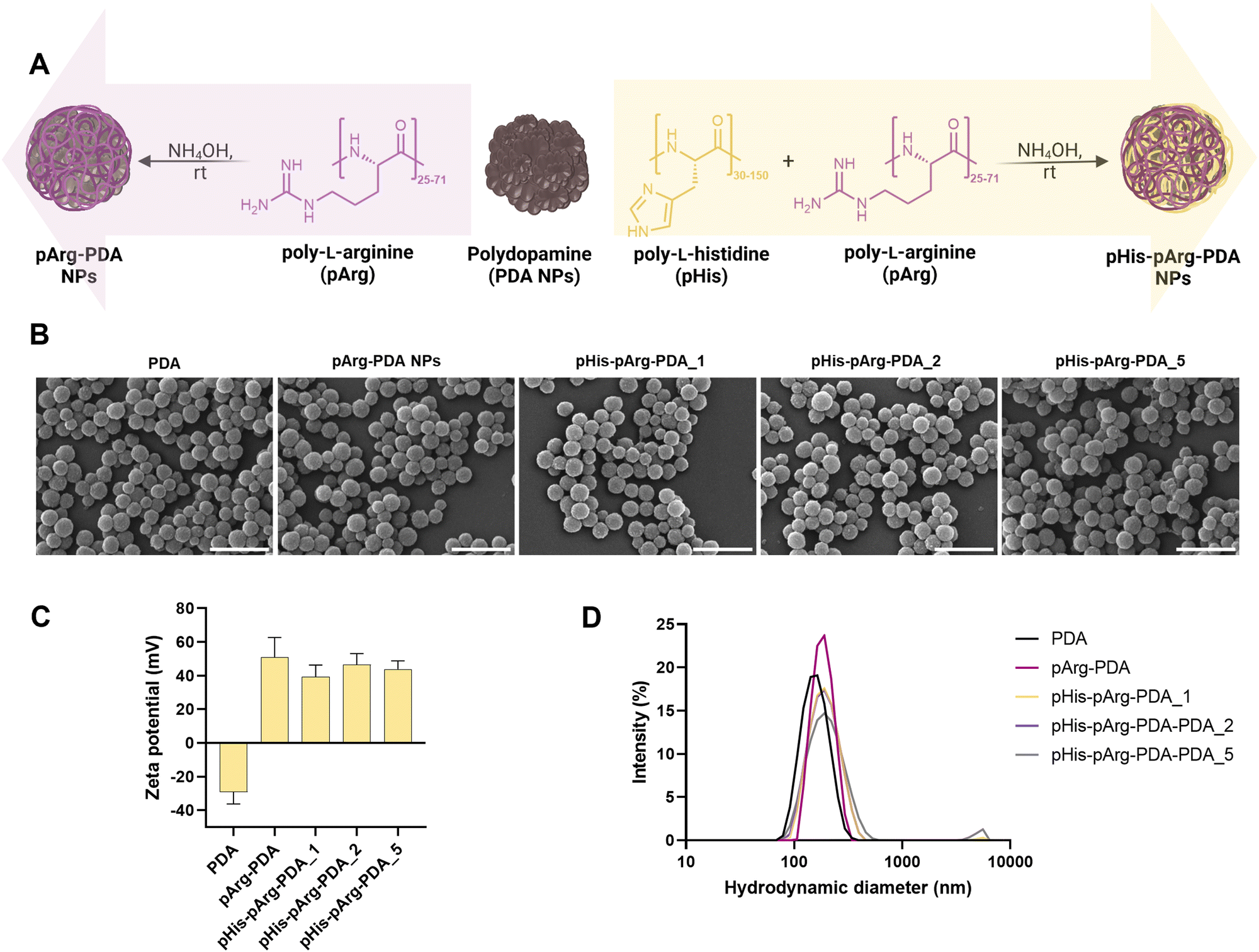 | ||
| Fig. 1 Synthesis and characterisation of pArg-PDA and pHis-pArg-PDA NPs. (A) Scheme showing the functionalisation of PDA NPs with pArg and pHis-pArg. (B) SEM micrographs of PDA, pArg-PDA, pHis-pArg-PDA_1, pHis-pArg-PDA_2 and pHis-pArg-PDA_5 NPs. The NPs were dispersed on a glass slide and coated with 10 nm sputtered Pt. Scale bar is 500 μm. (C) Zeta (ζ)-potential and (D) hydrodynamic size of PDA, pArg-PDA, pHis-pArg-PDA_1, pHis-pArg-PDA_2 and pHis-pArg-PDA_5 NPs obtained from dynamic light scattering measurements (DLS) in water. | ||
Functionalisation with pArg and pHis-pArg did not result in significant changes in NP morphology, shape nor size distribution, as observed from SEM micrographs (Fig. 1B and Fig. S1, ESI†). However, both pArg and pHis-pArg modifications resulted in strong positively charged PDA NPs (Fig. 1C and Table S2, ESI†). A high zeta (ζ)-potential of +51.6 ± 11.7 mV was obtained for pArg-PDA NPs, while the addition of pHis-pArg combinations resulted in a slight reduction of the ζ-potential for pHis-pArg-PDA NPs (+39.1 ± 7.2 to +46.5 ± 6.6 mV). Additionally, an increase in the hydrodynamic diameters for pArg- and pHis-pArg-functionalised NPs was observed compared to the unmodified PDA NPs (Fig. 1D and Fig. S1, Table S2, ESI†). This may be attributed to the strong cationic nature of the obtained particles and the resulting association of water molecules.38
In order to assure successful gene delivery, the carriers need to strongly bind a nucleic acid cargo.15 For this study, EGFP-encoding pDNA was selected, as its successful transfection could be precisely monitored using confocal microscopy and flow cytometry. The abilities of pArg-PDA NPs and pHis-pArg-PDA NPs to immobilise genetic material into nanostructured pDNA@NP complexes were evaluated by agarose gel electrophoresis. Formulations were prepared in different weight ratios (WR) of NPs to pDNA, ranging from 1:1 (WR1) to 50:1 (WR50), as displayed in Fig. 2A and Fig. S2, S3, ESI.† As expected, bare pArg polymer achieved complete pDNA binding below WR1, due to its strong cationic nature at neutral pH and the formation of hydrogen bonds between the guanidino moieties in pArg and the phosphate backbone of pDNA.39–42 Contrary to this, pHis alone was not able to fully bind pDNA, even at WR50, which might be partially attributed to the low solubility of pHis in water. Similarly to pArg, pArg-PDA NPs were immobilising pDNA at WR6. Interestingly, the addition of pHis did not significantly influence the binding affinity of pDNA for all prepared pHis-pArg-PDA NP formulations. A slight increase in binding affinity was observed for pHis-pArg-PDA_2 and pHis-pArg-PDA_5, which were able to immobilise pDNA at WR3. Similar effects were observed for dextran carriers modified with arginine and histidine peptides.43
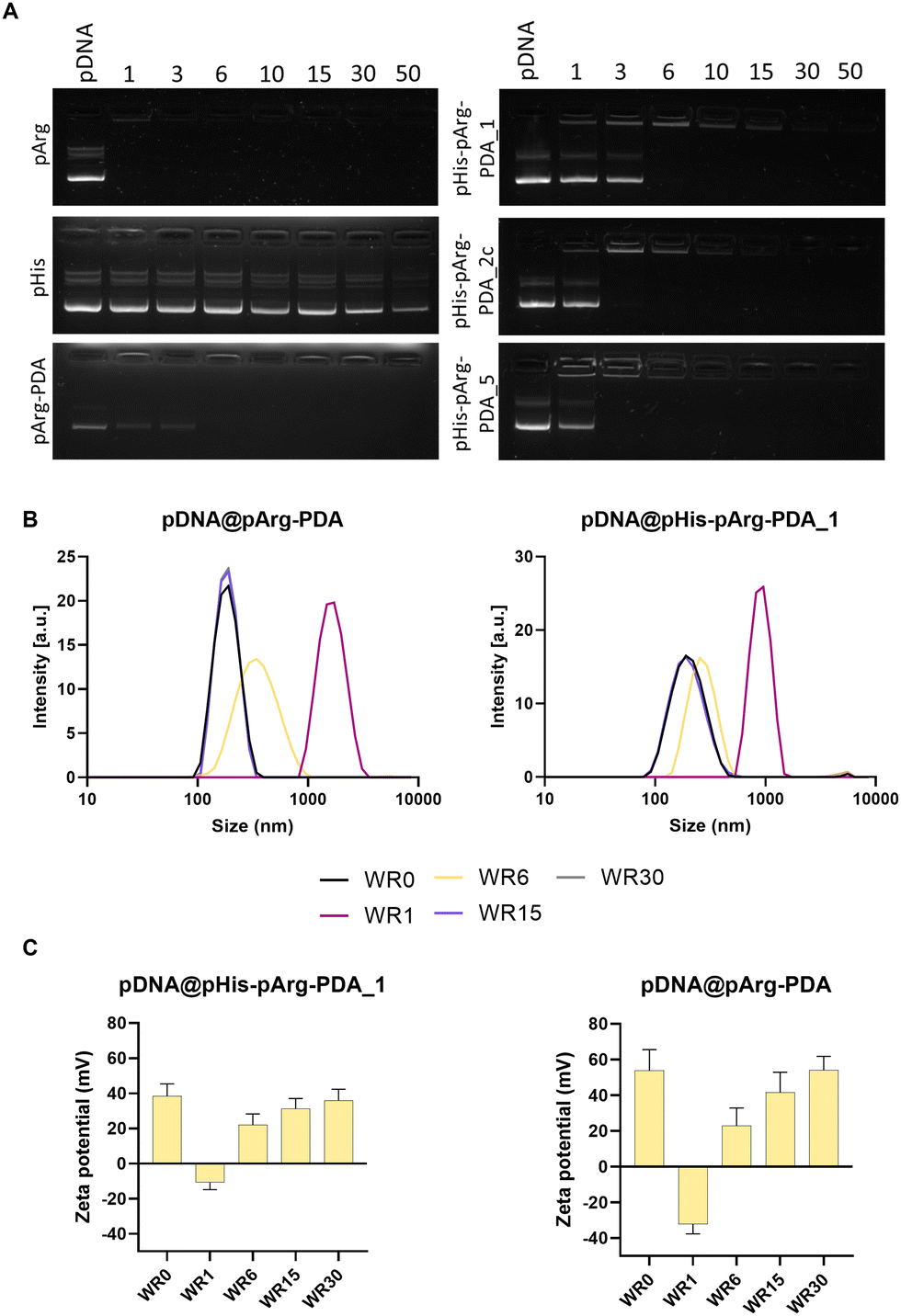 | ||
| Fig. 2 Binding studies of pDNA to pArg-PDA and pHis-pArg-PDA NPs. (A) Gel electrophoresis (1% agarose gel) of pDNA@pArg, pDNA@pHis. pDNA@pArg-PDA, pDNA@pHis-pArg-PDA_1, pDNA@pHis-pArg-PDA_2 and pDNA@pHisp-pArg-PDA_5 formulations ranging from WR1 – WR50. (B) Hydrodynamic size of pDNA@pArg-PDA and pDNA@pHis-pArg-PDA_1 measured for WR1, WR6, WR15 and WR30. (C) Zeta potential of pDNA@pArg-PDA and pDNA@pHis-pArg-PDA_1 obtained for WR1, WR6, WR15 and WR30. | ||
The size and surface charge of gene delivery systems play an important role in the cellular internalisation and cargo release.44–46 Therefore, the hydrodynamic diameter and surface charge were evaluated for different WRs of pDNA@pArg-PDA and pDNA@pHis-pArg-PDA NP formulations (Fig. 2B, C and Fig. S4, Table S3, ESI†). The pDNA@NP complexes with equivalent masses of NPs and pDNA (WR1) resulted in particle aggregation and negative surface potential. Interestingly, the surface charge of the pDNA@pArg-PDA formulation (−32.3 ± 5.4 mV) was significantly more negative compared to the pDNA@pHis-pArg-PDA (−10.8 ± 4.1 to −7.6 ± 4.2 mV). However, an increased relative amount of the NPs (WR > 6) facilitated the condensation and complexation of pDNA resulting in strong positive surface potential and particle size similar to their pDNA-free counterparts (WR0). Additionally, no change in shape and surface morphology was observed for WR > 6, while significant structural heterogeneity was noted with WR1 as shown for pDNA@pArg-PDA (Fig. S5, ESI†), which further corroborated the hydrodynamic size evaluation. This indicated that pDNA was tightly compacted and neutralised by pArg-PDA and pHis-pArg-PDA NP formulations at higher WRs (WR > 6), thereby mimicking pDNA packing in cells by arginine-rich peptides, such as protamines.29,47,48
Following successful binding of EGFP-encoding pDNA to the pArg and pHis-pArg functionalised PDA nanocarriers, their transfection efficacy was evaluated in HEK-293 cells after 24 h of administration. Since full pDNA immobilisation for all tested formulations was achieved above WR6, the EGFP expression was determined for WR6, WR15 and WR30 using confocal microscopy (Fig. 3 and Fig. S6, S7, ESI†). Lipofectamine 2000/pDNA was used as a positive control. Although pDNA was bound to bare pArg and pArg-PDA NPs, EGFP expression was not observed upon administration of these formulations to HEK-293 after 24 h. In contrast, pHis-pArg-PDA NPs successfully delivered pDNA and their administration resulted in strong EGFP expression. An increase in transfection efficacy was observed depending on pHis to pArg ratio with pHis-pArg-PDA_5 > pHis-pArg-PDA_2 > pHis-pArg-PDA_1. In addition, an increase in WRs resulted in higher transfection rates for all pHis-pArg-PDA formulations (WR30 > WR15 > WR6). To further elucidate the role of pHis in the formulation, CX-Rhodamine-labelled pDNA was immobilized on pArg-PDA NPs and the complex administered to HEK-293 cells. The rhodamine signal within the cells indicated successful pDNA cell internalisation. However, lack of EGFP fluorescence indicated that pArg-modified NPs were either not able to release pDNA from the carrier or escape the endosomes (Fig. S8 and S9, ESI†). This emphasized the importance of adding pHis to the gene-delivery formulation.
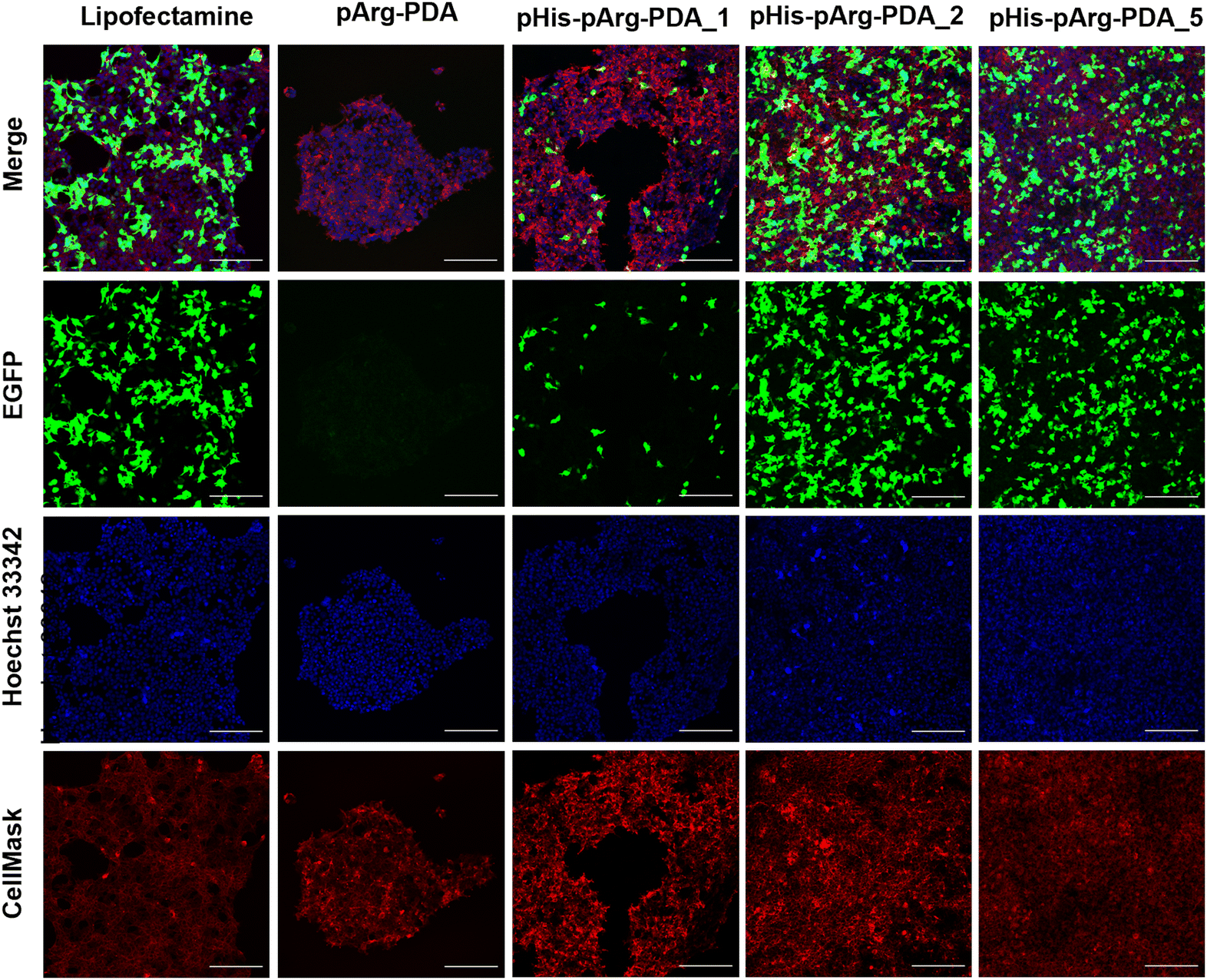 | ||
| Fig. 3 In vitro EGFP transfection study using pArg and pHis-pArg-functionalised PDA nanocarriers. Confocal microscopy images of pDNA@pArg-PDA, pDNA@pHis-pArg-PDA_1, pDNA@pHis-pArg-PDA_2 and pDNA@pHis-pArg-PDA_5 NP formulation (WR 30) 24 h after administration to HEK-293 cells. Lipofectamine was used as a positive control. Cells were incubated with CellMask (deep red) and Hoechst 33342 (blue) to stain cell membrane and nuclei, respectively. Green channel captured the fluorescence of EGFP. The scale bar is 200 μm. | ||
Flow cytometry (FACS) was used to quantify the transfection efficacy of the nanoformulations (Fig. 4A and Fig. S10, ESI†). The percentage of EGFP-positive cells was quantified for 5 biological repeats with Lipofectamine 2000/pDNA as a positive control (74.16 ± 9.41%) and untreated cells as a negative control (0.17 ± 0.12%). In agreement with the confocal microscopy studies, no significant EGFP expression was observed upon administration of pDNA@pArg-PDA NP formulations. In contrast, dosing of pDNA@pHis-pArg-PDA NPs resulted in strong transfection. The highest EGFP expression was obtained for pDNA@pHis-pArg-PDA_2 WR30 (59.93 ± 5.17%), pDNA@pHis-pArg-PDA_5 WR15 (65.58 ± 7.13%) and WR30 (58.35 ± 7.19%). FACS analysis further corroborated the general positive correlation between transfection efficacy and higher pHis to pArg ratio in pHis-pArg-PDA NPs, as well as an increased NP to pDNA ratio. However, a slight decline in the mean transfection efficacy was observed with pDNA@pHis-pArg-PDA_5 NPs WR30, compared to the corresponding WR15.
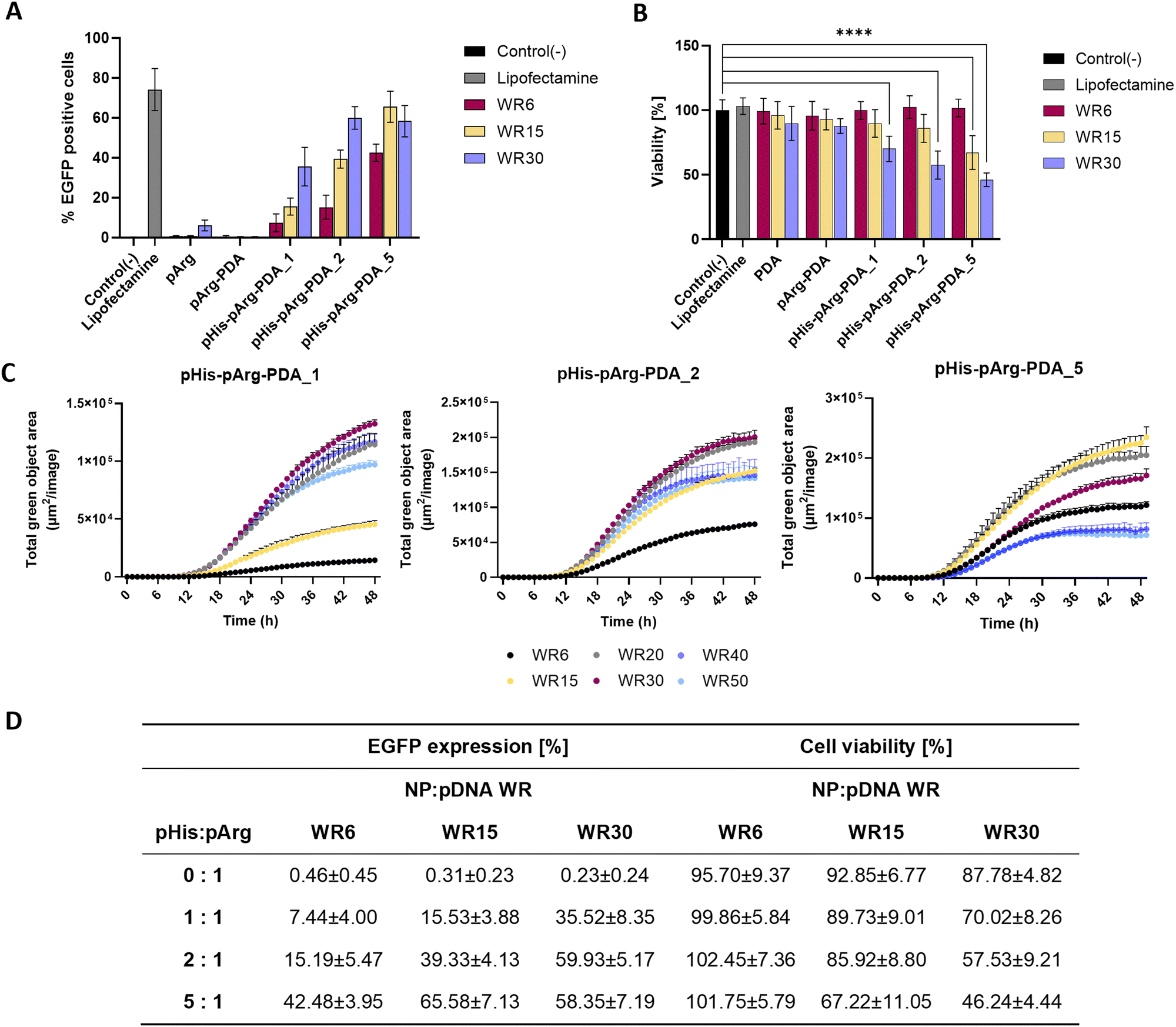 | ||
| Fig. 4 Quantification of the transfection efficacy and viability of pArg- and pHis-pArg-functionalized PDA NPs. (A) Percentage of EGFP expression determined by flow cytometry analysis of HEK-293 cells after treatment with pArg- and pHis-pArg-PDA NPs. Lipofectamine was used as a positive control. (B) Viability of HEK-293 cells treated with pDNA complexes of PDA, pArg-PDA and pHis-pArg PDA NPS. The effect on cell viability was evaluated for all NPs at WR6, WR15 and WR30 corresponding to 15, 37.5 and 75 μg mL−1 NP concentrations after 24 h treatment using MTS assay. As controls the viability of unmodified PDA NPs and lipofectamine were determined. Data are expressed as mean ± SD obtained from three separate measurements. One-way ANOVA was used to compare the viability to the untreated cells, control (−). Significance levels are defined as the following: ns for p > 0.05, * for p ≤ 0.05, ** for p ≤ 0.01, *** for p < 0.001, and **** for p < 0.0001. (C) Transfection efficacy of pHis-pArg PDA NPs monitored over 48 h using Incucyte live-cell imaging for WR6, WR15, WR20, WR40 and WR50. (D) Summary of EGFP expression and cell viability for the tested pArg-PDA and pHis-pArg-PDA formulations. | ||
Live-cell imaging, using the IncuCyte System (Sartorius, Germany), was conducted to monitor the EGFP expression over 48 h after treatment with pDNA@pHis-pArg-PDA NPs. In addition to WR6, WR15 and WR30, WR20, WR40 and WR50 were evaluated (Fig. 4C). The study of pDNA@pHis-pArg-PDA_5 indicated that transfection decrease occurs for WR20 and further decreases at higher WRs, with WR40 and WR50 showing even lower EGFP expression than the corresponding WR6. A similar trend was also found for the other pHis-pArg-PDA_1 and pHis-pArg-PDA_2 NPs, which showed a decrease in EGFP expression for ratios greater than WR30.
Additionally, the cell viability of HEK-293 was examined using an MTS endpoint assay in presence of different weight ratios of pDNA@pArg-PDA and pDNA@pHis-pArg-PDA nanocarriers (Fig. 4B). No significant differences in cell viability were observed comparing the pDNA@NP complexes (Fig. 4B) to their pDNA-free counterparts (Fig. S11, ESI†). While unmodified and pArg-PDA NPs show no impact on cell viability, a decrease in cell viability (29.98 ± 8.92 to 53.76 ± 4.80%) was observed at higher concentrations (WR30), with pHis-pArg-DA_5 showing significant decrease of 32.78 ± 11.93% already at WR15. The pHis-pArg-PDA_5 NPs at WR30 resulted in the lowest cell viability (46.2 ± 4.8%).
Higher ratios of NP-bound pHis to pArg and nanocarrier to pDNA generally resulted in abundant transgene expression, data analysis revealed that those samples impacted cell viability the most (Fig. 4D). A similar correlation between toxicity and transfection efficacy was previously noted for other gene delivery systems, such as PEI.49,50 However, the use of PDA nanocarriers enabled surface tailoring and facile adjustment of pHis to pArg ratios to achieve the highest transfection efficiency in combination with the lowest cell toxicity.
In conclusion, we have demonstrated that the modification of PDA NPs with pArg and pHis resulted in a catch-and-release system for the trafficking of pDNA to human cells. Using self-reporting EGFP-pDNA, we monitored high transfection efficacies, which were dependent on the ratio of pHis and pArg as well as the ratio of NPs to pDNA. Although it was previously demonstrated that the functionalisation of nanomaterials with Arg-containing oligomers and peptides significantly improved gene-binding properties, we showed that the introduction of pHis was crucial for efficient cargo release and subsequent protein expression. Furthermore, we employed commercially available blends of pArg and pHis, thus making this approach more cost-effective and scalable compared to using tailored cationic peptides. Due to the tunability and high transfection efficiency of our system, the synergistic effects between pHis and pArg blends for catching and releasing DNA cargo may be translated to other nanocarriers and surfaces, thus opening routes not only to new gene delivery strategies, but also to nucleic acid capture systems for biosensor design and liquid biopsy.
Author contributions
C. O. F. and A. B. P. contributed equally. C. O. F., A. B. P. and L. F. conceptualized the project, and C. O. F., A. B. P., I. A. and L. F. performed data curation. C. O. F., A. B. P and R. H. conducted formal analysis. A. B. P. conducted visualization. P. T. and L. F. supervised the work. C. O. F. and A. B. P. wrote the original draft. R. H., P. T. and L. F. helped with critical comments throughout the project and revising the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. O. F. would like to acknowledge the funding by the Engineering and Physical Sciences Research Council Centre for Doctoral Training in Sensor Technologies and Applications (EP/L015889/1) as well as the support by AstraZeneca in the framework of Project Beacon. A. B. P. would like to acknowledge funding from the Engineering and Physical Sciences Research Council Centre Interdisciplinary Research Collaboration (EPSRC IRC, EP/S009000/1). I. A. would like to acknowledge funding from HFSP programme grant RGP0004/2019 and L. F. the funding from the German Academic Scholarship Foundation.References
- A. J. Pollard and E. M. Bijker, Nat. Rev. Immunol., 2021, 21, 83–100 CrossRef CAS PubMed.
- I. Delany, R. Rappuoli and E. De Gregorio, EMBO Mol. Med., 2014, 6, 708–720 CrossRef CAS PubMed.
- K. Roy, H. Q. Mao, S. K. Huang and K. W. Leong, Nat. Med., 1999, 5, 387–391 CrossRef CAS PubMed.
- Y.-H. Chuang, Y.-H. Yang, S.-J. Wu and B.-L. Chiang, Curr. Gene Ther., 2009, 9, 185–191 CrossRef CAS PubMed.
- K. Bloom, F. van den Berg and P. Arbuthnot, Gene Ther., 2020, 28(3–4), 117–129 Search PubMed.
- M. A. Liu, Immunol. Rev., 2011, 239, 62–84 CrossRef CAS PubMed.
- H. Laroui, A. L. Theiss, Y. Yan, G. Dalmasso, H. T. T. Nguyen, S. V. Sitaraman and D. Merlin, Biomaterials, 2011, 32, 1218–1228 CrossRef CAS PubMed.
- S.-A. Shu, J. Wang, M.-H. Tao and P. S. C. Leung, Clin. Rev. Allergy Immunol., 2015, 49, 163–176 CrossRef CAS PubMed.
- D. Cross and J. K. Burmester, Clin. Med. Res., 2006, 4, 218 CrossRef CAS PubMed.
- E. Dolgin, Nature, 2021, 597, 318–324 CrossRef CAS PubMed.
- M. A. Liu, Vaccines, 2019, 7, 37 CrossRef CAS PubMed.
- S. H. T. Jorritsma, E. J. Gowans, B. Grubor-Bauk and D. K. Wijesundara, Vaccine, 2016, 34, 5488–5494 CrossRef CAS PubMed.
- B. Shi, M. Zheng, W. Tao, R. Chung, D. Jin, D. Ghaffari and O. C. Farokhzad, Biomacromolecules, 2017, 18, 2231–2246 CrossRef CAS PubMed.
- A. K. Sarkar, K. Debnath, H. Arora, P. Seth, N. R. Jana and N. R. Jana, ACS Appl. Mater. Interfaces, 2022, 14, 3199–3206 CrossRef CAS PubMed.
- C. O. Franck, L. Fanslau, A. Bistrovic Popov, P. Tyagi and L. Fruk, Angew. Chem., Int. Ed., 2021, 60, 13225–13243 CrossRef CAS PubMed.
- S. P. Kaur and V. Gupta, Virus Res., 2020, 288, 198114 CrossRef CAS PubMed.
- A. Golubovic, S. Tsai and B. Li, ACS Bio Med Chem Au, 2023, 114–136 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- D. Putnam, Nat. Mater., 2006, 5, 439–451 CrossRef CAS PubMed.
- N. S. K. Gowthaman, H. N. Lim, T. R. Sreeraj, A. Amalraj and S. Gopi, Biopolymers and Their Industrial Applications, 2021, pp. 351–372 Search PubMed.
- C. C. Ho and S. J. Ding, J. Mater. Sci. Mater. Med., 2013, 24, 2381–2390 CrossRef CAS PubMed.
- A. B. Popov, F. Melle, E. Linnane, C. González-López, I. Ahmed, B. Parshad, C. O. Franck, H. Rahmoune, F. M. Richards, D. Muñoz-Espín, D. I. Jodrell, D. Fairen-Jimenez and L. Fruk, Nanoscale, 2022, 14, 6656–6669 RSC.
- J. Liebscher, Eur. J. Org. Chem., 2019, 4976–4994 CrossRef CAS.
- L. Crocker, P. Koehler, P. Bernhard, A. Kerbs, T. Euser and L. Fruk, Nanoscale Horiz., 2019, 4, 1318–1325 RSC.
- C. L. Grigsby and K. W. Leong, J. R. Soc., Interface, 2010, 7, 67–82 CrossRef PubMed.
- P. Zhang, Q. Xu, J. Du and Y. Wang, RSC Adv., 2018, 8, 34596–34602 RSC.
- P. Zhang, X. Li, Q. Xu, Y. Wang and J. Ji, Colloids Surf., B, 2021, 208, 112125 CrossRef CAS PubMed.
- P. Zhang, Q. Xu, X. Li and Y. Wang, Mater. Sci. Eng., C, 2020, 108, 110396 CrossRef CAS PubMed.
- J. Derouchey, B. Hoover and D. C. Rau, Biochemistry, 2013, 52, 3000–3009 CrossRef CAS PubMed.
- M. A. Crosio, M. A. Via, C. I. Cámara, A. Mangiarotti, M. G. Del Pópolo and N. Wilke, Biomolecules, 2019, 9, 625 CrossRef CAS PubMed.
- A. Wahane, S. Malik, K. C. Shih, R. R. Gaddam, C. Chen, Y. Liu, M. P. Nieh, A. Vikram and R. Bahal, ACS Appl. Mater. Interfaces, 2021, 13, 45244–45258 CrossRef CAS PubMed.
- P. Bilalis, L. A. Tziveleka, S. Varlas and H. Iatrou, Polym. Chem., 2016, 7, 1475–1485 RSC.
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC.
- J. C. García-Mayorga, H. C. Rosu, A. B. Jasso-Salcedo and V. A. Escobar-Barrios, RSC Adv., 2023, 13, 5081–5095 RSC.
- R. Tejido-Rastrilla, S. Ferraris, W. H. Goldmann, A. Grünewald, R. Detsch, G. Baldi, S. Spriano and A. R. Boccaccini, Materials, 2019, 12, 500 CrossRef CAS PubMed.
- F. Bernsmann, B. Frisch, C. Ringwald and V. Ball, J. Colloid Interface Sci., 2010, 344, 54–60 CrossRef CAS PubMed.
- J. He, S. Xu and A. James Mixson, Pharmaceutics, 2020, 12, 1–31 CrossRef PubMed.
- A. Kumar and C. K. Dixit, Adv. Nanomed. Delivery Ther. Nucleic Acids, 2017, 44–58 CrossRef PubMed.
- M. Li, S. Schlesiger, S. K. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2015, 54, 2941–2944 CrossRef CAS PubMed.
- B. J. Calnan, S. B. Tidor, A. Biancalana, D. Hudson and A. D. Frankel, Science, 1991, 252, 1167–1171 CrossRef CAS PubMed.
- N. A. Alhakamy and C. J. Berkland, Mol. Pharm., 2013, 10, 1940–1948 CrossRef CAS PubMed.
- A. E. Aliaga, C. Garrido, P. Leyton, G. Diaz F, J. S. Gomez-Jeria, T. Aguayo, E. Clavijo, M. M. Campos-Vallette and S. Sanchez-Cortes, Spectrochim. Acta Part A: Mol. Biomol. Spectrosc., 2010, 76, 458–463 CrossRef CAS PubMed.
- Y. Hu, H. Wang, H. Song, M. Young, Y. Fan, F.-J. Xu, X. Qu, X. Lei, Y. Liu and G. Cheng, Biomater. Sci., 2019, 7, 1543–1553 RSC.
- M. Durymanov and J. Reineke, Front. Pharmacol., 2018, 9, 971 CrossRef PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- N. V. Hud, M. J. Allen, K. H. Downing, J. Lee and R. Balhorn, Biochem. Biophys. Res. Commun., 1993, 193, 1347–1354 CrossRef CAS PubMed.
- J. DeRouchey, V. A. Parsegian and D. C. Rau, Biophys. J., 2010, 99, 2608–2615 CrossRef CAS PubMed.
- V. Kafil and Y. Omidi, Bioimpacts, 2011, 1, 23 CAS.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00269a |
| ‡ Equally contributed. |
| This journal is © The Royal Society of Chemistry 2023 |