
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient light-harvesting, energy migration, and charge transfer by nanographene-based nonfullerene small-molecule acceptors exhibiting unusually long excited-state lifetime in the film state†
Tomokazu
Umeyama
 *a,
Kensho
Igarashi
a,
Daiki
Sasada
a,
Yasunari
Tamai
bc,
Keiichi
Ishida
a,
Tomoyuki
Koganezawa
d,
Shunsuke
Ohtani
b,
Kazuo
Tanaka
b,
Hideo
Ohkita
*b and
Hiroshi
Imahori
*ae
*a,
Kensho
Igarashi
a,
Daiki
Sasada
a,
Yasunari
Tamai
bc,
Keiichi
Ishida
a,
Tomoyuki
Koganezawa
d,
Shunsuke
Ohtani
b,
Kazuo
Tanaka
b,
Hideo
Ohkita
*b and
Hiroshi
Imahori
*ae
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: umeyama@scl.kyoto-u.ac.jp; imahori@scl.kyoto-u.ac.jp
bDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: ohkita@photo.polym.kyoto-u.ac.jp
cJapan Science and Technology Agency (JST), PRESTO, 4-1-8 Honcho Kawaguchi, Saitama 332-0012, Japan
dJapan Synchrotron Radiation Research Institute, 1-1-1, Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
eInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
First published on 5th March 2020
Abstract
Electron-acceptor small-molecules possessing a long exciton lifetime and a narrow energy band gap, opposing the energy gap law, are highly desirable for high-performance organic photovoltaics (OPVs) by realizing their efficient light-harvesting ability (LH), exciton diffusion (ED), and charge transfer (CT). Toward this goal, we designed an acceptor–donor–acceptor (A–D–A) type nonfullerene acceptor (NFA), TACIC, having an electron-donating, self-assembling two-dimensional (2D) nanographene unit, thienoazacoronene, at the center with electron-withdrawing groups at both ends. The TACIC film exhibited a narrow band gap (1.59 eV) with excellent LH. Surprisingly, the TACIC film showed an extremely long exciton lifetime (1.59 ns), suppressing undesirable nonradiative decay by its unique self-assembling behavior. When combined with a conjugated polymer donor, PBDB-T, slow ED and CT were observed (60 ps) with the excitation of TACIC owing to the large TACIC domain sizes. Nevertheless, the unusually high efficiencies of ED and CT (96% in total) were achieved by the long TACIC exciton lifetime. Additionally, unusual energy transfer (EnT) from the excited PBDB-T to TACIC was seen, demonstrating its dual LH role. The OPV device with PBDB-T and TACIC showed a high incident photon-to-current efficiency (IPCE) exceeding 70% at up to 710 nm and a power conversion efficiency of ∼10%. This result will open up avenues for a rational strategy of OPVs where LH, ED, and CT from the acceptor side as well as LH, EnT, ED, and CT from the donor side can be better designed by using 2D nanographene as a promising building block for high-performance A–D–A type NFAs.
Introduction
Organic photovoltaics (OPVs) have several advantages such as potential low cost, absorption tunability, mechanical flexibility, and roll-to-roll manufacture capability, which are absent in their inorganic counterparts.1 Prerequisites to achieving a high power conversion efficiency (PCE) of OPVs include (i) efficient light-harvesting (LH), (ii) exciton diffusion (ED) to the donor–acceptor (D–A) interface, (iii) charge transfer (CT), (iv) charge dissociation (CD), and (v) charge collection (CC) with small energy loss. Recently, considerable progress has been realized in OPVs owing to the rapid development of efficient nonfullerene acceptors (NFAs).2,3 Among NFAs, A–D–A type small-molecule acceptors, as represented by ITIC (Fig. 1),4 exhibit outstanding photovoltaic performance. The PCEs of single-junction OPVs based on A–D–A NFAs with conjugated polymer donors have reached up to 10–17%.2–5 Compared to conventional fullerene-based acceptors,6 NFAs show intense and broad optical absorption in the visible and near infrared (NIR) region as a result of their low band gaps, achieving high LH ability. In addition, the molecular energy levels of NFAs are readily tunable, enabling efficient CT and CD with minimal energy loss. Nevertheless, the nonradiative decay from the excited state to the ground state generally becomes faster when the energy difference is smaller,7 which is called energy gap law. Therefore, low band gap organic materials typically suffer from having a short exciton lifetime, especially in the film state, potentially hampering efficient ED to the D–A interface significantly far from the excited position. While triplet-state materials can provide a long exciton lifetime,8 their intersystem crossing from the singlet excited state to the triplet excited state usually causes an unfavorable energy loss. If NFAs could accomplish both the high efficiencies of LH and ED without such energy loss, we would achieve higher photovoltaic performance, e.g., by reducing the film thickness as well as optimizing the combination with donor polymers with complementary absorption. Overall, a rational molecular design for a new type of NFA is needed to combine the desirable long exciton lifetime and low band gap.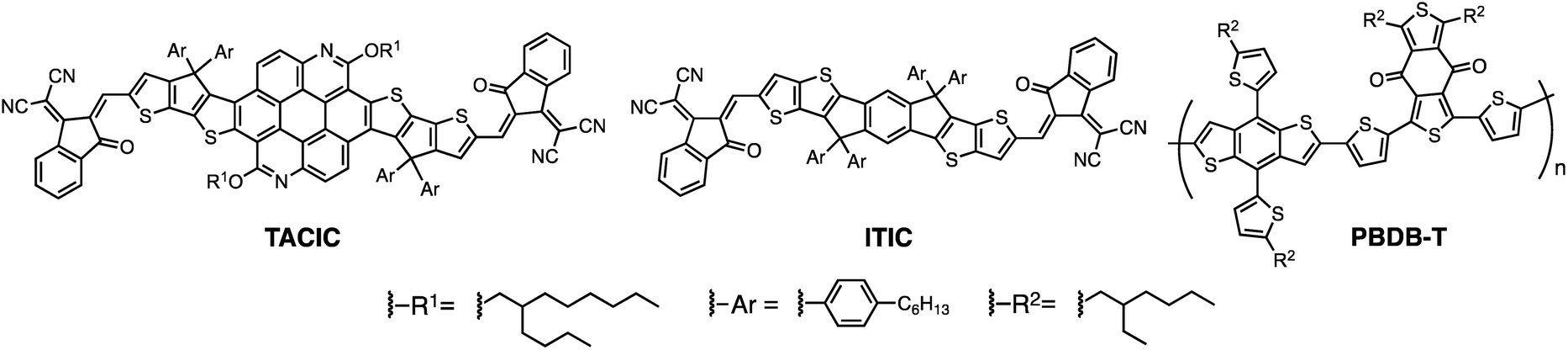 | ||
| Fig. 1 Chemical structures of TACIC, PBDB-T, and ITIC. | ||
A–D–A NFAs generally consist of a fused ring electron-donating core with side chains hanging out from the molecular plane and two compact strong electron-withdrawing units at both ends.3 The fused-ring donor structure at the center has an important influence on the electronic properties and intermolecular interactions of NFAs, which impact their physicochemical and photovoltaic properties. Most reported A–D–A NFAs are based on simple ladder-type aromatics such as indacenodithieno[3,2-b]thiophene,3,4,9 exhibiting a short exciton lifetime (≪1 ns)10 and moderate electron mobility (10−5 to 10−4 cm2 V−1 s−1).11 It has been established that the rectangular extension of the π-conjugation of the fused ring electron-donating core in NFAs enhanced their intermolecular interactions.3,12 Accordingly, it is highly promising to replace the fused ring electron-donating core with a new one showing unique intermolecular interactions to fulfill the above requirements.
Disk-shaped nanographene molecules are attractive candidates as the new core because their two-dimensional (2D), extended π-conjugation system offers unique optical and electronic properties and self-assembling behavior.13 Thus, the incorporation of nanographene molecules into NFAs is expected to enhance intermolecular interactions, which would improve the singlet exciton (S1) lifetime and electron mobility while retaining low band gap. Such studies, however, have not been carried out so far. In this study, a heterocyclic 2D nanographene, i.e., thienoazacoronene,14,15 was incorporated into the NFA structure. Because ITIC is a representative, well-investigated NFA with an A–D–A structure, thienoazacoronene was fused with two cyclo-pentadienylthiophene units to serve as the electron-donating core and paired with electron-withdrawing 1,1-dicyanomethylene-3-indanone units to yield a novel NFA, TACIC (Fig. 1), following the design motif of ITIC. The thienoazacoronene structure was chosen because it has a strong self-assembling character14,15 and electron-withdrawing conjugated nitrogen atoms that lower the HOMO level relative to the corresponding thienocoronene. In addition, the orientation-controlled bis-thiophene fused structure of thienoazacoronene is suited for incorporation into the A–D–A structure. The established facile synthetic accessibility of thienoazacoronene in a large quantity is also significantly beneficial.15 The 2- and 8-positions of thienoazacoronene, in addition to the annulated cyclopentadienyl rings, allow the introduction of substituents to tune solubility and aggregation behavior. Herein, the photophysical and electronic properties of TACIC and its photovoltaic performance, in combination with a middle-band gap conjugated polymer donor, PBDB-T (Fig. 1), were investigated in detail and compared with those of the representative ITIC.
Experimental
Femtosecond transient absorption
Femtosecond transient absorption data were collected with a pump and probe femtosecond transient spectroscopy system. This system consists of a regenerative amplified Ti:sapphire laser (Spectra-Physics, Hurricane) and a transient absorption spectrometer (Ultrafast systems, Helios). The amplified Ti:sapphire laser provided 800 nm fundamental pulses at a repetition rate of 1 kHz with an energy of 0.8 mJ and a pulse width of 100 fs (full-width at half-maximum), which were split into two optical beams with a beam splitter to generate pump and probe pulses. One fundamental beam was converted into white-light pulses employed as probe pulses in a wavelength region from 450 to 1300 nm. The other fundamental beam was used for pump pulses at 700 nm after conversion with an ultrafast optical parametric amplifier (Spectra-Physics, TOPAS). The pump pulses were modulated mechanically at a repetition rate of 500 Hz. Temporal evolution of the probe intensity was recorded with a CMOS linear sensor (Ultrafast Systems, SPEC-VIS) for visible measurements, and with an InGaAs linear diode array sensor (Ultrafast Systems, SPEC-NIR) for NIR measurements. Transient absorption spectra and decays were collected over a time range of −5 ps to 3 ns. Typically, 2500 laser shots were averaged at each delay time to obtain a detectable absorbance change as small as ∼10−4. To cancel out orientation effects on the dynamics, the polarization direction of the linearly polarized probe pulse was set at a magic angle of 54.7° with respect to that of the pump pulse. The sample films were encapsulated in an N2 filled glove box. Note that the transient absorption spectra and dynamics were highly reproducible even after several measurements. In other words, laser irradiation had negligible effects on sample degradation, at least under those experimental conditions.Fabrication of an organic photovoltaic device based on TACIC
OPV devices were prepared on patterned indium tin oxide (ITO) substrates which were cleaned by ultra-sonication in deionized water, CHCl3, acetone, and tetramethyl-ammonium hydroxide aqueous solution for 15 min each, and then in deionized water for 25 min, followed by 2-propanol and ethanol for 15 min each. They were subsequently dried under a nitrogen flow, and treated in a UV-ozone cleaner for 25 min. A solution of Zn(OAc)2·2H2O (55 mg), 2-methoxyethanol (1 mL), and ethanolamine (15.6 μL) was stirred at room temperature, and was spin-coated on the substrates at 3000 rpm for 20 s. The ZnO layer was dried at 200 °C for 30 min, and then transferred into a glove box filled with dried N2 gas to coat the active layer. A blend solution of PBDB-T with TACIC ([PBDB-T] = 6 mg mL−1 and [PBDB-T]:[TACIC] = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.9, w/w) in chloroform with 1-chloronaphthalene (0.4 vol%) was prepared and stirred on a hotplate at 50 °C for 3 h. The active layer was spin-coated at 2250 rpm for 20 s on the top of the ZnO layer, and the samples were annealed at 100 °C for 5 min. The thicknesses of the photoactive layers were ca. 100 nm. All samples were finally transferred to an evaporation chamber for MoO3 deposition (∼10 nm) at a rate of 0.1–0.3 Å s−1 and Ag deposition (∼100 nm) at a rate of 1–2 Å s−1 before obtaining their J–V characteristics (under AM1.5 conditions). OPV devices had the architecture of ITO/ZnO/PBDB-T:TACIC/MoO3/Ag.
:0.9, w/w) in chloroform with 1-chloronaphthalene (0.4 vol%) was prepared and stirred on a hotplate at 50 °C for 3 h. The active layer was spin-coated at 2250 rpm for 20 s on the top of the ZnO layer, and the samples were annealed at 100 °C for 5 min. The thicknesses of the photoactive layers were ca. 100 nm. All samples were finally transferred to an evaporation chamber for MoO3 deposition (∼10 nm) at a rate of 0.1–0.3 Å s−1 and Ag deposition (∼100 nm) at a rate of 1–2 Å s−1 before obtaining their J–V characteristics (under AM1.5 conditions). OPV devices had the architecture of ITO/ZnO/PBDB-T:TACIC/MoO3/Ag.
Results and discussion
The synthetic route for synthesis of TACIC (Scheme S1†) and the detailed synthetic procedures are provided in the ESI.† The chemical structures of the intermediates and TACIC were confirmed by 1H and 13C NMR spectroscopy (Fig. S1†), IR spectroscopy, and high-resolution mass spectra. The solubility of TACIC in chloroform (16 mg mL−1) is lower than that of ITIC (29 mg mL−1). In addition, dynamic light-scattering (DLS) measurements revealed the larger particle formation of TACIC (median size (MS) = 245 nm) than ITIC (MS = 16 nm) in chloroform (Fig. S2†). These results signify that the 2D extended π-conjugation system of the thienoazacoronene core enhances aggregation despite the long, branched alkoxy chains at the 2- and 8-positions. The decomposition temperature (Td, 5% weight loss) of TACIC recorded by thermogravimetric analysis (TGA) under nitrogen (Fig. S3a†) was 380 °C, showing excellent thermal stability. Additionally, no distinct glass transition or melting point was observed by differential scanning calorimetry (DSC) measurements of TACIC (Fig. S3b†).The UV-vis-NIR absorption spectra of TACIC and ITIC were measured in chloroform (Fig. 2a). Their absorptions were rather similar, but TACIC displayed intense bands in the 600–720 nm region with a maximum molar extinction coefficient (ε) of 3.2 × 105 cm−1 M−1. The ε value is 60% higher than that of ITIC (2.0 × 105 cm−1 M−1), showing the better light-harvesting property of TACIC. The optical band gap of TACIC in chloroform (1.70 eV) was estimated from the absorption onset. Relative to the solution, the TACIC film revealed a prominent redshift and broadening of the absorption bands, supporting significant intermolecular interaction in the film state (Fig. S4†). According to the absorption onset, the optical band gap of TACIC in the film state was estimated to be 1.59 eV.
 | ||
| Fig. 2 (a) UV-vis-NIR absorption spectra of TACIC and ITIC in chloroform. (b) HOMO and LUMO energy levels of PBDB-T, TACIC, and ITIC estimated by differential pulse voltammetry. | ||
The energy levels of TACIC were estimated through electrochemical measurements (Fig. S5†). In the cyclic voltammogram, TACIC exhibited a reversible reduction, whereas ITIC showed an irreversible reduction, indicating the higher stability of the reduced state of TACIC than ITIC. The HOMO and LUMO energy levels of TACIC were determined to be −5.44 eV and −3.72 eV. Thus, the calculated electrochemical band gap of TACIC (1.72 eV) is close to its optical band gap in chloroform (1.70 eV, Fig. S4†). The HOMO and LUMO energy levels of TACIC, ITIC, and PBDB-T are illustrated in Fig. 2b. The LUMO level of TACIC is higher than that of ITIC (−3.83 eV). Because the open-circuit voltage (VOC) of an OPV device is generally proportional to the energy difference between the LUMO of the electron-acceptor and the HOMO of the electron-donor, TACIC-based OPV devices are likely to have a higher VOC than ITIC-based ones. To gain insight into the molecular geometry and electronic structure of TACIC, density functional theory (DFT) calculations at the B3LYP/6-31G(d) level were performed, where the alkyl chains were simplified with methyl groups (Fig. S6†). TACIC possesses a highly planar backbone, which is suited for enhancing intermolecular π–π interactions. It is evident that the HOMO is mainly distributed over the electron-rich fused ring core and the LUMO is largely delocalized along the entire molecular backbone, likewise with ITIC (Fig. S6†). The calculated LUMO energy level (−3.19 eV) is higher than that of ITIC (−3.34 eV). The trend is consistent with the results from the electrochemical measurements (Fig. S5†).
Reflecting the self-assembling characteristics arising from the 2D π-extended structure of the thienoazacoronene unit, the electron mobility (μe) estimated by the space charge limited current (SCLC) method16 for the TACIC single-component film (6.2 × 10−4 cm2 V−1 s−1) is ca. two times higher than that of ITIC (2.6 × 10−4 cm2 V−1 s−1) (Table S1†).3a This verifies the intrinsic potential for the higher CC efficiency of TACIC than ITIC.
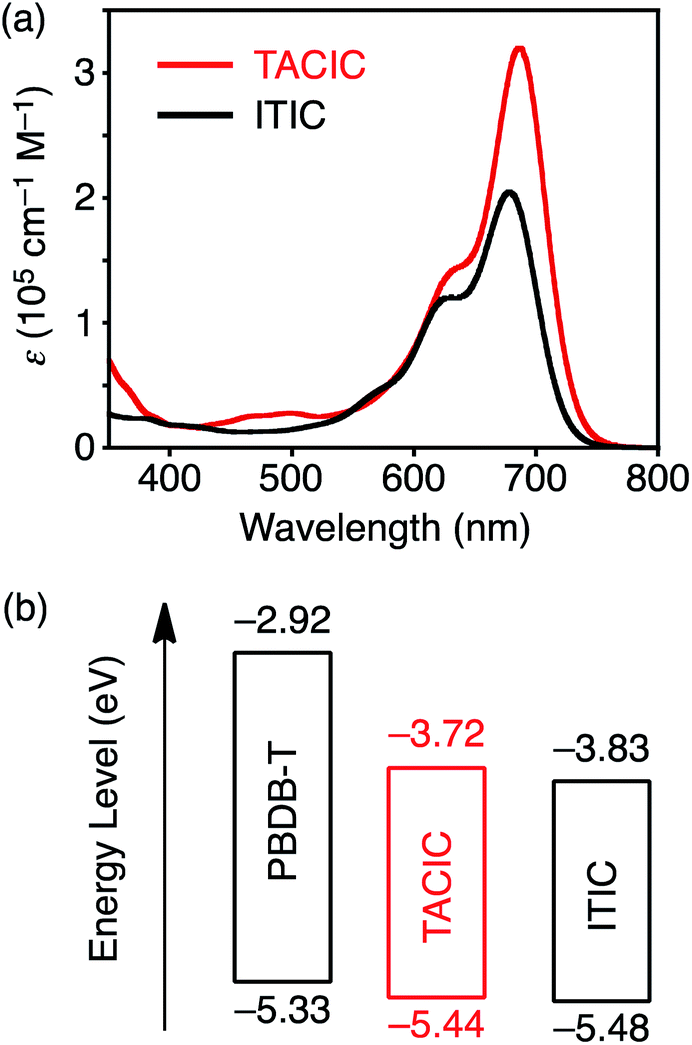
To examine the S1 lifetime, femto- to picosecond time-resolved transient absorption (TA) spectra of the TACIC solution and film were measured (Fig. 3). The detailed discussion on excitation intensity dependencies is provided in the Supporting Note in the ESI.† The S1 lifetime of TACIC in solution (220 ps, Fig. 3a, S7a and b†) is slightly longer than that of ITIC in solution (176 ps, Fig. S8a–c†). Notably, the S1 lifetime of the TACIC film (1.59 ns, Fig. 3b and S7c†) is ca. 7 times longer than that of TACIC in solution. This prolongation sharply contrasts with the S1 lifetimes of most organic molecules17 including ITIC where the S1 lifetime in the film state (ITIC: 29.2 ps, Fig. S8d and e†) is much shorter than that in solution. Fluorescence lifetime measurements of TACIC and ITIC in both film and solution states (Fig. S9†) exhibited results consistent with the TA spectra. Although the TACIC film does not show a clear characteristic of aggregate-induced emission,18 its absolute photoluminescence quantum yield, estimated by the integrated sphere method (ΦPL = 0.077), is slightly higher than that of the TACIC chloroform solution (ΦPL = 0.065). Furthermore, ΦPL of the ITIC film (0.022) is considerably low compared to that of the ITIC solution (0.063). In the 2D grazing incidence wide-angle X-ray scattering (GIWAXS) measurements, the TACIC and ITIC films displayed no significant difference (Fig. S10†). Nevertheless, the unique π–π interactions between the thienoazacoronene units in TACIC may induce efficient singlet exciton migration, remarkably suppressing undesirable nonradiative decay in the TACIC film (Table S2†). On the other hand, nonradiative decay is considerably accelerated in the ITIC film (Table S2†). To the best of our knowledge, this is the first example of the elongation of the S1 lifetime in the film state compared to that in a solution of NFAs with the A–D–A structure. In other words, TACIC is an unprecedented NFA that combines a long S1 lifetime and low band gap in the film state.
 | ||
| Fig. 3 Transient absorption spectra of (a) TACIC in chloroform in 0–500 ps and (b) TACIC film in 0–1000 ps. The excitation wavelength (λex) and intensity were (a) 650 nm and 26 μJ cm−2 and (b) 700 nm and 0.68 μJ cm−2. | ||
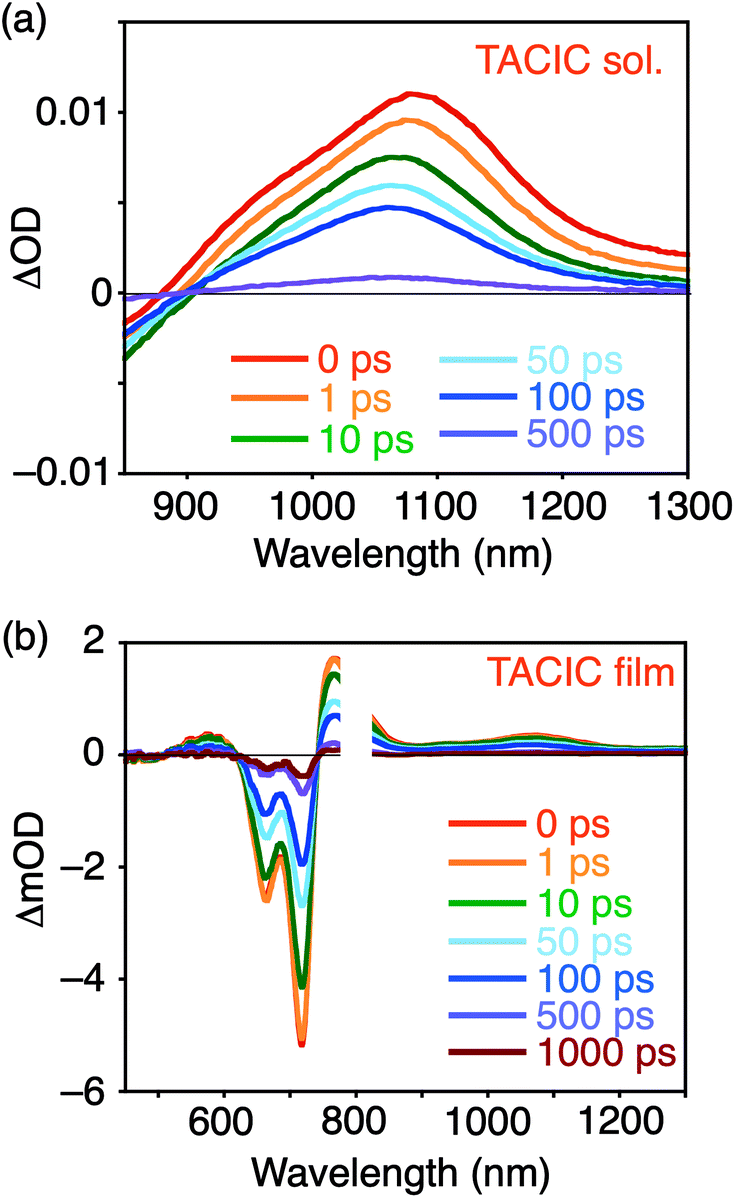
To investigate the photovoltaic performance of TACIC, we fabricated a photoactive layer of OPV devices by blending TACIC and PBDB-T, possessing complementary absorptions (Fig. S11†). The devices adopted a configuration of ITO/ZnO/PBDB-T:acceptor/MoO3/Ag. The detailed device fabrication procedures are described in Experimental for PBDB-T:TACIC and the ESI† for PBDB-T:ITIC. The characteristic current density–voltage curves are shown in Fig. 4a. The best and average photovoltaic parameters, short-circuit current density (JSC), VOC, fill factor (FF), and PCE, are listed in Table 1. The TACIC-based device achieved a PCE of 9.92%, which rivals or surpasses a PCE of a device with the representative ITIC (PCE = 9.71%, Table 1). Because of the higher LUMO level of TACIC than ITIC, a VOC of the TACIC-based device (0.982 V) is significantly higher than that of the ITIC-based device (0.885 V). However, an FF of the TACIC-based device (0.609 V) is lower than that of the ITIC-based device (0.685). In response to the broad absorption of PBDB-T:TACIC (Fig. S12†), the incident photon-to-current efficiency (IPCE) spectrum exhibited a broad photocurrent response in 400–800 nm (Fig. 4b). The IPCEs of the TACIC- and ITIC-based devices are comparable and are consistent with JSC. The IPCE value of the TACIC-based device reaches over 70% at a wavelength of up to 710 nm, where TACIC mainly absorbs incident photons. This indicates an efficient conversion of the photons absorbed by TACIC to photocurrent despite a small HOMO energy offset (0.11 eV) between TACIC and PBDB-T (Fig. 2b).
 | ||
| Fig. 4 (a) Current density–voltage curves of the best-performing OPV devices and (b) photocurrent action spectra of typical OPV devices based on PBDB-T:TACIC and PBDB-T:ITIC. The convolution of the spectral response with the photon flux of the AM1.5G spectrum provided the estimated JSC values of 15.8 and 15.4 mA cm−2, respectively, in (b). | ||
| NFA | J SC/mA cm−2 | V OC/V | FF | PCE/% |
|---|---|---|---|---|
| a Parameters of the best-performing devices are shown. In parentheses, the photovoltaic parameters were averaged from ten independent devices. | ||||
| TACIC | 16.59 (16.17 ± 0.23) | 0.982 (0.984 ± 0.004) | 0.609 (0.605 ± 0.005) | 9.92 (9.63 ± 0.12) |
| ITIC | 16.02 (15.81 ± 0.19) | 0.885 (0.880 ± 0.004) | 0.685 (0.683 ± 0.006) | 9.71 (9.50 ± 0.12) |
Photoluminescence (PL) quenching experiments were conducted to estimate the overall efficiencies of ED to the D–A interface and CT at the interface in the PBDB-T:TACIC and PBDB-T:ITIC blend films at λex = 710 nm for NFAs and λex = 520 nm for PBDB-T (Fig. S13†). The emissions from NFAs and PBDB-T were all quenched efficiently in the blend films (95–96%, Table 2). These data indicate effective ED and CT from the donor to acceptor and vice versa in PBDB-T:TACIC and PBDB-T:ITIC. In addition, energy transfer (EnT) from the excited PBDB-T to TACIC also contributed to the PL quenching in PBDB-T:TACIC at λex = 520 nm (vide infra).
| NFA | PL quenching/% | η CD/% | μ e /×10−4 cm2 V−1 s−1 | μ h /×10−4 cm2 V−1 s−1 | |
|---|---|---|---|---|---|
| NFAa | PBDB-Tb | ||||
| a The blend films were excited at 710 nm. b The blend films were excited at 520 nm. c Measured by the SCLC method with the device configuration of ITO/ZnO/PBDB-T:NFA/Al. d Device configuration: ITO/PEDOT:PSS/PBDB-T:NFA/MoO3/Au. | |||||
| TACIC | 96 | 96 | 80 | 1.2 | 2.6 |
| ITIC | 95 | 95 | 75 | 3.1 | 2.1 |
To further study photodynamics in the blend films, TA spectra of PBDB-T:TACIC and PBDB-T:ITIC within the pico- to nanoseconds time range19 were recorded (Fig. 5) where the NFA was predominantly excited at λex = 700 nm (Fig. S11†). The detailed assignments and discussion on excitation intensity and wavelength dependencies are described in the Supporting Note and in Fig. S14–S16 in the ESI.† In the spectrum of PBDB-T:TACIC, positive signals attributed to S1 of TACIC were observed at 550 and 1080 nm (Fig. 5a). The S1 decay constant of TACIC was as long as 60 ps (Fig. S15a†), which is much longer than that of typical high-performance OPV devices (<10 ps).10,20 The S1 of ITIC in PBDB-T:ITIC was found to have two components with much shorter lifetimes: <100 fs (beyond the time resolution of the instrument) and 7 ps (Fig. S16a†). The longer S1 lifetime of TACIC results from the ED to the D–A interface in the relatively large TACIC domains and/or the rather slow CT at the D–A interface. Generally, a long S1 lifetime is accompanied by low efficiencies in ED and/or CT,21 but the overall efficiency of ED and CT in PBDB-T:TACIC is close to unity (96%) in the PL quenching experiments with TACIC excitation (Table 2). The extremely long S1 lifetime of the TACIC single-component film (1590 ps, Fig. 3 and S7†), which originates from the unique self-assembling behavior of TACIC, is responsible for the slow but highly efficient ED and CT in the PBDB-T:TACIC blend film.
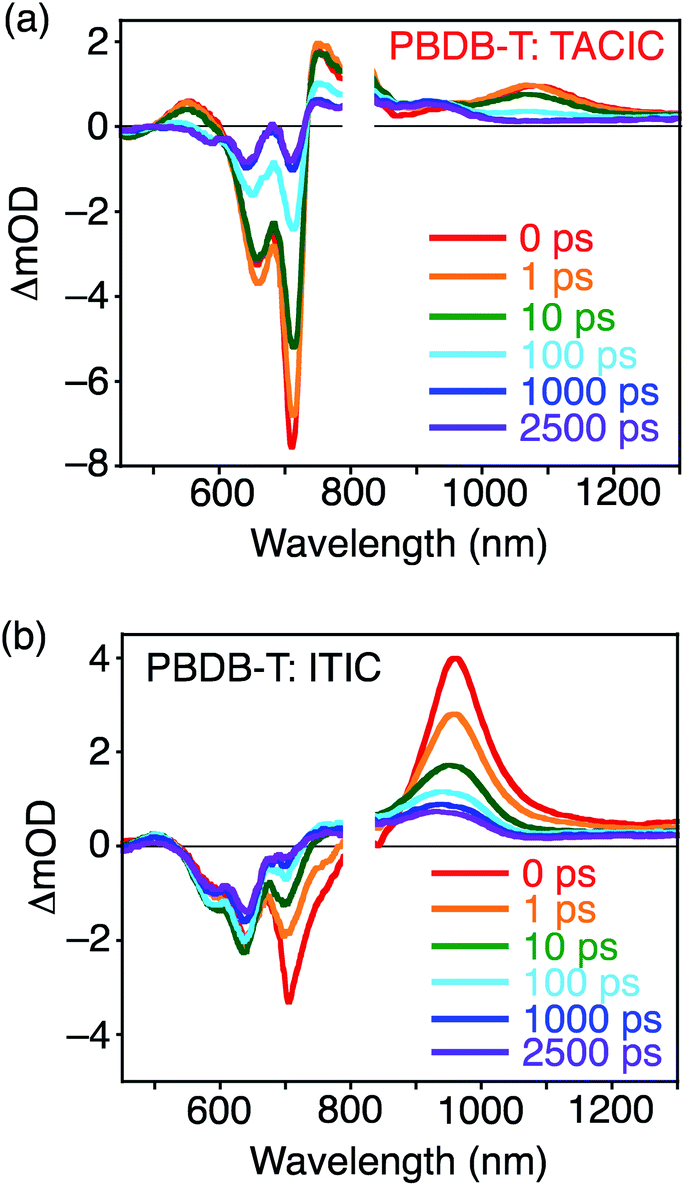 | ||
| Fig. 5 Transient absorption spectra of (a) PBDB-T:TACIC and (b) PBDB-T:ITIC blend films measured from 0–2500 ps. The excitation wavelength and intensity were 700 nm and 7.5 μJ cm−2, respectively. | ||
The TA spectrum of PBDB-T:TACIC was also measured where the PBDB-T was mainly excited at λex = 580 nm (Fig. S11†). Immediately after photoexcitation, an absorption band was observed at 1140 nm, which can be assigned to S1 of PBDB-T (Fig. S17†).22 If CT between S1 of PBDB-T and the ground state of TACIC occurred exclusively, the signal of S1 of PDBD-T would continue to exist for a while after photoexcitation until CT was completed. However, this is not the case. In addition to the signals of the PBDB-T hole polaron and TACIC radical anion at 930 nm, the signal of S1 of TACIC emerged at 1080 nm and the peak assignable to S1 of PBDB-T almost disappeared at 1 ps after photoexcitation. The emergence of S1 of TACIC unambiguously confirms that the efficient EnT from S1 of PBDB-T to TACIC occurred in addition to CT by the selective excitation of PBDB-T in the PBDB-T:TACIC blend film. This result emphasizes the importance of EnT from PBDB-T to TACIC for photocurrent generation. So far, there have been several reports referring to the photodynamics of the blend films of a conjugated polymer donor and NFA with the selective excitation of the polymer donor, where the band gap of the NFA is smaller than that of the polymer donor.10c,23 In all such previous reports, however, EnT was not observed, only CT was seen. Although the reasons for the occurrence of EnT only in the PBDB-T:TACIC blend film are not clear at the current stage, mutual molecular orientations that preferentially cause EnT may be formed at the D–A interface in PBDB-T:TACIC. On the other hand, we recently found that the hydrocarbon-based 2D nanographenes, i.e., hexa-peri-hexabenzocoronene and a short graphene nanoribbon, accelerate EnT and suppress CT in the covalently linked systems with electron-donating porphyrins.13d Likewise, the heterocyclic 2D structure of thienoazacoronene may accelerate EnT in PBDB-T:TACIC.
From the decay profile of the PBDB-T hole polaron monitored at 950 nm in the TA spectrum of PBDB-T:TACIC (Fig. S15b†), the CD efficiency from the CT state was estimated to be ca. 80% (Table 2), assuming the constant fraction at >2500 ps stemming from the dissociated free charges and the decayed component at <2500 ps derived from the geminate recombination.20a The CD efficiency of PBDB-T:ITIC (ca. 75%, Fig. S16b† and Table 2) is lower than that of PBDB-T:TACIC. Considering the higher CD efficiency of PBDB-T:TACIC than PBDB-T:ITIC, in addition to the similar IPCEs, absorption intensities, and fluorescence quenching ratios in PBDB-T:TACIC and PBDB-T:ITIC, the CC efficiency of the PBDB-T:TACIC-based device should be lower than that of the PBDB-T:ITIC-based device. This is seemingly contradicted by the fact that the μe of the TACIC single-component film is higher (6.2 × 10−4 cm2 V−1 s−1) than that of ITIC (2.6 × 10−4 cm2 V−1 s−1) (Table S1†). However, the μe of the PBDB-T:TACIC blend film (1.2 × 10−4 cm2 V−1 s−1) is much lower than that of the TACIC single-component film, whereas the μe of PBDB-T:ITIC (3.1 × 10−4 cm2 V−1 s−1) is comparable to that of the ITIC single-component film (Tables 2 and S1†). Overall, PBDB-T:ITIC shows an excellent balance between μe and hole mobility (μh = 2.1 × 10−4 cm2 V−1 s−1and μe/μh = 1.5) as compared to PBDB-T:TACIC (μh = 2.6 × 10−4 cm2 V−1 s−1 and μe/μh = 0.46) (Tables 2 and S1†). It is recognized that, if μe and μh do not match each other in the blend film, then the carriers with lower mobility will accumulate in the device.24 The accumulation may lead to an additional electric field in the device, which blocks the extraction of the carriers, thus reducing the FF. Therefore, more balanced charge mobility leads to a higher FF of the PBDB-T:ITIC-based device (0.685) than the PBDB-T:TACIC-based device (0.609) (Table 1). Although no significant differences are seen in the film surface morphologies and crystallinities of the PBDB-T:TACIC and PBDB-T:ITIC blend films through atomic force microscopy (AFM) and GIWAXS measurements (Fig. S18 and S19†), PBDB-T:TACIC may not form a well-balanced D–A bicontinuous structure, unlike PBDB-T:ITIC. The unique π–π interactions between the thienoazacoronene units may cause the growth of large TACIC domains in PBDB-T:TACIC, as suggested by the long S1 lifetime of TACIC in PBDB-T:TACIC (60 ps, Fig. S15a†). The large domain growth of TACIC results in the somewhat unfavorable formation of the electron transport pathway and in turn unbalanced charge mobilities in PBDB-T:TACIC, despite the potential for more efficient electron transport of TACIC than ITIC. Nevertheless, it should be emphasized that a high ED efficiency (>0.96) was achieved in PBDB-T:TACIC thanks to the extremely long S1 lifetime of the TACIC film (Fig. S20†).
Conclusions
In summary, we have successfully synthesized a 2D nanographene-based A–D–A type NFA, TACIC. Due to the introduction of thienoazacoronene into the D unit, TACIC possesses (i) a narrow energy band gap (1.59 eV) with a high ε value, (ii) a remarkably long exciton lifetime (1.59 ns), (iii) an excellent μe value in the film state, and (iv) high stability of the reduced state. In particular, the exciton lifetime of the TACIC film was significantly prolonged compared to TACIC in solution (220 ps) as a result of the unique self-assembling behavior of TACIC in the film state. This is highly advantageous for ED in OPV devices. Consistently, when combined with a conjugated polymer donor, PBDB-T, unusually slow (60 ps) but highly efficient ED and CT (96% in total) were achieved with the excitation of TACIC. Importantly, the unusual EnT from the excited PBDB-T to TACIC was also observed, demonstrating the dual role of TACIC in terms of LH: its direct LH and indirect LH through PBDB-T. The OPV device with PBDB-T and TACIC showed a PCE of 9.92%, which was comparable to that of the representative ITIC (9.71%). It is notable that the formation of large TACIC domains deteriorates the charge mobility balance (μe/μh) of the blend film with PBDB-T, mainly restricting photovoltaic performance. Although the PCE value is still not as high as those of state-of-the-art OPV devices, it is well-established that side-chain engineering has a large impact on the film structure of photoactive D–A layers and photovoltaic performance.25 Accordingly, there is still large room for improvement by modifying the side-chain and core structures of thienoazacoronene, thereby modulating the self-assembling behaviour and balancing the charge mobilities of the blend film. Thus, by using a thienoazacoronene-based NFA with such an optimized self-assembling nature, not only the JSC and VOC but also the FF can be improved. Furthermore, because TACIC has a long exciton lifetime and acts as an excellent energy acceptor from the polymer donor, the TACIC-based OPV devices are expected to show high ED efficiencies, even in the phase-separation structure with large domain sizes. Such characteristics of TACIC are advantageous to the future commercialization of OPV devices, because one of the problems that hamper the large-scale production of high-performance OPV devices is the difficulty in the highly reproducible formation of the D–A bicontinuous structures in the photoactive layers with domain sizes smaller than the exciton diffusion length. This work will open up avenues for a new rational strategy of OPVs where all the photocurrent generation processes, LH, ED, and CT starting from the acceptor side as well as LH, EnT, ED, and CT starting from the donor side can be better designed by using a 2D heterocyclic nanographene core as a promising building block for high-performance A–D–A type NFAs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant Number 18H03898). The synchrotron radiation experiments for GIWAXS measurements were performed at the BU46XU of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2019A1824).Notes and references
- (a) B. Kippelen and J.-L. Brédas, Energy Environ. Sci., 2009, 2, 251 RSC; (b) L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642 CrossRef CAS PubMed; (c) H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821 CrossRef CAS PubMed.
- (a) Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC; (b) J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119 CrossRef CAS PubMed; (c) J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Nat. Energy, 2018, 3, 720 CrossRef CAS; (d) G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447 CrossRef CAS PubMed; (e) A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596 RSC.
- (a) D. He, F. Zhao, L. Jiang and C. Wang, J. Mater. Chem. A, 2018, 6, 8839 RSC; (b) F. Shen, J. Xu, X. Li and C. Zhan, J. Mater. Chem. A, 2018, 6, 15433 RSC; (c) S. Dey, Small, 2019, 15, 1900134 CrossRef PubMed; (d) H. Wang, J. Cao, J. Yu, Z. Zhang, R. Geng, L. Yang and W. Tang, J. Mater. Chem. A, 2019, 7, 4313 RSC.
- (a) Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170 CrossRef CAS PubMed; (b) W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734 CrossRef CAS PubMed.
- (a) S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423 CrossRef CAS PubMed; (b) W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148 CrossRef CAS PubMed; (c) H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743 CrossRef CAS PubMed; (d) J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140 CrossRef CAS; (e) B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746 CrossRef CAS; (f) K. Li, Y. Wu, Y. Tang, M.-A. Pan, W. Ma, H. Fu, C. Zhan and J. Yao, Adv. Energy Mater., 2019, 1901728 CrossRef; (g) X. Xu, K. Feng, Z. Bi, W. Ma, G. Zhang and Q. Peng, Adv. Mater., 2019, 31, 1901872 CrossRef PubMed; (h) Y. Lin, B. Adilbekova, Y. Firdaus, E. Yengel, H. Faber, M. Sajjad, X. Zheng, E. Yarali, A. Seitkhan, O. M. Bakr, A. El-Labban, U. Schwingenschlögl, V. Tung, I. McCulloch, F. Laquai and T. D. Anthopoulos, Adv. Mater., 2019, 31, 1902965 CrossRef CAS PubMed.
- T. Umeyama and H. Imahori, Acc. Chem. Res., 2019, 52, 2046 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145 CrossRef CAS.
- L. Yang, W. Gu, L. Lv, Y. Chen, Y. Yang, P. Ye, J. Wu, L. Hong, A. Peng and H. Huang, Angew. Chem., Int. Ed., 2018, 57, 1096 CrossRef CAS PubMed.
- (a) Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955 CrossRef CAS PubMed; (b) H. Yao, L. Ye, J. Hou, B. Jang, G. Han, Y. Cui, G. M. Su, C. Wang, B. Gao, R. Yu, H. Zhang, Y. Yi, H. Y. Woo, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1700254 CrossRef PubMed.
- (a) Y. Li, L. Zhong, B. Gautam, H.-J. Bin, J.-D. Lin, F.-P. Wu, Z. Zhang, Z.-Q. Jiang, Z.-G. Zhang, K. Gundogdu, Y. Li and L.-S. Liao, Energy Environ. Sci., 2017, 10, 1610 RSC; (b) N. D. Eastham, J. L. Logsdon, E. F. Manley, T. J. Aldrich, M. J. Leonardi, G. Wang, N. E. Powers-Riggs, R. M. Young, L. X. Chen, M. R. Wasielewski, F. S. Melkonyan, R. P. H. Chang and T. J. Marks, Adv. Mater., 2018, 30, 1704263 CrossRef PubMed; (c) X. Yi, B. Gautam, I. Constantinou, Y. Cheng, Z. Peng, E. Klump, X. Ba, C. H. Y. Ho, C. Dong, S. R. Marder, J. R. Reynolds, S.-W. Tsang, H. Ade and F. So, Adv. Funct. Mater., 2018, 28, 1802702 CrossRef.
- D. He, F. Zhao, J. Xin, J. J. Rech, Z. Wei, W. Ma, W. You, B. Li, L. Jiang, Y. Li and C. Wang, Adv. Energy Mater., 2018, 8, 1802050 CrossRef.
- (a) J. Zhu, Z. Ke, Q. Zhang, J. Wang, S. Dai, Y. Wu, Y. Xu, Y. Lin, W. Ma, W. You and X. Zhan, Adv. Mater., 2018, 30, 1704713 CrossRef PubMed; (b) T. Li, S. Dai, Z. Ke, L. Yang, J. Wang, C. Yan, W. Ma and X. Zhan, Adv. Mater., 2018, 30, 1705969 CrossRef PubMed.
- (a) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS PubMed; (b) E. Moulin, J.-J. Cid and N. Giuseppone, Adv. Mater., 2013, 25, 477 CrossRef CAS PubMed; (c) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC; (d) T. Umeyama, T. Hanaoka, H. Yamada, Y. Namura, S. Mizuno, T. Ohara, J. Baek, J. Park, Y. Takano, K. Stranius, N. V. Tkachenko and H. Imahori, Chem. Sci., 2019, 10, 6642 RSC.
- (a) B. He, A. B. Pun, L. M. Klivansky, A. M. McGough, Y. Ye, J. Zhu, J. Guo, S. J. Teat and Y. Liu, Chem. Mater., 2014, 26, 3920 CrossRef CAS; (b) B. He, B. A. Zhang, F. Liu, A. Navarro, M. P. Fernández-Liencres, R. Lu, K. Lo, T. L. Chen, T. P. Russell and Y. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 20034 CrossRef CAS PubMed.
- B. He, J. Dai, D. Zherebetskyy, T. L. Chen, B. A. Zhang, S. J. Teat, Q. Zhang, L. Wang and Y. Liu, Chem. Sci., 2015, 6, 3180 RSC.
- R. Tao, T. Umeyama, T. Higashino, T. Koganezawa and H. Imahori, ACS Appl. Mater. Interfaces, 2015, 7, 16676 CrossRef CAS PubMed.
- Photophysics of Aromatic Molecules, ed. J. B. Birks, Wiley, London, 1970 Search PubMed.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (b) M. Gon, K. Tanaka and Y. Chujo, Bull. Chem. Soc. Jpn., 2019, 92, 7 CrossRef CAS.
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, S. Tierney, W. Zhang, M. Heeney, I. McCulloch, J. Nelson, D. D. C. Bradley and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 3030 CrossRef CAS PubMed.
- (a) K. Kawashima, T. Fukuhara, Y. Suda, Y. Suzuki, T. Koganezawa, H. Yoshida, H. Ohkita, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 10265 CrossRef CAS PubMed; (b) Y. Tamai, Y. Fan, V. O. Kim, K. Ziabrev, A. Rao, S. Barlow, S. R. Marder, R. H. Friend and S. M. Menke, ACS Nano, 2017, 11, 12473 CrossRef CAS PubMed; (c) H. Cha, G. Fish, J. Luke, A. Alraddadi, H. H. Lee, W. Zhang, Y. Dong, S. Limbu, A. Wadsworth, I. P. Maria, L. Francàs, H. L. Sou, T. Du, J.-S. Kim, M. A. McLachlan, I. McCulloch and J. R. Durrant, Adv. Energy Mater., 2019, 1901254 CrossRef.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed.
- (a) B. Kan, J. Zhang, F. Liu, X. Wan, C. Li, X. Ke, Y. Wang, H. Feng, Y. Zhang, G. Long, R. H. Friend, A. A. Bakulin and Y. Chen, Adv. Mater., 2018, 30, 1704904 CrossRef PubMed; (b) O. M. Awartani, B. Gautam, W. Zhao, R. Younts, J. Hou, K. Gundogdu and H. Ade, J. Mater. Chem. A, 2018, 6, 12484 RSC; (c) T. R. Hopper, D. Qian, L. Yang, X. Wang, K. Zhou, R. Kumar, W. Ma, C. He, J. Hou, F. Gao and A. A. Bakulin, Chem. Mater., 2019, 31, 6860 CrossRef CAS.
- (a) X. Shi, L. Zuo, S. B. Jo, K. Gao, F. Lin, F. Liu and A. K.-Y. Jen, Chem. Mater., 2017, 29, 8369 CrossRef CAS; (b) L. Xue, Y. Yang, J. Xu, C. Zhang, H. Bin, Z.-G. Zhang, B. Qiu, X. Li, C. Sun, L. Gao, J. Yao, X. Chen, Y. Yang, M. Xiao and Y. Li, Adv. Mater., 2017, 29, 1703344 CrossRef PubMed.
- B. Qiab and J. Wang, Phys. Chem. Chem. Phys., 2013, 15, 8972 RSC.
- C. Zhang, S. Feng, Y. Liu, R. Hou, Z. Zhang, X. Xu, Y. Wu and Z. Bo, ACS Appl. Mater. Interfaces, 2017, 9, 33906 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06456g |
| This journal is © The Royal Society of Chemistry 2020 |