
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis of brominated-boron-doped PAHs by alkyne 1,1-bromoboration: mechanistic and functionalisation studies†‡
K.
Yuan
a,
R. J.
Kahan
b,
C.
Si
c,
A.
Williams
b,
S.
Kirschner
 a,
M.
Uzelac
a,
E.
Zysman-Colman
*c and
M. J.
Ingleson
*a
a,
M.
Uzelac
a,
E.
Zysman-Colman
*c and
M. J.
Ingleson
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: michael.ingleson@ed.ac.uk
bSchool of Chemistry, University of Manchester, Manchester, M13 9PL, UK
cOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK
First published on 27th February 2020
Abstract
The synthesis of a range of brominated-Bn-containing (n = 1, 2) polycyclic aromatic hydrocarbons (PAHs) is achieved simply by reacting BBr3 with appropriately substituted alkynes via a bromoboration/electrophilic C–H borylation sequence. The brominated-Bn-PAHs were isolated as either the borinic acids or B-mesityl-protected derivatives, with the latter having extremely deep LUMOs for the B2-doped PAHs (with one example having a reduction potential of E1/2 = −0.96 V versus Fc+/Fc, Fc = ferrocene). Mechanistic studies revealed the reaction sequence proceeds by initial alkyne 1,1-bromoboration. 1,1-Bromoboration also was applied to access a number of unprecedented 1-bromo-2,2-diaryl substituted vinylboronate esters directly from internal alkynes. Bromoboration/C–H borylation installs useful C–Br units onto the Bn-PAHs, which were utilised in Negishi coupling reactions, including for the installation of two triarylamine donor (D) groups onto a B2-PAH. The resultant D–A–D molecule has a low optical gap with an absorption onset at 750 nm and emission centered at 810 nm in the solid state.
Introduction
The incorporation of main group elements into conjugated organic scaffolds is a powerful strategy to tune electronic properties.1 Recently, this approach has been extended to the introduction of three-coordinate boron atoms into polycyclic aromatic hydrocarbons (PAHs).1,2 In these “B-doped”-PAHs interaction between the empty p orbital on boron and the extended π system often leads to molecules with low energy LUMOs.2 B-doped PAHs now are being explored for a range of applications including organic light-emitting diodes, thin film transistors, solar cells, lithium batteries and electrocatalysis.2 However, two factors have limited the wider uptake of purely B-doped PAHs (i.e. PAHs without co-doping elements e.g. N): (i) the limited simple synthetic routes to form Bn-PAHs, particularly for n > 1, which generally have the deeper LUMOs;2 (ii) the challenge associated with incorporating Bn-doped-PAHs into more complex materials, e.g. by coupling reactions to access donor–acceptor (D–A) structures.3 Some notable progress has been made addressing (i), with several routes to larger Bn-PAHs, (n ≥ 2, see A–D, Fig. 1) including examples with low LUMO energies, recently reported.4,5 However, the incorporation of Bn-PAHs into more complex materials currently requires the post synthetic introduction of additional functionality onto the Bn-PAH to enable subsequent steps (e.g. cross coupling).6 This has associated challenges (e.g. selectivity in halogenation) along with step-economy issues. Therefore, a simple route to form a range of Bn-doped PAHs with low LUMO energies that concomitantly installs a second useful functional group, such as halide, would be highly attractive and facilitate access to more complex materials with desirable properties.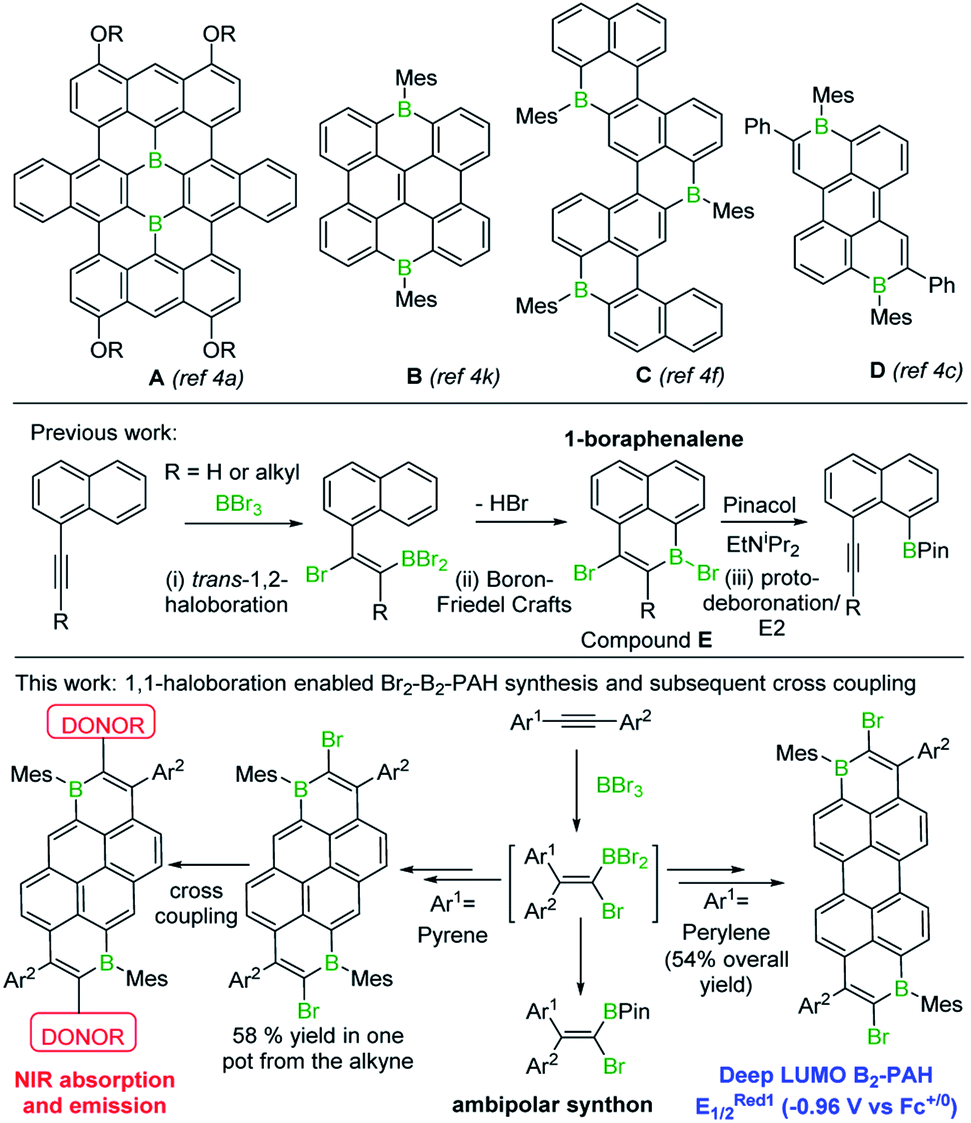 | ||
| Fig. 1 Top, select examples of some of the largest/deepest LUMO Bn-PAHs (n > 1) containing three coordinate C3B units. Middle, previous work forming boraphenalenes by bromoboration/SEAr. Bottom, this work accessing and utilising large, low LUMO energy dibrominated B2-PAHs. | ||
In our previous work, a one-pot synthesis of 1-boraphenalenes (E) via sequential bromoboration/intramolecular boron-Friedel–Crafts reaction of 1-alkynylnaphthalenes (Fig. 1, middle) was described.7 We envisaged that this methodology could be extended by: (i) using larger (than naphthalene) π-scaffolds to obtain B1 and B2 doped PAHs possessing deeper LUMOs; (ii) utilising the bromo substituent to access more complex materials with desirable properties via coupling reactions. Herein these two objectives are realised enabling access to the lowest LUMO energy ambient stable Bn-doped PAH reported to date, to the best of our knowledge, and a low-optical gap D–A–D material where the acceptor unit is a B2-PAH. In addition, mechanistic studies on the bromoboration/SEAr process revealed it proceeds by alkyne 1,1-bromoboration, a process not previously observed. Therefore 1,1-bromoboration also was applied to simple diarylalkynes demonstrating its utility for forming 1-bromo-2,2-diaryl substituted vinylboronate esters that are otherwise challenging to access.
Results and discussion
Synthesis of boron-doped PAHs and 1,1-bromoboration studies
Initially, 1-(pent-1-yn-1-yl)pyrene, 1, was combined with BBr3 in the presence of one equivalent of 2,4,6-tri-tert-butylpyridine (TBP), targeting an analogous trans-haloboration/SEAr process that previously afforded 1-boraphenalenes.7 Post protection at boron by reaction with dimesitylzinc, purification afforded two isomeric compounds, 1a and 1b (Scheme 1) in low yield, partly due to low stability of these compounds to the column chromatography. To provide enhanced stability, the bulkier 2,4,6-triisopropylphenyl (Tip) protecting group was used instead of mesityl (Mes). Using similar reaction conditions, compounds 1c and 1d were isolated in higher yields relative to 1a and 1b.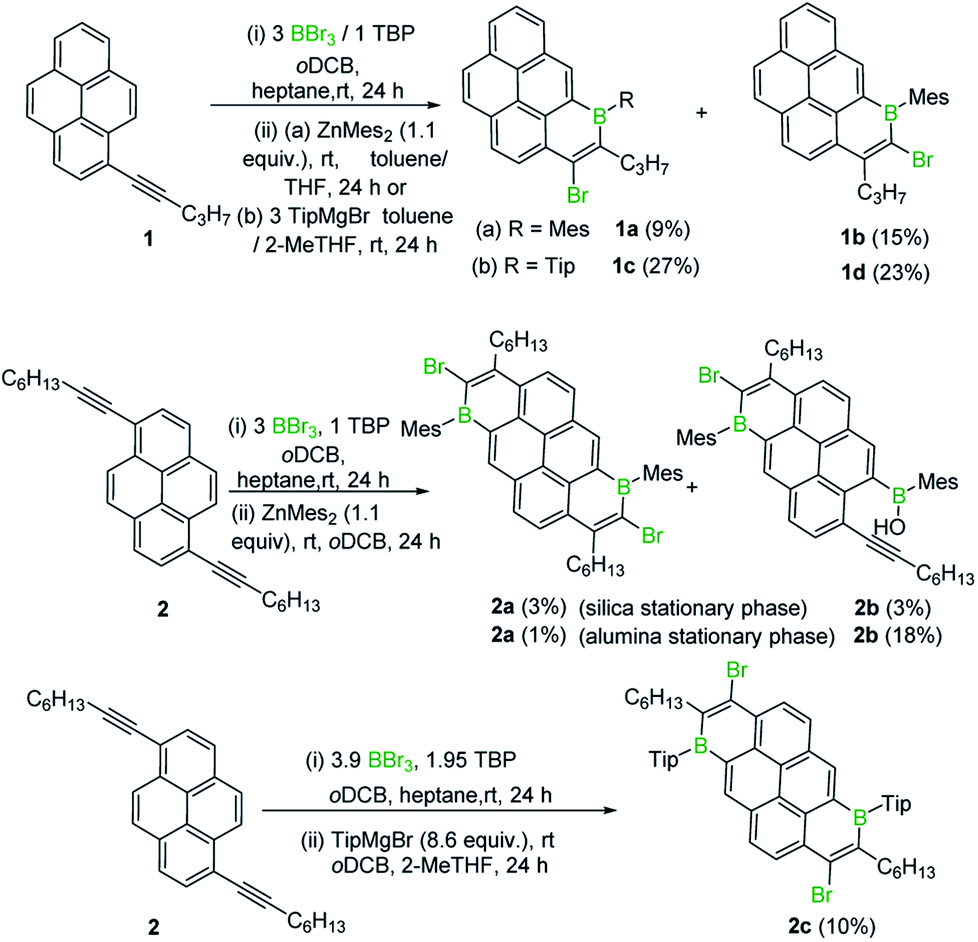 | ||
| Scheme 1 Isolated products from the reaction of alkyl-alkynyl-pyrenes with BBr3. | ||
B2-doped PAHs were next targeted through a two-fold bromoboration/SEAr sequence starting from 1,6-di(pent-1-yn-1-yl)pyrene, 2. However, post borylation/protection at boron, compound 2a was the only fully fused B2-PAH isolable via column chromatography in our hands and it was obtained in a very low yield alongside 2b. Compound 2b presumably derives from protodeboronation of a vinyl C–B unit (analogous to that in 1a) followed by an E2 elimination reaction as previously observed for the 1-boraphenalenes (see Fig. 1, middle right).7 Tip installation conditions also were applied to the product mixture derived from BBr3/2, however this led to the formation of compound 2c in 10% yield as the only isolable product in our hands. The structures of 1a, 2a and 2c were confirmed by single crystal X-ray diffraction analysis (see subsequent discussion). It should be noted that products derived from both 1,2 and 1,1-haloboration were observed (e.g. the B–Br analogues of 2a/2c) and this may explain the complex mixtures observed starting from 2 with lower symmetry (than 2a/2c) borylated products also observed spectroscopically (presumably containing one 1,1- and one 1,2-bromoborated boracycle in the same molecule).
In previous work 1-(arylethynyl)naphthalenes reacted with BBr3 to give different products to that starting from 1-(alkyl-ethynyl)naphthalenes, with the former affording products from a 1,1-bromoboration/SEAr cyclisation process (e.g.F, inset Scheme 2, is formed instead of an analogue of E, Fig. 1 middle),7 with no 1,2-bromoboration derived products observed. Therefore, aryl-alkynyl-pyrene derivatives (e.g.3) were utilised in an attempt to avoid forming mixtures derived from competing 1,1 and 1,2-bromoboration. Starting from 3, the desired Tip protected product 3a was isolated in 75% yield with negligible 1,2-bromoboration products observed. In addition, the borinic acid 3b was accessible in good yield using simple aqueous workup conditions. 3b proved sufficiently stable for column chromatography and even towards 1 M HCl (aq.) for at least 0.5 h. Notably, all the aryl-substituted derivatives studied herein proved more resistant to protodeboronation than the alkyl congeners, with protection by mesityl also providing sufficient stabilisation for these derivatives. The NMR data for 3a/3b are comparable to that previously reported for the B–OH and B-aryl 1-boraphenalenes.7
 | ||
| Scheme 2 Formation of 3a and 3b and mechanistic implications. | ||
Earlier studies on the bromoboration of diphenylacetylene using BBr3 by Lappert and co-workers suggested that only the 1,2-cis-bromoboration product (4, Scheme 3 inset) forms.8 Furthermore, alkyne 1,1-haloboration using boron electrophiles was to the best of our knowledge unprecedented prior to this work, with 1,2-haloboration or 1,1-carboboration the general outcomes.9 Thus, in our previous report on the bromoboration of 1-(arylalkynyl)-naphthalenes,7 a mechanism was proposed for forming F that avoided 1,1-bromoboration. According to that previously proposed mechanism, compound 3c (inset Scheme 2) should be obtained starting from 3via cleavage of the pyrene C1–Calkyne bond. However, X-ray diffraction analysis of the haloboration products 3a and 3b confirmed that boron was attached to C10 of pyrene and that the C1–Calkyne bond remains intact. These observations indicate the reaction most likely proceeds through 1,1-bromoboration followed by electrophilic C–H borylation (Scheme 2, bottom right). Therefore, the bromoboration of other diarylalkynes, including diphenylacetylene, using BBr3 was revisited to probe the apparent discrepancy with earlier work.
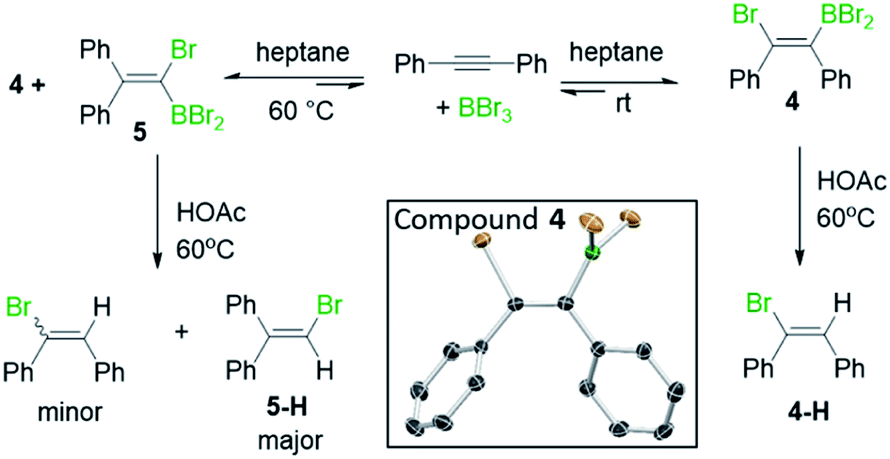 | ||
| Scheme 3 The observation of 1,1- and 1,2-bromoboration under different conditions. Inset, the solid state structure of 4, hydrogens omitted for clarity. | ||
On combining diphenylacetylene with BBr3 in heptane, crystals precipitated over the course of 5 minutes X-ray diffraction analysis on these revealed them to be the 1,2-cis-bromoboration product, 4 (inset Scheme 3). NMR studies under the same conditions revealed that only one product is observed in solution, assigned as compound 4. Protodeboronation of this reaction mixture afforded 1-bromo-1,2-diphenylethene, 4-H with no 1,1-bromoboration isomer (5-H, Scheme 3) observed. While these observations are consistent with previous work,8 upon heating the reaction mixture in heptane at 60 °C an additional product, assigned as 5, was observed (by 13C{1H} and 11B NMR spectroscopy). Protodeboronation of this reaction mixture afforded 1-bromo-2,2-diphenylethene, 5-H, derived from 1,1-haloboration as the major product along with minor amounts of 1-bromo-1,2-diphenylethene (4-H). Notably, when bromoboration was performed in DCM, both 1,2- and 1,1-bromoboration products were observed after short reaction times at 20 °C (ca. 10 minutes by NMR spectroscopy and by analysis of products post protodeboronation). These observations suggest that 1,1-bromoboration proceeds through a polar transition state(s), and potentially a vinyl cation type intermediate(s), more stabilised by the polar solvent DCM. Identical product distributions also were observed using 3 equiv. of BBr3 in place of 1.5 equiv. of BBr3.
Similar outcomes were observed with di-p-tolylacetylene (6) and 1,2-bis(4-fluorophenyl)ethyne with the 1,1-haloboration product being the major species observed in DCM in both cases. Exact ratios of the BBr2 1,1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1,2-haloboration (both cis and trans isomers) products in DCM are challenging to determine in situ by NMR spectroscopy due to uncertainty in isomer assignment. Furthermore, ratios of BPin/vinyl C–H products post work-up and isolation are not indicative of in situ ratios due to competing pinacol protection/protodeboronation/retrohaloboration (see ESI‡ Section 4 for further discussion). For di-p-tolylacetylene a mixture of bromoboration products (ca. 3:10) was formed in DCM 10 minutes after the addition of BBr3 at room temperature. To determine the identity of the major isomer the fact that alkyne 1,2-bromoboration products (e.g.4) are converted rapidly back into the starting alkyne on adding pyridine,8 (concomitantly forming pyridine–BBr3) was exploited. Upon addition of pyridine to the 3:10 bromoboration reaction mixture, the minor bromoboration product, assigned as 6a (Scheme 4), was converted into the starting alkyne and pyridine–BBr3, while the major bromoboration product, assigned as 6b, formed an adduct, 7, with pyridine at room temperature. Upon heating at 60 °C, compound 7 also converted into di-p-tolylacetylene and pyridine–BBr3. Addition of excess BBr3 to the mixture containing 7 to sequester all pyridine as pyridine–BBr3 regenerated the mixture of 1,2- and 1,1-haloboration products in an identical 3:10 ratio to that originally observed. Combined these results indicate that 1,1-haloboration dominates in DCM, that this reaction reaches its equilibrium position rapidly in DCM, and that both 1,2- and 1,1-bromoboration are reversible. DFT calculations (at the M06-2X/6-311G(d,p) PCM (DCM) level) revealed the 1,1- and 1,2-bromoboration products (e.g.4 and 5) were effectively isoenergetic (all pairs of isomers had ΔE < 1.3 and ΔG < 0.8 kcal mol−1) consistent with the observation of mixtures on reaching equilibrium. Finally, 1,1-bromoboration is not limited to diarylalkynes, with 1-phenyl-1-propyne also leading to 1,1-bromoboration products.
:1,2-haloboration (both cis and trans isomers) products in DCM are challenging to determine in situ by NMR spectroscopy due to uncertainty in isomer assignment. Furthermore, ratios of BPin/vinyl C–H products post work-up and isolation are not indicative of in situ ratios due to competing pinacol protection/protodeboronation/retrohaloboration (see ESI‡ Section 4 for further discussion). For di-p-tolylacetylene a mixture of bromoboration products (ca. 3:10) was formed in DCM 10 minutes after the addition of BBr3 at room temperature. To determine the identity of the major isomer the fact that alkyne 1,2-bromoboration products (e.g.4) are converted rapidly back into the starting alkyne on adding pyridine,8 (concomitantly forming pyridine–BBr3) was exploited. Upon addition of pyridine to the 3:10 bromoboration reaction mixture, the minor bromoboration product, assigned as 6a (Scheme 4), was converted into the starting alkyne and pyridine–BBr3, while the major bromoboration product, assigned as 6b, formed an adduct, 7, with pyridine at room temperature. Upon heating at 60 °C, compound 7 also converted into di-p-tolylacetylene and pyridine–BBr3. Addition of excess BBr3 to the mixture containing 7 to sequester all pyridine as pyridine–BBr3 regenerated the mixture of 1,2- and 1,1-haloboration products in an identical 3:10 ratio to that originally observed. Combined these results indicate that 1,1-haloboration dominates in DCM, that this reaction reaches its equilibrium position rapidly in DCM, and that both 1,2- and 1,1-bromoboration are reversible. DFT calculations (at the M06-2X/6-311G(d,p) PCM (DCM) level) revealed the 1,1- and 1,2-bromoboration products (e.g.4 and 5) were effectively isoenergetic (all pairs of isomers had ΔE < 1.3 and ΔG < 0.8 kcal mol−1) consistent with the observation of mixtures on reaching equilibrium. Finally, 1,1-bromoboration is not limited to diarylalkynes, with 1-phenyl-1-propyne also leading to 1,1-bromoboration products.
 | ||
| Scheme 4 Bromoboration in DCM, and reactivity of products towards pyridine. | ||
The preparation of fully-substituted 1-bromo-1-alkenyl boronate esters currently is challenging (the major route to these types of compounds proceeds via hydroboration of alkynyl-halides thus is limited to forming tri-substituted alkenes).10 Indeed diaryl-derivatives (such as 8a–fScheme 5), have not been previously reported to the best of our knowledge. While 1,1-bromoboration represents a simple approach to these compounds to be useful protection at boron without (or with minimal) competing formation of the starting alkyne by retro-bromoboration is required. In the presence of excess triethylamine, the 1,1-bromoboration products (e.g.5) were converted into the pinacol boronate esters, 8a–g demonstrating that ortho, meta and para substituted aryl-alkynes are amenable to this procedure. The yields are moderate at best due to the presence of the 1,2-isomer in the reaction mixture and the propensity of the Br-vinyl-BBr2 isomers (e.g.4 and 5) to undergo retrohaloboration in competition to pinacol protection (see ESI‡ for more details). Indeed, under these conditions all the 1,2-bromoboration products (and the mass balance of the 1,1-haloboration products) were converted into the starting alkyne and Et3N–BBr3. Attempts at pinacol installation in the absence of base led to competitive protodeboronation (presumably due to the HBr by-product from pinacol installation onto the BBr2 moiety) and lower yields of 8x in most cases. The pinacol boronate esters were fully characterised and a number confirmed by X-ray diffraction analysis (e.g. inset Scheme 5). It should be noted that substrates containing CF3 were not amenable to this process due to B–F/C–Br bond formation on addition of BBr3. Furthermore, ether cleavage occurs concomitantly to haloboration, with substrate 8f isolated from the 4-methoxyaryl precursor. In addition, attempts to bromoborate 2,2′-bis(2-thienyl)ethyne with BBr3 were successful to some extent, but protection with pinacol under a range of conditions was unsuccessful. This instead led to protodeboronation under a range of conditions. Finally an electronically biased diaryl alkyne was subjected to haloboration in DCM, however pinacol protection revealed that multiple haloborated isomers, e.g.8h, had formed that could not be separated in our hands.
 | ||
| Scheme 5 1,1-Haloboration and subsequent pinacol protection. Inset right, solid state structure of 8b, hydrogens omitted for clarity, ellipsoids at 50% probability. a = from the p-MeO derivative with ether cleavage occurring concomitantly to haloboration. b = pinacol protected without Et3N. | ||
With an understanding of the bromoboration process in hand, the extension of 1,1-haloboration/SEAr to other substrates was explored. Using 1,6-bis((4-(tert-butyl)phenyl)ethynyl)pyrene, 9 (an analogue of 2) and BBr3 this sequential transformation worked effectively and post mesityl installation afforded 9a in 58% yield in a one-pot reaction starting from 9. Compound 9a proved bench stable (as 9–12a/b all are) and was purified via column chromatography. The borinic acid 9b could also be prepared from 9 in 62% yield via a simple hydrolytic work-up. Starting from 7-(tert-butyl)-1,3-bis((4-(tert-butyl)phenyl)-ethynyl)pyrene, 10, both the Mes-protected product 10a and the borinic acid 10b also were accessible, albeit isolated in lower yields. 1,1-Haloboration/SEAr also was applicable to perylene substrates. For 3-(p-tolylethynyl)perylene, 11, the Tip-protected product 11a (50%) and the borinic acid 11b (88%) were isolated post column chromatography. The bromoboration/SEAr cyclisation reaction also worked with 3,9-bis((4-(tert-butyl)phenyl)ethynyl)perylene, 12, to yield the Mes-protected B2-PAH 12a (54% yield) and the borinic acid 12b (82% yield) depending on the work-up protocol (Scheme 6).
 | ||
| Scheme 6 The formation of extended Bn-PAHs by bromoboration/SEAr. | ||
Solid-state structures
The solid-state structures of B1-PAHs 1a, 1c and 3b revealed effectively planar six-membered boracycles with boron atoms adopting a trigonal planar geometry (Σ(C–B–C) ≈ 360°). Notably, the C12–C13 bond length (1.355(5) Å in 1a) is shorter than the C11–C12, C1–C10 and C10–C11 bond lengths (1.462(5), 1.448(5) and 1.414(4) Å, respectively in 1a) as is the case in 1c and 3b. The bond length alternation indicates the lack of significant π delocalisation within the boracycle as was observed with the 1-boraphenalenes.7 The fusion of a boracycle onto the PAH core does have some impact, for example on the C–C bond in the K-region of pyrene. For 1a the bond length of C1–C2 (1.367(4) Å) is longer than that of C14–C15 (1.337(5) Å). The elongation of C1–C2 vs. C14–C15 may indicate some delocalisation of this π-electron density into the boron centre.11a For compound 3b, the B–O bond length (1.369(4) Å) is close to the reported values for other related borinic acids, indicating some multiple bond character between boron and oxygen. The Mes/Tip and tert-butylphenyl groups are all almost orthogonal to the boracycle, which precludes close face to face π stacking involving the boracycles in the intermolecular structure (Fig. 2). | ||
| Fig. 2 Solid-state structures of 1a (left) and 3b (right), most hydrogens omitted for clarity. Ellipsoids at the 50% probability level. | ||
The solid-state structures of the B2-doped PAHs 2a, 2c, 9a, 10b and 12a also were determined (Fig. 3, 4 and ESI‡). All five compounds show planar π-conjugated cores with the peripheral substituents effectively orthogonal to the PAH core. As a result, no face to face π-stacking is observed in the extended structures for these compounds (excluding 10b, see ESI‡). For example, the face to face intermolecular separation of the cores of two parallel molecules is found to be 6.67 Å for 2c. The boracycle metrics are similar to the mono-borylated compounds with C–C bond length alternation within the boracycles also observed (for example a C11–C12 vinylic distance in 12a of 1.359(7) Å and long endocyclic B–C bonds 1.534(6) and 1.543(6) Å indicate minimal π delocalisation consistent with previous NICS calculations).7 Notably, for the diborylated pyrene compounds, the length of the C–C bonds in the pyrene K-region is longer than that in the mono-borylated compound (e.g. the bond length of C7–C10 is 1.379(3) Å for compound 2c). The elongation of C7–C10 bond length may indicate a greater delocalisation of these π-electrons into the boron centres for the di-borylated compounds relative to the mono-borylated compounds.
 | ||
| Fig. 3 Solid state structures of 2c (two views) and 9a, with hydrogens omitted for clarity. Ellipsoids are at the 50% probability level. | ||
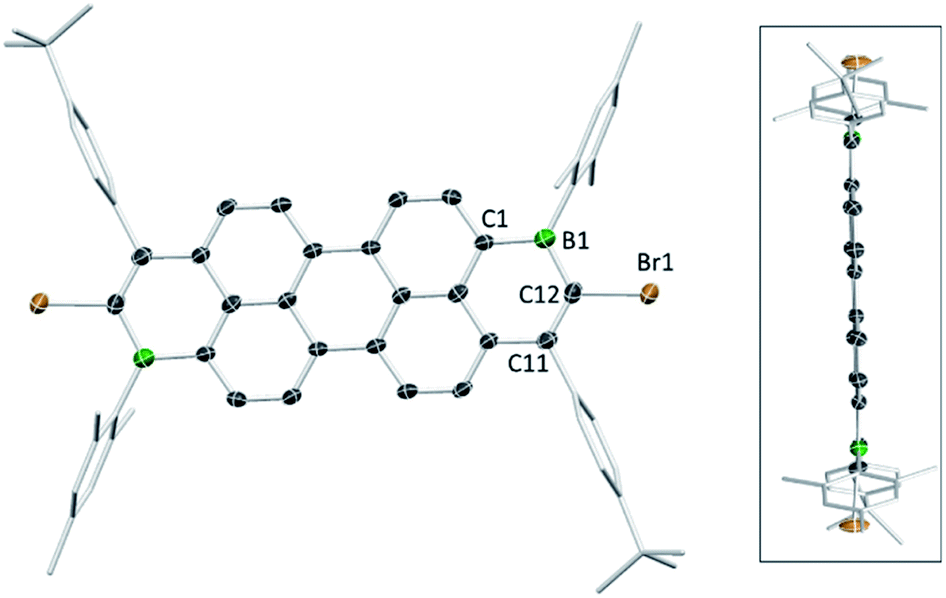 | ||
| Fig. 4 Solid state structure of 12a (two views), with hydrogens omitted for clarity. Ellipsoids are at the 50% probability level. | ||
Cyclic voltammetry
The first reduction potentials ranged from −1.91 V (3b) to −0.96 V (12a, Table 1). Within the scan window, the B1-doped PAHs showed one reversible reduction wave except for compound 11a, for which two reduction waves were observed. All B2-PAHs showed two reversible reduction waves; however, no oxidation waves were observed within the solvent window for these compounds. The hydroxyl group on boron reduced the electron-accepting abilities of the boron-doped PAHs compared with the Tip/Mes group as previously observed.4c Also as noted by Würthner and co-workers,4c the boron doping pattern also significantly influences the redox properties with the first reduction potential of 9a more positive than that of 10a by 0.15 V (for 9bvs.10b ΔEred11/2 = 0.32 V). Notably, the reduction potential of 9a is more positive than bis-(BMes2)-pyrenes (4,9 isomer Ered11/2 = −2.31 V, 1,6 isomer Ered11/2 = −1.81 V)11 and the products from the electrophilic borylation of 1,6-(2-pyridyl)2-pyrene (when the boron moiety = BPh2Ered11/2 = −2.08 V)14 this is ascribed to greater π delocalization in 9a as a result of planarization and three-coordinate B centres. Finally, 9a has a more positive reduction potential than its all carbon analogue (for which Ered11/2 = −2.11 V),15 further confirming boron-doping as an effective strategy to obtain molecules with deep LUMOs due to their electron-deficient nature (B2-PAHs are isoelectronic to dicationic carbon analogues).| E red11/2 (V) | E red21/2 (V) | LUMOexpb (eV) | λ abs [ε/10−3 M−1 cm−1] (nm) | λ PL (nm) | Φ PL (%) | E g (eV) | HOMOf (eV) | LUMOf (eV) | |
|---|---|---|---|---|---|---|---|---|---|
| a Measurements carried out at 298 K in THF with 0.1 M [nBu4N][PF6] as the supporting electrolyte. Electrochemical reduction potentials were calibrated with ferrocene as internal standard and referenced vs. Fc+/Fc. b E LUMO = −Ered1/2 − 5.15 eV.12 c Optical measurements carried out at 298 K in toluene at 1 × 10−5 M. d Under aerated conditions using [Ru(bpy)3](PF6)2 in CH3CN as the reference (ΦPL: 1.8%). e E g were determined at the energy corresponding to 10% of the lowest energy absorption band. f From calculations at the M06-2X/6-311G(d,p) level. g From ref. 4c. h PDI refers to the perylene diimide with N-Dip substituents from ref. 13 (Dip = 2,6-diisopropylphenyl). | |||||||||
| 3a | −1.66 | — | −3.49 | 375[18.04], 395[15.12], 417[13.16], 491[17.98] | 553, 592 | 9.2 | 2.26 | −7.04 | −2.29 |
| 3b | −1.91 | — | −3.24 | 359[3.81], 383[3.62], 404[5.34], 449[7.86] | 500, 532 | 8.7 | 2.51 | −6.97 | −2.05 |
| 9a | −1.03 | −1.49 | −4.12 | 345[3.23], 362[4.74], 396[2.64], 536[24.13], 567[18.00] | 599, 648 | 2.0 | 2.07 | −7.26 | −2.98 |
| 9b | −1.42 | −1.73 | −3.73 | 340[2.41], 355[3.69], 390[1.56], 501[32.13],528[25.05] | 557, 598 | 6.8 | 2.23 | −6.97 | −2.60 |
| 10a | −1.30 | −2.11 | −3.85 | 332[20.75], 386[21.72], 406[35.98], 435[15.45], 508[3.64], 545[4.88], 588[3.52] | 614, 668, 729 | 5.9 | 2.00 | −7.19 | −2.67 |
| 10b | −1.61 | −2.25 | −3.54 | 380[34.50], 417[3.93], 443[6.04], 476[8.37], 507[11.57], 543[8.22] | 560, 605, 657 | 46.9 | 2.19 | −7.00 | −2.43 |
| 11a | −1.43 | −2.06 | −3.72 | 314[57.13], 360[8.01], 405[10.53], 431[15.93], 456[26.37], 500[16.68], 534[33.55], 573[43.71] | 591, 628 | 0.6 | 2.08 | −6.76 | −2.50 |
| 11b | −1.57 | — | −3.58 | 312[28.42], 340[5.37], 410[7.71], 433[14.85], 456[13.10], 487[29.85],521[31.05] | 532, 570, 618 | 0.4 | 2.31 | −6.68 | −2.29 |
| 12a | −0.96 | −1.28 | −4.19 | 336[5.91], 392[5.49], 528[18.02], 568[44.48], 614[68.41] | 634, 686 | 1.6 | 1.93 | −7.06 | −3.08 |
| 12b | −1.28 | −1.54 | −3.87 | 370[1.48], 494[4.90], 530[12.52], 571[19.61] | 582, 630 | 3.8 | 2.10 | −6.86 | −2.74 |
| D | −1.07 | −1.41 | −4.08 | 611, 417 | 668 | 74 | — | — | |
| PDI | −1.02 | −4.13 | 527 | 534 | 99 | −7.21 | −2.89 | ||
Comparing pyrene and perylene core structures the reduction potentials of the borylated perylene compounds are positively shifted relative to the pyrene substrates. For example, the monoborylated compound 11a exhibited the first reduction wave at −1.43 V more positive than 3a by 0.23 V. Indeed among all the compounds investigated, 12a shows the least negative first reduction potential with Ered11/2 = −0.96 V, which is also much more positive than the related all carbon peropyrenes.16 To the best of our knowledge, compound 12a has the least negative reduction potential of all ambient stable three coordinate at boron B-doped PAHs reported to date, with compound D being the ambient stable Bn-PAH with the next least negative reduction potential (Table 1).4c
Photophysical properties
By varying the polycyclic core and the boron substituent, a series of Bn-doped PAHs (n = 1, 2) are obtained forming solutions with colours ranging from yellow to dark blue. UV-vis absorption and fluorescence spectra were recorded and the optical energy gap estimated (Table 1). All compounds showed well resolved absorption/emission profiles with small Stokes shifts. Consistent with the observed solution colours, the obtained spectra showed the lowest energy absorption band spanning 449 nm (3b, light yellow solution) to 614 nm (12a, dark blue solution). The doubly borylated compound 9a showed λmax at 567 nm, which is red-shifted by 82 nm compared with the mono-borylated compound 3a (λmax = 491 nm). The same trend was also observed for 11a/12a, due to the extended conjugation and double boracycle annulation leading to narrower HOMO–LUMO gaps. In addition, all borinic acids exhibited hypsochromic shifts of their lowest absorption band by ca. 40 nm compared with the Tip/Mes protected analogues. The photoluminescence quantum yield (ΦPL) of the brominated compounds studied herein were relatively low, which might be attributed to the heavy atom effect of bromine, as related heavy atom free analogues reported by Würthner and co-workers are notably more emissive (ΦPLs up to 95%).4cComputational studies
To provide more insight into the trends observed in the experimental data, DFT calculations were performed at the M06-2X/6-311G(d,p) level with a polarisable continuum model (PCM) of DCM, calculations emulated the full compound (except for 2c, see ESI‡). The trend in the LUMO energies from the computational results were consistent with the trends in the cyclic voltammetry data with compound 3b, that has the most negative reduction potential, being calculated to have the highest LUMO energy (−2.05 eV) among the compounds studied, while compound 12a with the least negative reduction potential was predicted to have the deepest LUMO level (−3.08 eV, see Fig. 5). It is noteworthy that the computed LUMO energy level of 12a is lower than that of a perylene diimide (PDI, red, Fig. 5) calculated at the same level of theory. Indeed comparison of the CV data for 12a and a PDI compound (containing all C–H and two N-DIP groups), confirms that 12a has the deeper LUMO,13 while 9a has very similar frontier orbital energies to this PDI (Table 1). This further indicates that both these diborylated pyrene and perylene compounds have potential as strong acceptor moieties in organic electronics. | ||
| Fig. 5 Calculated Frontier orbitals (shown at iso-values of 0.03) for several B2-PAHs and for comparison an N–Me substituted PDI. Ar = p-tBu-phenyl. | ||
As shown in Fig. 5 and the ESI‡, the LUMO and HOMO of all molecules are delocalised over the whole π-backbone and involve boron contributions to both. Interestingly, the frontier molecular orbital distributions of 12a and 12b are very similar to that of PDI. Furthermore, the frontier orbitals of all the B2-PAHs studied herein (and indeed the related compounds reported by Würthner et al.)4c are related closely to the calculated HOMO and LUMO of the parent π core molecule (e.g. the HOMO and LUMO of pyrene and perylene)17 with some additional contributions from the appended B–C unit. For completeness, the anthracene-based compound G (blue, Fig. 5) was also calculated, which revealed that the HOMO and LUMO of this compound also was related in character to the HOMO and LUMO of anthracene. Note, attempts to synthesise G failed in our hands, possibly due to the additional peri C–H group inducing steric clash with the proximal aryl group (with some structural distortion observed in the calculated structure of G). It is notable that the trend in the relative HOMO/LUMO energy and optical gap of 9b, G and 12b mirrors that of the parent PAH core,18 with perylene (deepest LUMO/highest HOMO) < anthracene < pyrene (highest LUMO/deepest HOMO) for optical gap. This indicates that access to even lower LUMO energy B2-PAHs for this series maybe possible starting from lower energy LUMO core PAH structures.
Functionalisation of boron doped PAHs by cross coupling
One attraction of the compounds reported herein is the potential utility of the bromide unit, derived from the concomitant installation of B/Br in the initial step of Bn-PAH formation. While cross coupling reactions can be performed using the C–B units as the nucleophilic coupling partner,19 this removes the electronic effect of B-doping, thus utilising the C–Br unit in cross-coupling reactions offers a way to construct complex functional electronic materials that retain the C3B units that impart the deep LUMO character.3 Initially compound 3a containing the sterically more demanding Tip unit, was explored and found to be amenable to Negishi cross coupling reaction conditions. Specifically, in the presence of Pd(PPh3)4, 3a reacted with the in situ generated aryl-zinc reagent affording the desired product 13 (Scheme 7) in moderate 33% isolated yield. We attribute the modest yield to the highly sterically encumbered vinyl-Br position present in these B-PAHs, thus the B-Mes derivatives were explored.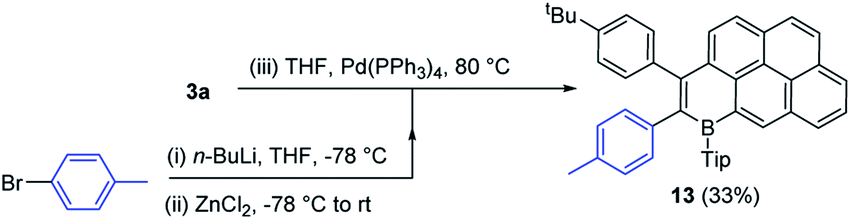 | ||
| Scheme 7 Negishi cross coupling to form compound 13. | ||
The same methodology could be applied to B-Mes derivatives to access a D–A–D molecule with a narrow HOMO–LUMO energy gap. Using the procedure established for 13, 9a was coupled with the organozinc compound derived in situ from 4-bromotriphenylamine to construct compound 14 (Fig. 6). The desired double cross coupled product, 14, was isolated by column chromatography in 21% yield. Cyclic voltammetry measurements on compound 14 showed one oxidation wave within the solvent window (peak-current Eox = 0.49 V vs. Fc/Fc+) and two reduction processes (Ered11/2 = −1.23 V, Ered21/2 = −1.62 V). The first reduction process is shifted cathodically by 0.2 V relative to 9a, presumably due to the replacement of inductively electron-withdrawing bromine for the triphenylamine groups. DFT calculations (Fig. 6, bottom) were performed on compound 14 at a lower theory level due to its size (M06-2x/6-31G(d)), 9a was calculated at the same level for direct comparison (which confirmed effectively identical HOMO/LUMO distributions for 9a using the two basis sets). Consistent with a D–A molecule the HOMO/LUMO are spatially separated, with the HOMO being predominantly localised on the triaryl amine unit thus being 0.91 eV higher in energy than the HOMO of 9a. The LUMO in 14 is localised on the B2-PAH acceptor unit and is closely related in character to the LUMO of 9a. Notably, the calculated energy for the LUMO of 14 is 0.19 eV higher than that for 9a in excellent agreement with the CV data; again this is attributed to the exchange of inductively withdrawing Br groups for triarylamine units.
 | ||
| Fig. 6 CV data, for compound 14 (1 mM solution in THF with 0.1 M [NBu4][PF6]. Inset bottom, HOMO and LUMO of compound 14 (iso-value 0.025) at the M06-2X/6-31G(d) level. | ||
The photophysical properties of 14 in solution, solid-state and dispersed in a PMMA film were investigated. Compound 14 displayed a broad absorption band stretching up to 750 nm in dichloromethane, with the onset corresponding to an optical gap of 1.65 eV (comparable to ΔEredox determined by electrochemistry). Thus, the strong acceptor character of the B2-PAH unit combined with the triphenylamine donor units results in a small HOMO–LUMO energy gap. We employed time-dependent DFT (TDDFT) calculations using the Tamm-Dancoff approximation at the PBE0/6-31G(d,p) level of theory to assign the spectral features observed in the absorption spectrum of 14. The lowest energy absorption features found within the tail of the band showing a maximum at 530 nm and a shoulder at 600 nm are assigned to be charge transfer (CT) states from the Ph2NPh group to the B2-PAH core (S0 → S1, S0 → S2). The principal distinguishable absorption features at 444 nm originate from mixed CT/LE (LE = locally excited) transitions, but this time the CT contribution implicates the mesityl groups as donors to the B2-PAH core (S0 → S3, S0 → S4, see ESI‡ for more details); the LE contribution is localized on the B2-PAH acceptor. Higher energy bands involve increasing contributions of LE transitions of the B2-PAH core. Recognizing the propensity of large acenes to aggregate in solution, even at very dilute concentrations we next investigated the effect of concentration on both the absorption and emission spectra. We note that both the ε values and the spectral features in the absorption spectra were independent of concentration, precluding aggregation of the molecule in its ground state in DCM at the concentrations studied (see ESI‡). In contrast, the emission in DCM was highly concentration dependent. At 0.15 μmol mL−1 the emission is broad and unstructured and centred at 479 nm. As the concentration is increased to 63 μmol mL−1, there is a decrease in intensity of this band coupled with a progressive and significant red-shift of the emission. At the highest concentrations studied, the emission narrows but remains unstructured (Fig. 7). We attribute this complex behaviour to the formation of mixtures of different excimers as the concentration increases, presumably with higher order aggregates forming at the higher concentrations.
 | ||
| Fig. 7 Top, photophysical data for compound 14 in DCM solution and in the solid state (neat film and dispersed in PMMA). Bottom, variation in the emission spectra on change of concentration of 14 in DCM solution. λexc = 370 nm. | ||
The ΦPL value in DCM for 14 at 0.52 μmol mL−1 is 4% and this value is not sensitive to oxygen. The excited state emission decays measured at both dilute (0.52 μmol mL−1) and concentrated (33 μmol mL−1) solutions are multi-exponential in nature but remain in the nanosecond regime, consistent with a fluorescence mechanism. In order to mitigate these non-radiative pathways, we next investigated the solid-state photophysical properties as thin films in PMMA at 0.1, 1 and 10 wt% doping concentrations, and as neat films. Generally, the same evolution in emission profiles is observed in the solid-state as was observed in DCM. As a 0.1 wt% PMMA doped film two unstructured emission bands are observed, one of higher relative intensity at high energy at λPL = 442 nm that we ascribe to the same monomer emission observed in dilute DCM, and a lower intensity and broader low energy band at λPL = 638 nm that we ascribe to excimeric emission. The ΦPL of this film is 5%, which is essentially identical to that measured in DCM. This suggests that during the spin-coating process aggregation remains strong even at this low doping concentration. As a 1 wt% PMMA doped film, two unstructured emission bands remain, one at λPL = 472 nm and one at λPL = 726 nm; here, we note the large red-shift and significant enhancement in intensity of the low-energy band. The ΦPL of this film is slightly lower at 3%. The 10 wt% doped PMMA film also shows two emission bands where the relative intensity of the high energy band at λPL = 434 nm is reduced further compared to the low energy band at λPL = 793 nm. The ΦPL of this film is <1%. The neat film shows only a single very weak and red-shifted emission band at λPL = 824 nm with a ΦPL < 1%. There is a systematic shortening of the τPL with increasing doping concentrations that mirrors the decrease in ΦPL (see ESI‡). Irrespective of the complexity of the emission spectra the successful formation of the low optical gap material 14 demonstrates the utility of these dibrominated B2-PAHs in accessing complex organic materials.
Conclusions
In conclusion, sequential bromoboration/intramolecular electrophilic C–H borylation enables formation of a range of brominated Bn-doped PAHs in useful yields via an operationally simple, one-pot route from readily available precursors (alkynes and BBr3). Cyclic voltammetry and photophysical studies revealed that these molecules have very low LUMO energies thus are attractive acceptor units for use in organic electronic applications. In particular, compound 12a has the least negative reduction potential among all reported ambient stable B-doped PAHs to the best of our knowledge. The C–Br units in the Bn-PAHs can be utilised directly in Negishi cross-coupling reactions, which enabled formation of a donor–acceptor–donor molecule displaying solution absorption up to 750 nm. Finally, mechanistic studies on the bromoboration reaction indicated it proceeds through the 1,1-bromoboration of the diarylalkynes, a reaction not previously observed using just boron electrophiles. 1,1-Bromoboration can be applied to functionalise other diarylalkynes, enabling access to unprecedented 1-bromo-2,2-diaryl substituted vinylboronate esters direct from internal alkynes. The utility of the 1,1-bromoboration reaction is being studied further in our laboratory as are the brominated-B2-PAHs, particularly to access useful functional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the European Research Council under the Horizon 2020 Research and Innovation Program (Grant no. 769599), the Leverhulme Trust (RPG-2014-340) and the EPSRC (EP/P010482/1). C. Si thanks the China Scholarship Council (201806890001). Dr G. Nichol is thanked for the collection of X-ray diffraction data, Dr G. Whitehead for the structure of 2a and Dr A. Woodward for collecting photophysical data on compound 2c (see ESI‡).Notes and references
- Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jäkle, John Wiley & Sons, Ltd., Chichester, 1st edn, 2018 Search PubMed.
- For recent reviews at least partly focused on PAHs containing C3B units see: (a) Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat’ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201911108; (b) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291 CrossRef CAS PubMed; (c) X.-Y. Wang, X. Yao and K. Müllen, Sci. China: Chem., 2019, 62, 1099 CrossRef CAS; (d) S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77 CrossRef; (e) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37 CrossRef CAS; (f) M. Stepien, E. Gonka, M. Zyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef CAS PubMed; (g) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846 RSC; (h) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257 RSC; (i) A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357 CrossRef CASFor their use in electrocatalysis see: (j) R. J. Kahan, W. Hirunpinyopas, J. Cid, M. J. Ingleson and R. A. W Dryfe, Chem. Mater., 2019, 31, 1891 CrossRef CAS.
- For rare example of halogenated-Bn-PAH formation, in this case starting from halo-aromatic precursors and their utility in cross coupling, see: (a) C. Reus, S. Weidlich, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2013, 135, 12892 CrossRef CAS PubMed; (b) A. Shuto, T. Kushida, T. Fukushima, H. Kaji and S. Yamaguchi, Org. Lett., 2013, 15, 6234 CrossRef CAS PubMed.
- For seminal work in the area of forming solely Bn-doped PAHs with three coordinate B centres see: (a) C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206 CrossRef CAS PubMed; (b) Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529 CrossRef CAS PubMedFor select recent examples on this topic see: (c) J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096 CrossRef CAS PubMed; (d) J. Radtke, K. Schickedanz, M. Bamberg, L. Menduti, D. Schollmeyer, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Sci., 2019, 10, 9017 RSC; (e) A. John, S. Kirschner, M. K. Fengel, M. Bolte, H.-W. Lerner and M. Wagner, Dalton Trans., 2019, 48, 1871 RSC; (f) R. J. Kahan, D. L. Crossley, J. Cid, J. E. Radcliffe, A. W. Woodward, V. Fasano, S. Endres, G. F. S. Whitehead and M. J. Ingleson, Chem. Commun., 2018, 54, 9490 RSC; (g) S. Kirschner, J.-M. Mewes, M. Bolte, H.-W. Lerner, A. Dreuw and M. Wagner, Chem.–Eur. J., 2017, 23, 5104 CrossRef CAS PubMed; (h) A. John, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2017, 56, 5588 CrossRef CAS PubMed; (i) D. L. Crossley, R. J. Kahan, S. Endres, A. J. Warner, R. A. Smith, J. Cid, J. J. Dunsford, J. E. Jones, I. Vitorica-Yrezabal and M. J. Ingleson, Chem. Sci., 2017, 8, 7969 RSC; (j) J. M. Farrell, D. Schmidt, V. Grande and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 11846 CrossRef CAS PubMed; (k) V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800 CrossRef CAS PubMed.
- There are also boron containing polymers that are Bn-PAHs with n > 2, however, while chemically well-defined the value of n is not as they are polydisperse. For select examples see: (a) S. Kawai, S. Saito, S. Osumi, S. Yamaguchi, A. S. Foster, P. Spijker and E. Meyer, Nat. Commun., 2015, 6, 8098 CrossRef CAS PubMed; (b) R. R. Cloke, T. Marangoni, G. D. Nguyen, T. Joshi, D. J. Rizzo, C. Bronner, T. Cao, S. G. Louie, M. F. Crommie and F. R. Fischer, J. Am. Chem. Soc., 2015, 137, 8872 CrossRef CAS PubMed.
- For examples of post synthetic modification of Bn-PAHs and subsequent functionalisation by cross coupling see: (a) T. Kushida, C. Camacho, A. Shuto, S. Irle, M. Muramatsu, T. Katayama, S. Ito, Y. Nagasawa, H. Miyasaka, E. Sakuda, N. Kitamura, Z. Zhou, A. Wakamiya and S. Yamaguchi, Chem. Sci., 2014, 5, 1296 RSC; (b) C. Hoffend, K. Schickedanz, M. Bolte, H.-W. Lerner and M. Wagner, Tetrahedron, 2013, 69, 7073 CrossRef CAS.
- R. J. Kahan, D. L. Crossley, J. Cid, J. E. Radcliffe and M. J. Ingleson, Angew. Chem., Int. Ed., 2018, 57, 8084 CrossRef CAS PubMed.
- (a) M. F. Lappert and B. Prokai, J. Organomet. Chem., 1964, 1, 384 CrossRef CAS; (b) Related findings were subsequently published: J. J. Eisch and L. J. Gonsior, J. Organomet. Chem., 1967, 8, 53 CrossRef CAS.
- For recent studies combining alkynes and R2B-X see: (a) K. Skoch, C. Pauly, C. G. Daniliuc, K. Bergander, G. Kehr and G. Erker, Dalton Trans., 2019, 48, 4837 RSC; (b) J. R. Lawson, V. Fasano, J. Cid, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2016, 45, 6060 RSCFor an early example see: (c) R.-J. Binnewirtz, H. Klingenberger, R. Welte and P. Paetzold, Chem. Ber., 1983, 116, 1271 CrossRef CAS.
- In contrast tri-substituted 1,1-bromo-BPin-alkenes derivatives are known, for a recent example see: Y. Pang, R. Kojima and H. Ito, Org. Biomol. Chem., 2018, 16, 6187 RSC . For an early example see: H. C. Brown and T. Imai, Organometallics, 1984, 3, 1392 CrossRef CAS.
- (a) L. Ji, I. Krummenacher, A. Friedrich, A. Lorbach, M. Haehnel, K. Edkins, H. Braunschweig and T. B. Marder, J. Org. Chem., 2018, 83, 3599 CrossRef CAS PubMed; (b) S.-B. Zhao, P. Wucher, Z. M. Hudson, T. M. McCormick, X.-Y. Liu, S. Wang, X.-D. Feng and Z.-H. Lu, Organometallics, 2008, 27, 6446 CrossRef CAS.
- Adapted from: C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- A. Nowak-Krol, K. Shoyama, M. Stolete and F. Würthner, Chem. Commun., 2018, 54, 13763 RSC.
- M. Vanga, R. A. Lalancette and F. Jäkle, Chem.–Eur. J., 2019, 25, 10133 CrossRef CAS PubMed.
- Y. Gu, X. Wu, T. Y. Gopalakrishna, H. Phan and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 6541 CrossRef CAS PubMed.
- W. Yang, R. R. Kazemi, N. Karunathilake, V. J. Catalano, M. A. Alpuche-Aviles and W. A. Chalifoux, Org. Chem. Front., 2018, 5, 2288 RSC.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, T. Wiesner, F. Laquai and M. O. Senge, ChemPhotoChem, 2018, 2, 606 CrossRef CAS.
- A. P. Davis and A. J. Fry, J. Phys. Chem. A, 2010, 114, 12299 CrossRef CAS PubMed.
- (a) J. M. Farrell, V. Grande, D. Schmidt and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 16504 CrossRef CAS PubMed; (b) E. Dimitrijevic, M. Cusimano and M. S. Taylor, Org. Biomol. Chem., 2014, 12, 1391 RSC.
Footnotes |
| † The research data supporting this publication can be accessed at https://doi.org/10.17630/3764e0e8-38dc-42b9-a77d-152eabdfd4db. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details, NMR spectra, crystallographic data, optoelectronic data and Cartesian coordinates for all calculations. CCDC 1961365–1961375. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05404a |
| This journal is © The Royal Society of Chemistry 2020 |