
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEpimerization-free access to C-terminal cysteine peptide acids, carboxamides, secondary amides, and esters via complimentary strategies†
Christine A.
Arbour
 ,
Thilini D.
Kondasinghe
,
Hasina Y.
Saraha
,
Teanna L.
Vorlicek
and
Jennifer L.
Stockdill
*
,
Thilini D.
Kondasinghe
,
Hasina Y.
Saraha
,
Teanna L.
Vorlicek
and
Jennifer L.
Stockdill
*
Wayne State University, Department of Chemistry, Detroit, MI, USA 48202. E-mail: stockdill@wayne.edu
First published on 9th November 2017
Abstract
C-Terminal cysteine peptide acids are difficult to access without epimerization of the cysteine α-stereocenter. Diversification of the C-terminus after solid-phase peptide synthesis poses an even greater challenge because of the proclivity of the cysteine α-stereocenter to undergo deprotonation upon activation of the C-terminal carboxylic acid. We present herein two general strategies to access C-terminal cysteine peptide derivatives without detectable epimerization, diketopiperazine formation, or piperidinylalanine side products.
C-Terminal cysteine peptides, including prenylated and farnesylated peptides,1 disulfide linked peptide toxins,2 and insulinotropic peptides,3,4 comprise an important but synthetically challenging class of biologically active peptides. Many of these peptides are modified at the C-terminus. C-terminal modifications such as esters and amides can be critical to maintaining a peptide's active conformation,5in vivo activity, and pharmacokinetics;6 therefore, the ability to vary the peptide structure in this location is crucial to drug development efforts.7 Although several methods have been reported for C-terminal functionalization after solid-phase peptide synthesis (SPPS) is complete,8 these approaches either result in epimerization when applied to C-terminal Cys peptides9 or the applicability of the method to C-terminal Cys peptides is not addressed.10,11 While activation of the C-terminal carboxylic acid can induce epimerization via oxazolone formation in most amino acids,12 cysteine is also prone to epimerization via direct deprotonation during its attachment to the resin13 and upon prolonged or repeated exposure to base (i.e., during peptide elongation via Fmoc SPPS).14 Therefore, even the preparation of simple carboxylic acids or carboxamides of C-terminal cysteine peptides can be fraught with contamination by epimerized products,1f,g,13a,15 reducing the overall yield and complicating the purification of the target peptides. A method for the epimerization-free synthesis and subsequent C-terminal modification of C-terminal Cys peptides would be highly impactful.
In this work, we report the first mild and convenient method for the epimerization-free diversification of peptides bearing a C-terminal cysteine.16 Carboxylic acids, primary and secondary amides, and esters are accessed without epimerization or formation of diketopiperazine and piperidinyl-alanine side products.17 We apply this strategy to the total synthesis of the nicotinic acetylcholine receptor (nAChR) antagonist α-conotoxin ImI.18 Additionally, we include an alternate strategy employing N-deprotected cysteine derivatives as nucleophiles, and we demonstrate its utility via the synthesis of the insect pheromone α-factor.1
In the context of our ongoing efforts toward the synthesis of disulfide-linked α- and μ-conotoxins,19,20 we were concerned about possible epimerization of the C-terminal cysteine during the SPPS. We recently reported a strategy for C-terminal functionalization of non-cysteine peptides involving activation of the methyl-diaminobenzoyl (MeDbz) linker (1 → 2)21 followed by nucleophilic cleavage of the N-acyl urea (MeNbz) group22 to yield various protected (3) or unprotected (4) peptides (Scheme 1).23 If this approach were to prove mild enough to enable preparation of challenging C-terminal cysteine peptide derivatives, it would establish the MeNbz group as one of the mildest known activated carbonyl intermediates.24 We were report herein the exploitation of this reactivity to access C-terminal cysteine peptide acids, primary and secondary amides, and esters without epimerization.
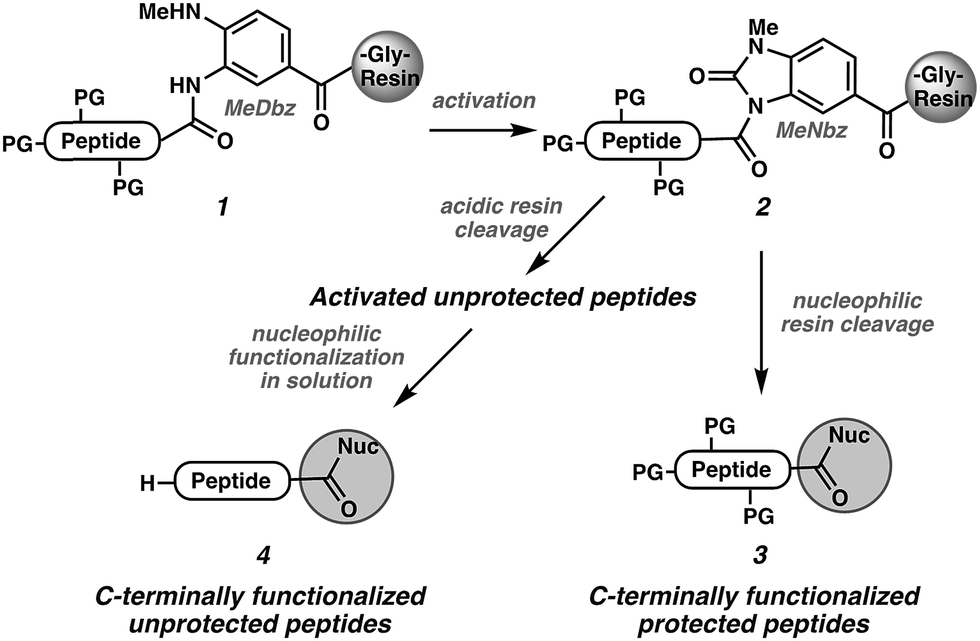 | ||
| Scheme 1 Our strategy for C-terminal functionalization of non-Cys terminated peptides. | ||
For epimerization-free functionalization at Cys, the attachment of the first amino acid,21b peptide elongation, linker activation, and nucleophilic attack all must occur without epimerization of the unusually acidic25 Cys α-stereocenter.
We expected that the parent diaminobenzoyl group would not be sufficiently activating to cause epimerization during prolonged piperidine exposure. Thus, we sought to establish the stereochemical integrity of the Cys residue under these conditions unequivocally. We selected Cys(Trt) for these experiments because of its extreme tendency toward epimerization.14 Thus, we synthesized tripeptide Boc-Ala-Trp(Boc)-Cys(Trt)-MeDbzGly-Wang and exposed it to 20% piperidine/DMF over 2, 4, and 24 h. The peptides were then cleaved under acidic conditions to afford H-AWC-MeDbz-Gly-OH. As expected, no epimerization was detected immediately following SPPS or after piperidine exposure at any time point (Fig. SI-2 and SI-3†).26 Importantly, this is the first report of a linker for which no epimerization is detected at a C-terminal Cys(Trt) residue after treatment with 20% piperidine for 24 h.
With this result in hand, we were poised to evaluate the ability of the activated MeNbz linker to undergo nucleophilic displacement without inducing epimerization of the C-terminal cysteine. We first evaluated epimerization-prone Cys(Trt)-terminated peptides with N and O nucleophiles (5). We began with displacement by ammonia because of its small size and the relatively low pKa of NH4+. We were pleased to observe formation of the target peptide (6, Nuc = NH2) with complete conversion and no detectable epimerization in 54% isolated yield (Table 1, entry 1). We next evaluated benzylamine, which has a similar pKa, but found that treatment of the activated linker with neat BnNH2 led to 16% epimerization (entry 2). However, using only 5 equiv. benzylamine in MeCN, the product was formed with no detectable epimerization (entry 3). We next tested neat butylamine, which is slightly more basic, and 8% epimerization was observed. Reducing the amount of amine and varying the solvent did not improve epimerization in this case (entry 4–6). However, when we reduced the amount of butylamine to 1.1 equiv., we retained reactivity while eliminating epimerization of C-terminal Cys(Trt) (entry 7).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | PG | Nuc-H | Base (5 equiv.) | Solvent | % conversion | Epimerizationb (% D-X) |
| a All reactions were performed on 20 mg of resin containing all L amino acids in 200 μL of solvent at ambient temperature (24 ± 1 °C). b All PGs were removed prior to epimerization assay unless otherwise noted. c Cys(PG) was intact during epimerization assay. d 1.1 equiv. of BuNH2 was used. e 0.7 equiv. KOtBu. f Reaction was conducted for 3 h. g Na2HPO4/NaH2PO4 buffer at pH = 8. | ||||||
| 1c | Trt | NH3 | — | DMF | >99 | <1 |
| 2 | Trt | PhCH2NH2 | — | PhCH2NH2 | >99 | 16 |
| 3 | Trt | PhCH2NH2 | — | MeCN | >99 | <1 |
| 4 | Trt | BuNH2 | — | BuNH2 | >99 | 8 |
| 5 | Trt | BuNH2 | — | DMF | >99 | 10 |
| 6 | Trt | BuNH2 | — | MeCN | >99 | 9 |
| 7d | Trt | BuNH2 | — | MeCN | >99 | <1 |
| 8c | Acm | BuNH2 | — | MeCN | >99 | <1 |
| 9c | Mob | BuNH2 | — | MeCN | >99 | <1 |
| 10c | Bn | BuNH2 | — | MeCN | >99 | <1 |
| 11 | StBu | BuNH2 | — | MeCN | >99 | <1 |
| 12c | tBu | BuNH2 | — | MeCN | >99 | <1 |
| 13e | Trt | MeOH | KOtBu | MeOH | >99 | 42 |
| 14f | Trt | MeOH | DIEA | MeOH | >99 | <1 |
| 15g | Trt | MeOH | — | MeOH/Na2HPO4(aq) | >99 | <1 |
| 16 | Trt | H2O | DIEA | H2O/MeCN | 56 | <1 |

Other commercially available Cys PGs should be less prone to epimerization than Trt. Therefore, we used 5 equiv. BuNH2 (i.e., entry 6 conditions) for the remaining protecting groups. First, we evaluated the Acm group in MeCN (entry 8), finding <1% epimerization in the formation of the C-terminal Cys(Acm) butylamide. Next, we tested Mob, Bn, StBu, and tBu with BuNH2 in MeCN. In all cases, no epimerization was detected (entries 9–12). Turning our attention to alcohol nucleophiles, we tested MeOH/KOtBu with Cys(Trt) as a benchmark9 and found 42% epimerization (entry 13). However, in the presence of 5 equiv. Hünig's base (DIEA) in MeOH, no epimerization was observed (entry 14). Because of the lower conversion in this case, we also investigated the use of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH/phosphate buffer solvent mixture (pH 8). In this case, complete conversion was observed while maintaining no detectable epimerization (entry 15). Even carboxylic acid derivatives with a C-terminal Cys(Trt) can be difficult to access without epimerization.27 Therefore, water was investigated as a nucleophile in the presence of Hünig's base. In this case, the reaction was slower, but the product acid was observed with <1% epimerization (entry 16).28 Thus, all nucleophiles tested react with the activated C terminus without any observed epimerization, regardless of the protecting group on cysteine.
:1 MeOH/phosphate buffer solvent mixture (pH 8). In this case, complete conversion was observed while maintaining no detectable epimerization (entry 15). Even carboxylic acid derivatives with a C-terminal Cys(Trt) can be difficult to access without epimerization.27 Therefore, water was investigated as a nucleophile in the presence of Hünig's base. In this case, the reaction was slower, but the product acid was observed with <1% epimerization (entry 16).28 Thus, all nucleophiles tested react with the activated C terminus without any observed epimerization, regardless of the protecting group on cysteine.
Next, we sought to demonstrate the viability of this cleavage strategy in the context of more complex peptides. Because of our interest in disulfide-linked neuroactive peptides,29 we targeted the C-terminal carboxamide α-conotoxin ImI (10), a sub-type selective nicotinic acetylcholine receptor antagonist30 isolated from the venom of Conus imperialis marine snails (Scheme 2).31,32 Both the C-terminal carboxamide and the correct disulfide bond connectivity are important to the bioactivity of α-ImI (10).31,33 Thus, peptide 7 was activated and cleaved with ammonia to yield the fully protected peptide carboxamide. Acidic removal of protecting groups and HPLC purification gave the reduced peptide 8 in 25% isolated yield.34 The first disulfide was formed in the presence of air in 1% DMSO in phosphate buffer at pH 8 (peptide 9). Subsequent iodine treatment removed both Acm groups and induced oxidation to form the native conotoxin (10) in 52% isolated yield over both folding steps. Co-injection of with a commercially available standard confirmed the correct folding.26 Alternatively, on-resin folding with iodine could be followed by MeDbz activation, cleavage with NH3, and side-chain deprotection to afford conotoxin 10 in 43% isolated yield; however, this approach gives a mixture of folded products, as expected.35
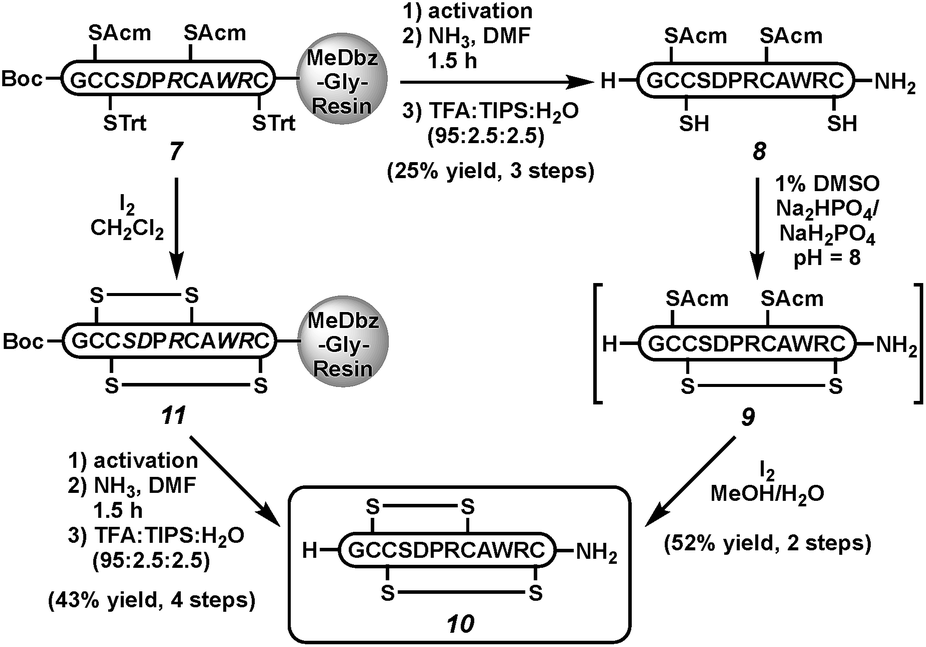 | ||
| Scheme 2 Synthesis of conotoxin α-ImI (10). | ||
We envisioned that situations might exist wherein it would prove advantageous to access the target peptides by using various pre-functionalized cysteine derivatives as nucleophiles for cleavage of MeNbz from the resin.27,36 For example, this would avoid the need to screen epimerization for each new derivative, it would enable access to authentic standards for peptides generated using direct C-terminal modification approaches, and it would allow incorporation of a very poor or hindered nucleophile. Recently, C-terminal cysteine peptide acids were synthesized using an N-(2-hydroxy-5-nitrobenzyl)-cysteine (N-Hnb-Cys) crypto-thioester approach.27 Although this method enabled access to a challenging Pro-Cys linkage at the C terminus, elevated temperatures and long reaction times were required to generate the C-terminal thioester, leading to undesired side products. The stability of esters to this approach was not determined. We envisioned that application of the cysteine elongation tactic to our MeNbz-based C-terminal modification would lead to a convenient alternative method to access challenging targets at ambient temperature and with short reaction times. However, there was no report of an intermolecular37 NCL-like reaction being conducted with the C-terminally linked peptide still attached to the resin.
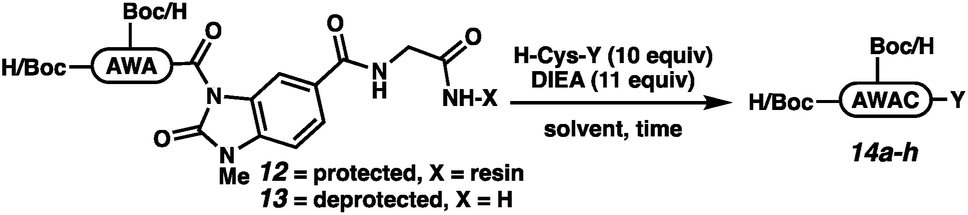
The elongation approach was tested with H-AWA-MeNbz-Gly-Rink peptides (12 and 13), which were treated with free cysteine, H-Cys-OEt, H-Cys-NH2, or H-Cys-NHBu in the presence of Hünig's base (Table 2). Protected peptides Boc-AW(Boc)AC-OH (14a), Boc-AW(Boc)AC-OEt (14b), and Boc-AW(Boc)AC-NH2 (14c) were formed with complete conversion (entries 1–3), while Boc-AW(Boc)AC-NHBu (14d) was formed with 38% conversion. The elongation was more efficient in solution,21 and unprotected peptides 14e–h were accessed with quantitative conversion (entries 5–8). We assumed that the mildly basic reaction conditions would result in rapid S to N acyl transfer upon cysteine thiol addition either on resin or in solution. In situ generation of the backbone amide was confirmed by independent synthesis of H-AWAC-OH followed by co-injection with 14e.26 The extent of product peptide epimerization was evaluated for the ethyl ester (14f), which is the most epimerization-prone derivative. Comparison to a co-injection of H-AWA(D-Cys)-OEt confirmed that the product peptides are not epimerized to any observable extent under the reaction conditions (Fig. SI-057†).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Substrate | Y | Solvent | Time (h) | Conversionb (%) |
| a Unless noted, on resin reactions were performed on 20 mg resin in 500 μL solvent, solution-phase reactions were performed on 20 mg crude peptide in 200 μL solvent, 100 μL of H2O was added as indicated, rt = 24 ± 1 °C. b Conversion based on integration of relevant peaks in HPLC/MS data. c Cystine formation was observed. d Performed on 3.8 mg of 13 using 520 μL MeCN:H2O. | |||||
| 1 | 12 | OH (14a) | (5:1) DMF:H2O |
4 | >99 |
| 2 | 12 | OEt (14b) | DMF | 4 | >99 |
| 3 | 12 | NH2 (14c) | DMF | 4 | >99 |
| 4c | 12 | NHBu (14d) | DMF | 4 | >38 |
| 5 | 13 | OH (14e) | (2:1) MeCN:H2O |
0.5 | >99 |
| 6 | 13 | OEt (14f) | MeCN | 0.5 | >99 |
| 7 | 13 | NH2 (14g) | MeCN | 0.5 | >99 |
| 8c,d | 13 | NHBu (14h) | (25:1) MeCN:H2O |
0.5 | >99 |
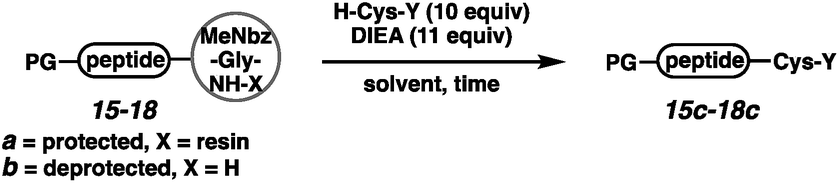
We next executed the cysteine elongation of a series of peptides varying in length and hydrophobicity both on the resin and in solution (Table 3). The unprotected peptide H-AKTWA-MeNbz-Gly (15b) was functionalized in solution to afford H-AKTWAC-OH (15c) with complete conversion in 30 min with no observed side-chain macrocyclization.23 To enable comparison with the crypto-thioester approach,27 C-terminal proline-containing peptide 16a was cleaved from resin using H-Cys-OH to afford protected H-AKTWPC-OH (16c) with 10% conversion over 4 h.38 Repeating this reaction in solution on unprotected peptide (16b) led to complete conversion after 1 h at ambient temperature. Elongation of Boc-LYRAGLRAY (17a) proceeded with resin cleavage and complete conversion in the presence of DMF and NCL buffer. Hydrophobic peptide 18, a fragment of amyloid β (Aβ(36–42)),39 was elongated both on resin (entry 5) and in solution (entry 6). On-resin elongation proved challenging for this substrate (10% conversion), while complete conversion was observed in solution. Overall, for shorter or non-hydrophobic peptides, this chemistry could be executed on resin and in the absence of added thiol. In challenging cases, resin cleavage and then in solution native chemical ligation40 afforded the target peptides.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | PG | Substrate | Peptide | Y | Solvent | Time (h) | Conversionb (%) |
| a Unless noted, on-resin reaction were performed on 20 mg peptide/resin in 500 μL solvent, rt = 24 ± 1 °C, NCL buffer at pH 7.2. b Based on integration of relevant peaks in HPLC/MS data. c Used 600 μL solvent. d Performed on 10 mg of 16b using 250 μL solvent. e Performed on 67 mg of 18b using 250 μL solvent. | |||||||
| 1c | H | 15b | AKTWA (15) | OH | (5:1) MeCN:H2O |
0.5 | >99 |
| 2 | Boc | 16a | AKTWAP (16) | OH | (1:1) DMF:NCL buffer |
4 | 10 |
| 3d | H | 16b | AKTWAP (16) | OH | NCL buffer | 1 | >99 |
| 4 | Boc | 17a | LYRAGLRAY (17) | Nh2 | (1:1) DMF:NCL buffer |
4 | >99 |
| 5 | Boc | 18a | VGGVVI (18) | OMe | (1:1) DMF:NCL buffer |
4 | 10 |
| 6e | H | 18b | VGGVVI (18) | OMe | NCL buffer | 0.5 | >99 |
To confirm the viability of this approach in the context of a complex natural product, we executed the total synthesis of the insect pheromone α-factor (21, Scheme 3), which requires both the C-terminal ester and the prenyl moiety for bioactivity.1e–h,41 Protected des-farnesyl α-factor was generated by displacement of peptide 19 with cysteine methyl ester. The elongation was conducted in NCL buffer in an effort to reduce cystine-functionalized by-products. However, even under these reducing conditions, cystine-appended α-factor was still observed. Side-chain deprotection, cystine reduction, and HPLC purification afforded peptide 20 in 41% yield over 3 steps. Alkylation per the reported conditions afforded α-factor in 9% yield (21).41
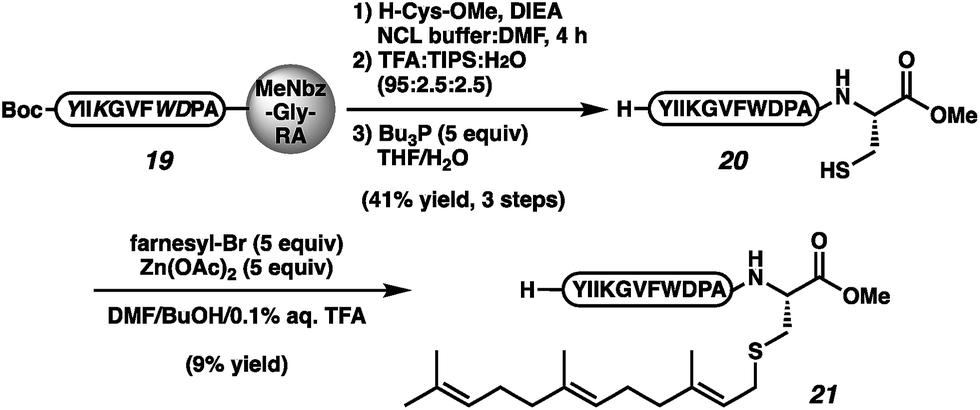 | ||
| Scheme 3 Synthesis of α-factor by cysteine elongation. | ||
In summary, we have developed two broadly applicable strategies for the epimerization-free preparation of C-terminal cysteine peptides. The first approach exploits the exceptionally mild activating nature of the N-acyl urea group for the direct diversification of the C terminus. Additionally, an alternative strategy wherein cysteine derivatives serve as the nucleophile in a resin-cleaving elongation reaction was also effective. For both strategies, the target peptides are prepared without observation of either diketopiperazine or piperidinylalanine side products. The utility of these methods was demonstrated in the preparation of the disulfide-linked conotoxin α-ImI, bearing a C-terminal cysteine carboxamide and the insect pheromone α-factor, which possesses a C-terminal cysteine methyl ester. Notably, no previous report has demonstrated successful functionalization of C-terminal cysteine peptides to access carboxylic acids, carboxamides, and other C-terminal derivatives without detectable epimerization of the α-stereocenter.
Conflicts of Interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank the National Institutes of Health (R00-GM097095), the National Science Foundation (CAREER Award supporting undergrad researcher TLV: CHE-1554752), and Wayne State University for generous financial support (startup funds to JLS, Rumble-Schaap Fellowships to CAA and TDK, Knoller Fellowship to HYS). We thank Alomone Labs for providing an analytical sample of α-conotoxin ImI free of charge. We also gratefully acknowledge the staff of the WSU Lumigen Instrument Center and Shimadzu for a grant supporting the mass spectrometer.References
- (a) S. Clarke, Annu. Rev. Biochem., 1992, 61, 355–386 CrossRef CAS PubMed; (b) J. A. Glomset, M. H. Gelb and C. C. Farnsworth, Trends Biochem. Sci., 1990, 15, 139–142 CrossRef CAS PubMed; (c) C. A. Hrycyna and S. Clarke, Mol. Cell. Biol., 1990, 10, 5071–5076 CrossRef CAS PubMed; (d) F. L. Zhang and P. J. Casey, Annu. Rev. Biochem., 1992, 65, 241–269 CrossRef PubMed; (e) D. G. Mullen, K. Kyro, M. Hauser, M. Gustavsson, G. Veglia, J. M. Becker, F. Naider and M. D. Distefano, Bioorg. Med. Chem., 2011, 19, 490–497 CrossRef CAS PubMed; (f) V. Diaz-Rodriguez, D. G. Mullen, E. Ganusova, J. M. Becker and M. D. Distefano, Org. Lett., 2012, 14, 5648–5651 CrossRef CAS PubMed; (g) V. Diaz-Rodriguez, E. Ganusova, T. M. Rappe, J. M. Becker and M. D. Distefano, J. Org. Chem., 2015, 80, 11266–11274 CrossRef CAS PubMed; (h) F. R. Naider and J. M. Becker, Pept. Sci.: Present Future, Proc. Int. Pept. Symp., 1st, 1997, 3–14 CAS.
- (a) D. J. Craik and D. J. Adams, ACS Chem. Biol., 2007, 2, 457–468 CrossRef CAS PubMed; (b) M. C. Inserra, S. N. Kompella, S. N. Vetter, A. Brust, N. L. Daly, H. Cuny, D. J. Craik, P. F. Alewood, D. J. Adams and R. J. Lewis, Biochem. Pharmacol., 2013, 86, 791–799 CrossRef CAS PubMed; (c) A. Gould and J. A. Camarero, ChemBioChem, 2017, 18, 1350–1363 CrossRef CAS PubMed.
- W. Shang, X. Yang, X. Ju, Y. Xie, Y. Zhang and W.-H. Lee, J. Pept. Sci., 2017, 23, 707–715 CrossRef CAS PubMed.
- J. M. Conlon, V. Musale, S. Attoub, M. L. Mangoni, J. Leprince, L. Coquet, T. Jouenne, Y. H. A. Abdel-Wahab, P. R. Flatt and A. C. Rinaldi, J. Pept. Sci., 2017, 23, 769–776 CrossRef CAS PubMed.
- T. S. Kang, S. Vivekanandan, S. D. S. Jois and R. M. Kini, Angew. Chem., Int. Ed., 2005, 44, 6333–6337 CrossRef CAS PubMed.
- (a) D. J. Merkler, Enzyme Microb. Technol., 1994, 16, 450–456 CrossRef CAS PubMed; (b) H. Bultmann, J. Teuton and C. R. Brandt, Antimicrob. Agents Chemother., 2007, 51, 1596–1607 CrossRef CAS PubMed.
- D. Goodwin, P. Simerska and I. Toth, Curr. Med. Chem., 2012, 19, 4451–4461 CrossRef CAS PubMed.
- J. Alsina and F. Albericio, Biopolymers, 2003, 71, 454–477 CrossRef CAS PubMed.
- J. Hansen, F. Diness and M. Meldal, Org. Biomol. Chem., 2016, 14, 3238–3245 CAS.
- (a) H. E. Elashal, Y. E. Sim and M. Raj, Chem. Sci., 2017, 8, 117–123 RSC; (b) H. E. Elashel, R. D. Cohen and M. Raj, Chem. Commun., 2016, 52, 9699–9702 RSC.
- (a) J. A. Camarero, B. J. Hackel, J. J. de Yoreo and A. R. Mitchell, J. Org. Chem., 2004, 69, 4145–4151 CrossRef CAS PubMed; (b) Y. Kwon, K. Welsh, A. R. Mitchell and J. A. Camarero, Org. Lett., 2004, 6, 3801–3804 CrossRef CAS PubMed; (c) R. A. Turner, R. J. Weber and R. S. Lokey, Org. Lett., 2010, 12, 1852–1855 CrossRef CAS PubMed; (d) E. Nicolás, J. Clemente, M. Perelló, F. Albericio, E. Pederoso and E. Giralt, Tetrahedron Lett., 1992, 33, 2183–2186 CrossRef; (e) A. A. Vinogradov, M. D. Simon and B. L. Pentelute, Org. Lett., 2016, 18, 1222–1225 CrossRef CAS PubMed.
- Oxazolone formation: B. Henkel, L. Zhang and E. Bayer, Liebigs Ann./Recl., 1997, 10, 2161–2186 CrossRef.
- (a) Y. Fujiwara, K. Akaji and Y. Kiso, Chem. Pharm. Bull., 1994, 42, 724–726 CrossRef CAS PubMed; (b) Y. M. Angell, J. Alsina, F. Albericio and G. Barany, J. Pept. Res., 2002, 60, 292–299 CrossRef CAS PubMed; (c) H. Hibino, Y. Miki and Y. Nishiuchi, J. Pept. Sci., 2014, 20, 30–35 CrossRef CAS PubMed.
- (a) Y. Han, F. Albericio and G. Barany, J. Org. Chem., 1997, 62, 4307–4312 CrossRef CAS PubMed; (b) I. Ramos-Tomillero, H. Rodríguez and F. Albericio, Org. Lett., 2015, 17, 1680–1683 CrossRef CAS PubMed; (c) A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed; (d) H. Hibino and Y. Nishiuchi, Org. Lett., 2012, 14, 1926–1929 CrossRef CAS PubMed; (e) H. Hibino, Y. Miki and Y. Nishiuchi, J. Pept. Sci., 2014, 20, 30–35 CrossRef CAS PubMed; (f) J. Lukszo, D. Patterson, F. Albericio and S. Kates, Lett. Pept. Sci., 1996, 3, 157–166 CrossRef CAS.
- (a) U. Boas, J. Brask and K. J. Jensen, Chem. Rev., 2009, 109, 2092–2118 CrossRef CAS PubMed and references therein; (b) G. Barany, Y. Han, B. Hargittai, R.-Q. Liu and J. T. Varkey, Biopolymers, 2003, 71, 652–666 CrossRef CAS PubMed; (c) V. Juvekar and Y. D. Gong, Org. Lett., 2016, 18, 836–839 CrossRef CAS PubMed; (d) Z. Huang, D. J. Derksen and J. C. Vederas, Org. Lett., 2010, 12, 2282–2285 CrossRef CAS PubMed.
- (a) A. Selvaraj, H.-T. Chen, A. Y. Huang and C.-L. Kao, Chem. Sci. 10.1039/c7sc03229c; (b) B. H. Gless, P. Peng, K. D. Pedersen, C. H. Gotfredsen, H. Ingmer and C. A. Olsen, Org. Lett., 2017, 19, 5276–5279 CrossRef CAS PubMed.
- (a) P. Cherkupally, G. A. Acosta, S. Ramesh, B. G. de la Torre, T. Govender, H. G. Kruger and F. Albericio, Amino Acids, 2014, 46, 1827–1838 CrossRef CAS PubMed; (b) J. Lukszo, D. Patterson, F. Albericio and S. A. Kates, Lett. Pept. Sci., 1996, 3, 157–166 CrossRef CAS; (c) V. Juvekar, K.-T. Kim and Y.-D. Gong, Bull. Korean Chem. Soc., 2017, 38, 54–62 CrossRef CAS.
- (a) D. S. Johnson, J. Martinez, A. B. Elgoyhen, S. F. Heinemann and J. M. McIntosh, Mol. Pharmacol., 1995, 48, 194–199 CAS; (b) E. F. R. Pereira, M. Alkondon, J. M. McIntosh and E. X. Albuquerque, J. Pharmacol. Exp. Ther., 1996, 278(3), 1472–1483 CAS; (c) S. Luo, T. A. Nguyen, G. E. Cartier, B. M. Olivera, D. Yoshikami and J. M. McIntosh, Biochemistry, 1999, 38, 14542–14548 CrossRef CAS PubMed; (d) M. Ellison, J. M. McIntosh and B. M. Olivera, J. Biol. Chem., 2003, 278, 757–764 CrossRef CAS PubMed; (e) M. Ellison, F. Gao, H.-L. Wang, S. M. Sine, J. M. McIntosh and B. M. Olivera, Biochemistry, 2004, 43, 16019–16026 CrossRef CAS PubMed.
- (a) B. M. Olivera, Mol. Biol. Cell, 1997, 8, 2101–2109 CrossRef CAS PubMed; (b) H. Terlau and B. M. Olivera, Physiol. Rev., 2004, 84, 4–68 CrossRef PubMed; (c) B. M. Olivera, J. S. Imperial and G. Bulaj, in Perspectives in Molecular Toxinology, 2002, pp. 143–158 Search PubMed; (d) O. Buczek, G. Bulaj and B. M. Olivera, Cell. Mol. Life Sci., 2005, 62, 3067–3079 CrossRef CAS PubMed; (e) M. C. Inserra, S. N. Kompella, I. Vetter, A. Brust, N. L. Daly, H. Cuny, D. J. Craik, P. F. Alewood, D. J. Adams and R. J. Lewis, Biochem. Pharmacol., 2013, 86, 791–799 CrossRef CAS PubMed.
- (a) K. B. Akondi, M. Muttenthaler, S. Dutertre, Q. Kaas, D. J. Craik, R. J. Lewis and P. F. Alewood, Chem. Rev., 2014, 114, 5815–5847 CrossRef CAS PubMed; (b) M. Gońgora-Benítez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2013, 114, 901–926 CrossRef PubMed.
- (a) J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed; (b) J. B. Blanco-Canosa, B. Nardone, F. Albericio and P. E. Dawson, J. Am. Chem. Soc., 2015, 137, 7197–7209 CrossRef CAS PubMed; (c) S. K. Mahto, C. J. Howard, J. C. Shimko and J. J. Ottesen, ChemBioChem, 2011, 12, 2488–2494 CrossRef CAS PubMed.
- R. Pascal, D. Chauvey and R. Sola, Tetrahedron Lett., 1994, 35, 6291–6294 CrossRef CAS.
- C. A. Arbour, H. Y. Saraha, T. F. McMillan and J. L. Stockdill, Chem.–Eur. J., 2017, 23, 12484–12488 CrossRef CAS PubMed.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- Y. M. Angell, J. Alsina, F. Albericio and G. Barany, J. Pept. Res., 2002, 60, 292–299 CrossRef CAS PubMed.
- See the ESI for details.†.
- D. Lelièvre, V. P. Terrier, A. F. Delmas and V. Aucagne, Org. Lett., 2016, 18, 920–923 CrossRef PubMed.
- Extended exposure to H2O/DIEA (18 h) increased conversion to 82%; however, 3% epimerization of Cys(Trt) was observed.
- T. D. Kondasinghe, H. Y. Saraha, S. B. Odeesho and J. L. Stockdill, Org. Biomol. Chem., 2017, 15, 2914–2918 CAS.
- (a) J. M. McIntosh, D. Yoshikami, E. Mahe, D. B. Nielsen, J. E. Rivier, W. R. Gray and B. M. Olivera, J. Biol. Chem., 1994, 269, 16733–16739 CAS; (b) D. S. Johnson, J. Martinez, A. B. Elgoyhen, S. F. Heinemann and J. M. McIntosh, Mol. Pharm., 1995, 48, 194–199 CAS; (c) M. Ellison, F. Gao, H.-L. Wang, S. M. Sine, J. M. McIntosh and B. M. Olivera, Biochemistry, 2004, 43, 16019–16026 CrossRef CAS PubMed.
- (a) I. V. Maslennikov, Z. O. Shenkarev, M. N. Zhmak, V. T. Ivanov, C. Methfessel and A. S. Arseniev, FEBS Lett., 1999, 444, 275–280 CrossRef CAS PubMed; (b) J. Gehrmann, N. L. Daly, P. F. Alewood and D. J. Craik, J. Med. Chem., 1999, 42, 2364–2372 CrossRef CAS PubMed; (c) J. S. Nielsen, P. Buczek and G. Bulaj, J. Pept. Sci., 2004, 10, 249–256 CrossRef CAS PubMed.
- (a) J. M. McIntosh, D. Yoshikami, E. Mahe, D. B. Nielsen, J. E. Rivier, W. R. Gray and B. M. Olivera, J. Biol. Chem., 1994, 269, 16733–16739 CAS; (b) K. B. Akondi, M. Muttenthaler, S. Dutertre, Q. Kaas, D. J. Craik, R. J. Lewis and P. F. Alewood, Chem. Rev., 2014, 114, 5815–5847 CrossRef CAS PubMed.
- C. J. Armishaw, N. L. Daly, S. T. Nevin, D. J. Adams, D. J. Craik and P. F. Alewood, J. Biol. Chem., 2006, 281, 14136–14143 CrossRef CAS PubMed.
- 70% crude yield, 86% HPLC purity.
- R. Söll and A. G. Beck-Sickinger, J. Pept. Sci., 2000, 6, 387–397 CrossRef.
- (a) B. Dang, T. Kubota, A. M. Correa, F. Bezanilla and S. B. H. Kent, Angew. Chem., Int. Ed., 2014, 53, 8970–8974 CrossRef CAS PubMed; (b) C. Sun, G. Luo, S. Neravelta, S. S. Ghosh and B. Forood, Bioorg. Med. Chem. Lett., 2013, 23, 5203–5208 CrossRef CAS PubMed.
- G. A. Acosta, M. Royo, B. G. de la Torre and F. Albericio, Tetrahedron Lett., 2017, 58, 2788–2791 CrossRef CAS.
- (a) L. Raibaut, P. Seeberger and O. MeInyk, Org. Lett., 2013, 15, 5516–5519 CrossRef CAS PubMed; (b) L. Raibaut, M. Cargoët, N. Ollivier, Y. M. Chang, H. Drobecq, E. Boll, R. Desmet, J.-C. M. Monbaliu and O. MeInyk, Chem. Sci., 2016, 7, 2657–2665 RSC.
- (a) B. Bacsa, S. Bösze and C. O. Kappe, J. Org. Chem., 2010, 75, 2103–2106 CrossRef CAS PubMed; (b) M. Quibell, W. G. Turnell and T. Johnson, J. Org. Chem., 1994, 59, 1745–1750 CrossRef CAS; (c) M. M. Condron, B. H. Monien and G. Bitan, Open Biotechnol. J., 2008, 2, 87–93 CrossRef CAS PubMed.
- (a) P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS; (b) E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- D. G. Mullen, K. Kyro, M. Hauser, M. Gustavsson, G. Veglia, J. M. Becker, F. Naider and M. D. Distefano, Bioorg. Med. Chem., 2007, 15, 931–938 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03553e |
| This journal is © The Royal Society of Chemistry 2018 |