
Reduced graphene oxide/silver hybrid with N,N-dimethyl formamide for oxygen reduction reactions and surface enhanced Raman scattering†
Xiu-Zhi Tangab,
Narasimalu Srikanthb,
Xi-Qiao Fengc,
Chee Kai Chuaa and
Kun Zhou*a
aSchool of Mechanical and Aerospace Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: kzhou@ntu.edu.sg
bEnergy Research Institute@NTU, 1 CleanTech Loop, Singapore 639798, Singapore
cDepartment of Engineering Mechanics, Tsinghua University, Beijing 100084, China
First published on 11th October 2016
Abstract
A reduced graphene oxide (RGO)/Ag hybrid for oxygen reduction reaction and surface enhanced Raman scattering was prepared and a reasonable reaction path towards the reduction of graphene oxide (GO) was investigated. The structures and properties of RGO and RGO/Ag were characterized by various methods, such as Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy and thermal gravity analysis. The results indicated that the reduction time for GO treated by N,N-dimethyl formamide (DMF) only had slight effect on the reduction degree of RGO; however, the hydrophobicity of RGO increased dramatically with the increasing reaction time, owing to the introduction of hierarchical structures. Instead of the traditional opinion on the DMF involved hydrolysis, we proposed a new possible reaction path rendering the chemical structure of the obtained RGO more reasonable. Furthermore, the obtained RGO/Ag demonstrated to be useful for surface enhanced Raman scattering and an effective catalyst for oxygen reduction reaction. This work was expected to provide inspiration for promoting the synthesis and applications of inorganic particles-decorated RGO hybrids.
1 Introduction
Graphene, a unique two-dimensional carbon material with one-atom thickness, has attracted tremendous attention since it was firstly exfoliated from bulk graphite by Geim et al.1 Recently, various methods have been exploited for achieving large-scale production of graphene. As the chemical oxidation product of nature graphite (NG), graphene oxide (GO) is widely accepted as a promising precursor for the scalable preparation of chemically converted graphene, whose chemical structure is very similar to that of pristine graphene. Therefore, for the investigation of graphene-based materials, it is not unexpected that the transformation method of GO to reduced graphene oxide (RGO) has been regarded as one of most important research topics.2–4With the extensive efforts from many research groups, different methods for the preparation of RGO were explored.4–9 Among these strategies, solvothermal synthesis of RGO is promising due to its ease of operation, high efficiency, and relative low cost.10–12 In our previous investigation, we showed that maintaining a fully suspension state of reactants during the entire transformation process is critical to obtain high-quality RGO.13 In addition, in view of the residual oxygen-containing groups, the heterogeneous RGO structure actually consists of both graphitic and oxidation domains.14 Therefore, for solvothermal strategy, it is of great importance for the correct selection of solvent, which is capable of maintaining both exfoliated GO and RGO nanosheets.
N,N-Dimethyl formamide (DMF), as a common organic solvent with high boiling point, has been confirmed to be an ideal solvent for both GO and RGO.15–18 Recently, the utilization of DMF as an effective reductant for the preparation of RGO has also been demonstrated.11,19,20 By heating GO/DMF suspension, GO can be partially reduced. As reported by Lin et al.,19 DMF could transfer GO to RGO at 100 or 150 °C, and they found that higher temperature was able to accelerate the reduction rate significantly, which was later confirmed by Ai et al.11 Additionally, Ai et al. found that DMF used here could also serve as an effective stabilizer for the final RGO nanosheets. A further work by Zhou et al. showed that dimethylamine, a pyrolysis product of DMF, was the true stabilizer.20 Despite different reaction mechanisms between DMF and GO as proposed by Ai et al. and Zhou et al., they both agreed that dimethylamine was the final modifier attached on the RGO nanosheets.11,20 In addition to the confirmed reduction effect on GO, DMF was also utilized for the synthesis of silver particles from Ag+,21,22 and silver-based materials have got widely applications due to their unique properties, such as antibacterial materials and electrochemical sensors.23–28 Inspired by this point, the preparation of GO/Ag and RGO/Ag in DMF was carried out by Dutta et al. and Yang et al.29,30 Although many studies relating to the reduction of RGO by DMF have been conducted, the reduction reaction mechanism during the treatment of GO remains unclear and few works have been reported on RGO/Ag hybrids prepared in one-step way at a temperature as high as 153 °C.
In this study, the preparation of RGO/Ag hybrid with DMF and the reduction reaction between DMF and GO were investigated. A different reaction path was proposed to explain the chemical structure of RGO prepared in DMF. Different from the previously proposed reactions, methylamine, rather than dimethylamine, was considered as the final modifier for the RGO. Moreover, RGO/Ag was prepared at the boiling point of DMF and demonstrated to be a promising material for surface enhanced Raman scattering (SERS) and an effective catalyst for oxygen reduction reaction (ORR).
2 Experimental
To increase the reduction degree of RGO and obtain silver particles decorated RGO, GO and silver ions were simultaneously reduced at the boiling point of DMF.Preparation of RGO and RGO/Ag with N,N-dimethyl formamide
GO was fabricated from NG powders (Huadong Graphite Factory, Pingdu, China) via a modified Hummer method and the details were described in our previous works.31,32 For the synthesis of RGO by the solvothermal method, GO powders (50 mg) were dispersed in 100 mL DMF and exfoliated with the aid of a probe sonicator for 30 min to form a stable suspension. Under vigorous stirring, the slurry was heated up to 153 °C and maintained for different durations (1, 5, 7 and 9 h). Afterwards, the black solids were obtained by vacuum filtration using a polypropylene membrane with a 0.22 μm pore size. After being repeatedly washed with excess deionized water and ethanol, the products were dried in a vacuum oven at 80 °C overnight. The final products were named as RGO-1, RGO-5, RGO-7 and RGO-9, according to their reaction durations, respectively.For the preparation of the RGO/silver hybrid, GO powders (150 mg) were firstly dispersed in 300 mL DMF (provided by Sigma-Aldrich, Singapore) with the aid of a probe sonicator, and then 600 mg AgNO3 (>99.8%, Sigma-Aldrich, Singapore) together with 1.2 g poly(N-vinyl-2-pyrrolidone) (molecular weight: 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich, Singapore) as a protect agent were added. After stirring at 153 °C for 1 h, the final product was obtained by vacuum filtration and dried at 60 °C for 12 h.
000, Sigma-Aldrich, Singapore) as a protect agent were added. After stirring at 153 °C for 1 h, the final product was obtained by vacuum filtration and dried at 60 °C for 12 h.
Electrochemical measurements
Electrochemical measurements were performed in a standard three-electrode glass cell system. A CHI 660C workstation using the saturated calomel electrode (SCE) as a reference electrode and the Pt wire as a counter electrode was utilized to record cyclic voltammetry curves. RGO/Ag or RGO powders (20 mg) with 100 μL of Nafion were added into 10 mL ethanol and sonicated for 20 min. 10 μL obtained catalyst ink was then pipetted onto a pre-cleaned glassy carbon electrode and dried under the ambient condition. Potassium hydroxide aqueous solution (0.1 M) was used as the electrolyte. To obtain O2- or N2-saturated solution, the electrolyte was bubbled by O2 or N2 for at least 30 min before the test, and the gas was maintained during the whole procedure. A typical scanning speed for electrochemical test was set at 50 mV s−1. ORR test was operated in the 0.1 M KOH electrolyte (Ag/AgCl as the reference electrode) on a rotating-disk electrode system, while the rotating speed varied from 225 to 2500 rpm with a scanning rate of 10 mV s−1. O2 was maintained during the whole test procedure. The samples for ORR test were firstly dispersed in DMF with a concentration of 2 mg mL−1. Then the slurry was coated onto the glassy-carbon glassy using Nafion (0.5 wt%) as the binder. To investigate the electrons transferred number (n), the linear sweep voltammetry (LSV) curves were collected. According to the Koutecky–Levich (K–L) equation:33
 | (1) |
| B = 0.2nFD2/3ν−1/6C | (2) |
485 C mol−1), D is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1), ν is the kinetic viscosity (0.01 cm2 s−1), and C represents the bulk concentration of O2 (1.2 × 10−6 mol L−1).
Characterizations
Field emission scanning electron microscopy (FE-SEM, JEOL JSM-7600F) images were done to study the surface morphologies of GO and RGO films. With an attenuated total reflection mode, Fourier transform infrared (ATR-FTIR) spectra of GO and RGO in the range of 600–4000 cm−1 were collected using a Perkin Elmer Instruments Spectrum GX FTIR spectrometer. Thermogravimetric analysis (TGA) was performed in a thermogravimetric analyzer Q500 and the heating rate was set at 10 °C min−1 under N2 atmosphere. X-ray diffraction (XRD) patterns of GO and RGO were recorded on a Bruker D8 Focus automated X-ray diffractometer system, where X-ray excitation was inspired in a Cu Kα radiation (λ = 1.5418 Å) source. Raman spectra were conducted at a Witec alpha 300 SR spectrometer and the wavelength of the laser excitation source was 633 nm. A drop of Rhodamine B (RhB) solution (in ethanol) with a concentration of 1 × 10−5 M was added onto RGO/Ag hybrid materials and then washed by deionized water repeatedly. SERS curves were recorded by a Renishaw inVia Raman microscope while laser excitation source was 514 nm. The laser power was set as 10% while its maximum value was 20 mW. The diameter of the facula was 1 μm. The objective lens was set as 100×. The exposure time was set as 1 s under the scanning mode.3 Results and discussion
To obtain well-exfoliated GO nanosheets, the chemical structure of NG was unavoidably damaged in the harsh oxidation environment and a number of oxygen-containing groups were introduced during the oxidation of graphite. Owing to the hybridization state of many carbon atoms converting from sp2 to sp3, the electronic transmission capacity of GO nanosheets decreased remarkably when compared to that of NG. Nevertheless, the conversion of GO to RGO could partially recover its electrical conductivity as shown in Fig. 1a, and the electric conductivity increased greatly from 3.5 × 10−7 S m−1 for GO to 24.4 S m−1 for RGO-1 h. The significantly improved electrical conductivity could be ascribed to the recovery of graphitic sp2 structure, which was subsequently confirmed by XPS data. Fig. 1b shows that the atomic ratios of carbon to oxygen increased from 2.50 to 4.89, implying the removal of oxygen-containing groups. Furthermore, from C 1s curves of GO and RGO reflected in Fig. 1c, the higher binding energy peak corresponding to sp3 carbon atoms decreased dramatically and only one main peak remained, confirming the obvious recovery of sp2 domains. Although those peaks corresponding to sp3 carbon atoms can still be fitted in the RGO C 1s curve, there is no doubt that the partial recovery of the graphitic area on RGO nanosheets has taken place.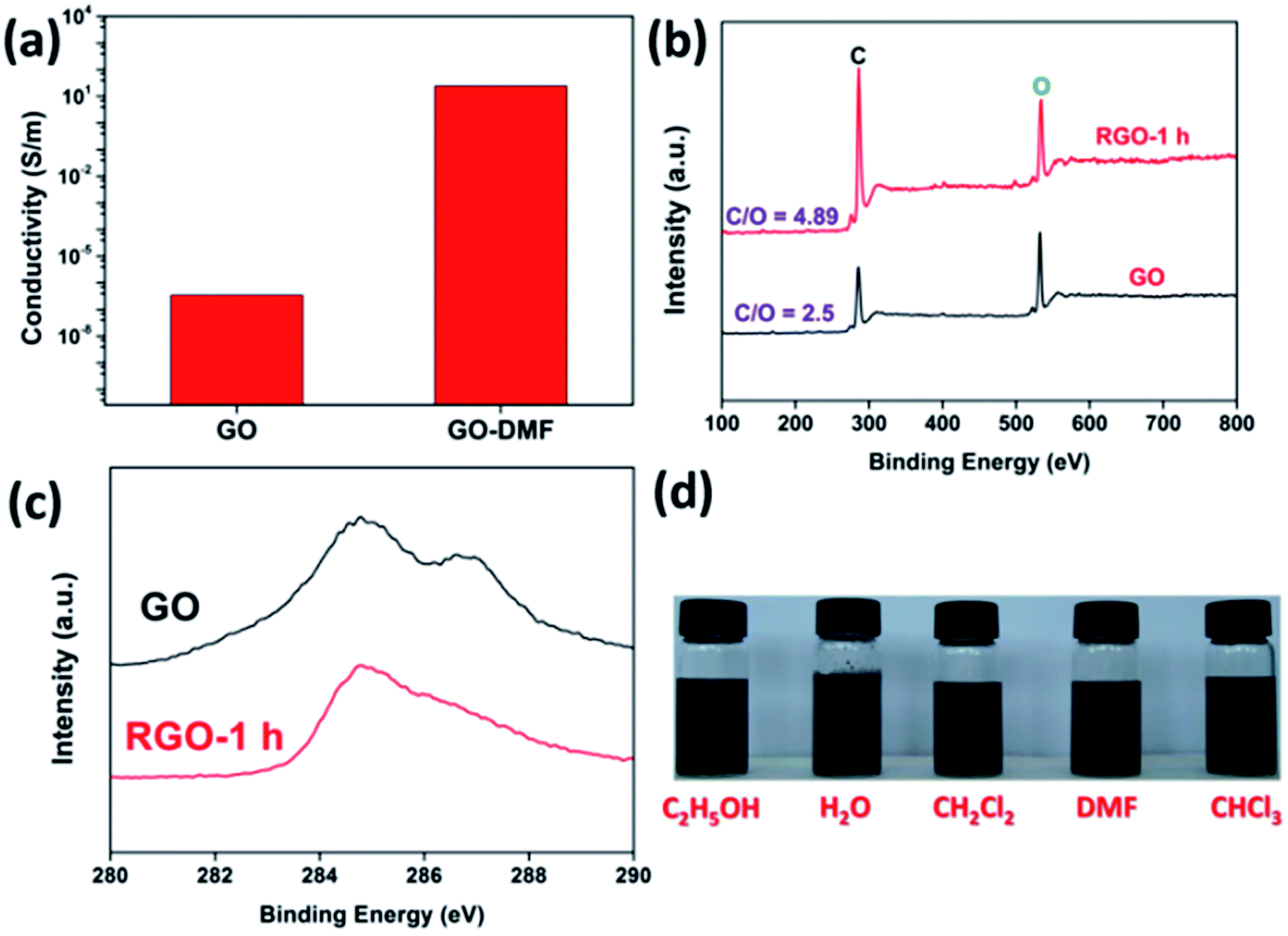 | ||
| Fig. 1 (a) Electrical conductivity of GO and RGO-1 h; (b) wide-scan XPS curves of GO and RGO-1 h, (c) C 1s curves of GO and RGO-1 h; (d) dispersion of RGO in ethanol, water, dichloromethane, DMF and chloroform (photos were taken after 1 hour stationary). | ||
The dispersion state of RGO is critical for its following application by the wet-chemistry process. Therefore, the dispersion test in water, ethanol, dichloromethane, DMF and chloroform was carried out. We found that the RGO-1 h can be well dispersed in all of these five solvents with a concentration of 0.5 mg mL−1 (as shown in the inset of Fig. 1d). No obvious layered separation is visible even the suspensions were stationary for more than 1 hour. The stable dispersion of RGO in both polar solvents and non-polar solvents should originate from the heterogeneous chemical structure of RGO and the gentle reduction ability of DMF.14 Moreover, these solvents with distinct polarities applied here suggested a convenience of RGO processing for its further processing.34,35
During the process of solvothermal reduction, the oxygen-containing groups on GO nanosheets were removed gradually and some exotic molecules were also decorated onto the RGO nanosheets simultaneously. Therefore, ATR-FTIR spectra were utilized to monitor the evolution of oxygen-containing groups on GO sheets. As shown in Fig. 2a, those typical peaks attributed to carboxyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 1730 cm−1), carboxyl groups (C–O, 1353 cm−1) and alkoxy groups (C–O, 1038 cm−1) can be observed from the FTIR curves of GO. For the case of RGO, those peaks corresponding to oxygen-containing groups became weak obviously as a result of the solvothermal reduction. Two peaks located at 1637 cm−1 and 1252 cm−1 should be ascribed to –CO (amide) and C–N, separately. Thus, it can be concluded that some organic molecules had been grafted onto RGO by amide groups, especially for the peak located at 1580 cm−1 was believed to be caused by N–H asymmetric scissoring vibration.36 Unexpectedly, the peak corresponding to carboxyl group, which usually disappeared in RGO curves, was still visible, indicating the relative weak reduction of DMF.
O, 1730 cm−1), carboxyl groups (C–O, 1353 cm−1) and alkoxy groups (C–O, 1038 cm−1) can be observed from the FTIR curves of GO. For the case of RGO, those peaks corresponding to oxygen-containing groups became weak obviously as a result of the solvothermal reduction. Two peaks located at 1637 cm−1 and 1252 cm−1 should be ascribed to –CO (amide) and C–N, separately. Thus, it can be concluded that some organic molecules had been grafted onto RGO by amide groups, especially for the peak located at 1580 cm−1 was believed to be caused by N–H asymmetric scissoring vibration.36 Unexpectedly, the peak corresponding to carboxyl group, which usually disappeared in RGO curves, was still visible, indicating the relative weak reduction of DMF.
 | ||
| Fig. 2 (a) ATR-FTIR spectra of GO, RGO-1 h, RGO-5 h, RGO-7 h and RGO-9 h; (b) a proposed reaction between GO and DMF. | ||
Although the dimethyl amine was widely accepted as a modifier and reductant for DMF in many previous reports on RGO,37,38 they still failed to explain the –N–H bonds on RGO based on the following two facts: (1) dimethyl amine is a compound that is easily volatile or washed away; (2) no N–H groups can be maintained if the amidation reaction occurred between dimethyl amine groups and carboxyl groups. To explain the chemical structures of RGO based on the FTIR spectra, we propose a new possible reaction path for DMF treated RGO. As illustrated in Fig. 2b, the dimethylamine can further transform into the methylamine which possesses primary amine bond.37 Although it is hard to detect the volatile methylamine directly, the FTIR curves of RGO, in return, were actually providing indirect evidence for that. And, many studies had confirmed the methylamine was the degradation product of methylamine.39,40 The reaction between –NH2 (from methylamine) and –COOH (from GO) can form amide bonds and thus introduce N–H groups onto GO, which is consistent with FTIR results (Fig. 2a).
As a result of the solvothermal reduction, unstable oxygen-containing groups on GO sheets would be gradually removed. Thus, the chemical constitutions and properties of GO were expected to change with the increasing reaction time. Interestingly, as shown in Fig. 3a, the weight loss occurred in the range of 100 to 400 °C for RGO-9 h decreased by only 1.37% when compared to RGO-1 h, confirming the impact of the reaction time on the content of oxygen-containing groups is negligible because most of labile groups had been removed within 1 hour.
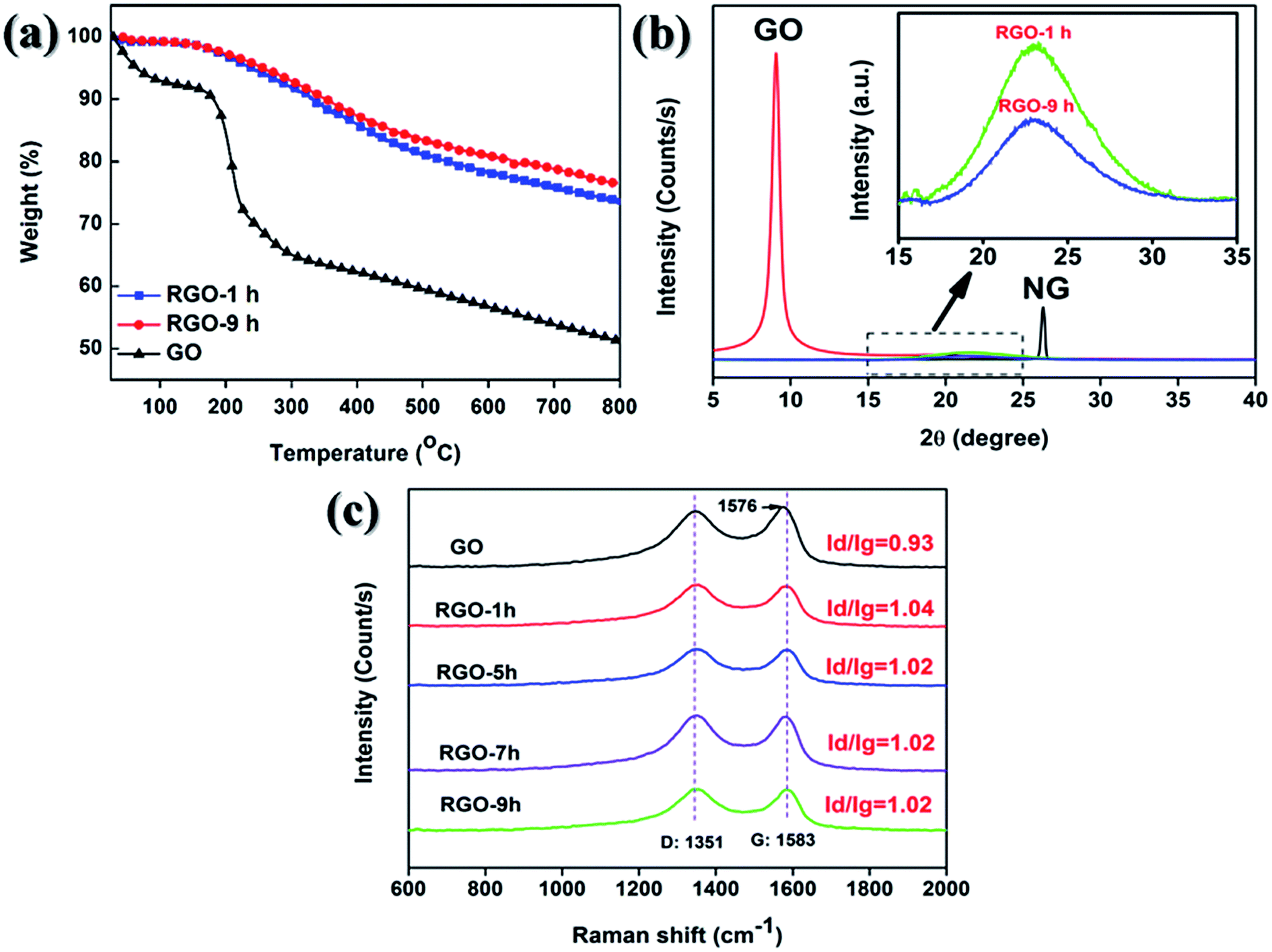 | ||
| Fig. 3 TGA curves (a) and XRD patterns (b) of GO, RGO-1 h and RGO and RGO-9 h; (c) Raman spectra GO, RGO-1 h, RGO-5 h, RGO-7 h and RGO-9 h. | ||
Owing to the remove of oxygen-containing groups, the interlayer spacing of RGO nanosheets decreased according to the XRD pattern in Fig. 3b. Compared to NG, the XRD peak of GO shifted from 26.6 to 12.4°, indicating the increase of interlayer spacing from 0.34 to 0.70 nm. After the reduction, the sharp peak for GO disappeared and a bump-like diffraction peak appeared, indicating that the crystalline structure of GO was damaged during the reduction and some RGO nanosheets re-stacked into the disordered structure.4,41–43
The structural evolution of GO was subsequently investigated by Raman spectra. As exhibited in Fig. 3c, the D band located at 1351 cm−1 indicates the structural defects of GO nanosheets had formed during the oxidation.44 While the G band located at 1577 cm−1 was ascribed to the sp2 carbon atom. After reduction, the intensity ratios of D peak (Id) to that of G peak (Ig) increased from 0.93 for GO and to 1.04 for RGO, suggesting that the average size of in-plane sp2 domains decreased. Moreover, neither Id/Ig ratios of RGO nor the location of G peak changed with the increasing reaction time. Therefore, it can be concluded that the reaction time exert negligible effect on the chemical structure of RGO.
However, the slight effect on the chemical structures of RGO caused by the reaction time did not mean that the same situation would reproduce on the microstructures of RGO. As presented in Fig. 4a, contact angles of GO, RGO-1 h and RGO-9 h films were measured with 13 μL deionized water and the results were shown in the histogram. Because of the oxygen-containing groups attached, the GO film exhibited obvious hydrophilicity that gave a low contact angle at 55°. After treatment in the DMF for 1 hour, the contact angle of RGO-1 h increased to 91° because of the removal of oxygen-containing groups. And the contact angle further increased to 111° for RGO-9 h.
 | ||
| Fig. 4 (a) Contact angles of GO, RGO-1 h and RGO-9 h; SEM images reflect surface morphologies of (b) GO, (c) RGO-1 h and (d) RGO-9 h. Inserts: digital photos for water contact angle test. | ||
Based on many previous investigations,45,46 the improved hydrophobicity is usually attributed to two facts: the change of chemical constituents, such as the introduction of some hydrophobic groups; the evolution of microstructures, such as the formation of hierarchical structure on the surface of the materials. According to the comparative analysis on FTIR and Raman data as described above, the chemical components and chemical structures of those RGO with different reaction time were found to be very similar. Thus, the variation of microstructure would be responsible for the different wettability of those RGO. To study the surface morphologies, SEM images are captured to characterize GO, RGO-1 h and RGO-9 h films. As shown in Fig. 4b, the surface of GO film was crumpled, and wrinkled edges of GO sheets can be observed. This hierarchical structure with hydrophilic GO surface led to a low contact angle of 55° for GO film. While in the case of RGO-1 h, the remove of oxygen-containing groups made RGO nanosheets much more hydrophobic and a larger contact angle of 91° was observed. A smoother surface with several wrinkles for RGO-1 h was shown in Fig. 4c. With longer DMF treatment, as shown in Fig. 4d, much more nano-scaled crumples formed on RGO-9 h. Thus, we can conclude that the formation of hydrophobic and micro–nano dual-structure is the main reason for the increased contact angle.
Silver is a typical noble metal and has been intensively investigated even in the emerging field of additive manufacturing.47–50 For the RGO/Ag hybrid, DMF is an effective reductant for both GO and silver ions. In this work, the hybrid material was prepared at the boiling point of DMF in one step way, while PVP was utilized as a protect agent. As exhibited in Fig. 5a, the typical peaks at 17.7°, 38.1°, 44.3°, 64.4°, 77.5°, and 82.5°, corresponding to typical (111), (200), (220), (311) and (222) planes of silver, were obviously observed, confirming the successful attachment of face-centered cubic silver particles on RGO.14,51 In addition to characteristic peaks corresponding to silver particles, the crystallinity evolution of RGO substrate can also be seen. Compared to XRD curve of GO, the peak located at 12.2° disappeared for RGO/Ag, suggesting the exfoliation of RGO nanosheets.
 | ||
| Fig. 5 (a) XRD pattern of GO and RGO/Ag; (b) UV-Vis spectra of GO, RGO-1 h and RGO/Ag; (c) TEM images of RGO/Ag hybrid; insert: selected area diffraction image of RGO/Ag; (d) partial enlarged view of silver nanoparticles distributed on RGO/Ag. | ||
Furthermore, the UV-Vis spectra as shown in Fig. 5b provide some useful informations for RGO/Ag hybrid. The peaks at ∼230 nm caused by π to π* transitions of the CC can be observed from UV-Vis curves of GO.52 After reduction, the peak shifted to 270 nm for both RGO-1 h and RGO/Ag. Based on above facts, it can be concluded that silver particles were successfully decorated onto RGO substrates. Furthermore, the silver particles on RGO can also be observed from the UV-Vis curve of RGO/Ag, where a broad peak around 430 caused by the surface plasmon resonance of silver particles was found. Furthermore, the TEM image, as show in Fig. 5c, provided a direct evidence for the formation and distribution of silver particles on RGO substrate. It can be observed that the sizes of most deposited silver particles were different. According to the Mie theory, the inhomogeneity of silver sizes was consistent with the asymmetric peak of UV-Vis curve for RGO/Ag.53 Although there was some aggregations in RGO/Ag, most of silver particles were still uniformly distributed on RGO nanosheets. The insert image in Fig. 5c displayed the selected area diffraction of RGO/Ag. It can be observed that the crystalline structure of both RGO and Ag were well maintained. From Fig. 5d, a lattice spacing of 0.236 nm was observed, corresponding to those typical face-centered cubic silver particles, which were consistent with XRD data in Fig. 5a.54 Moreover, the preparation of RGO/Ag with equal sizes and improved distribution will be the emphasis of our future work.
Since the “hot-spot” effect from silver nanoparticles is known to enhance intensities of Raman signals, RGO/Ag was anticipated to be a promising candidate material for SERS applications, which is very important for analytical chemistry.55 RhB is a common dye which usually serves as a tracer dye within water that is widely used in biotechnology applications. In this work, RhB was chosen as the analyte to investigate SERS effect of RGO/Ag. As shown in Fig. 6a, without any decoration of silver nanoparticles, RGO-1 h did not exhibit any SERS activity. In contrast, for RGO/Ag, many visible peaks corresponding to RhB molecules appeared, indicating that RGO/Ag is an ideal SERS material with high sensitivity to detect fluorescent molecules at low-concentration.56,57
 | ||
| Fig. 6 (a) Raman spectra of RhB on RGO-1 h and RGO/Ag; (b) CV curves of RGO/Ag under nitrogen atmosphere at scanning speed of 50 mV s−1; (c) catalysis activity towards ORR for RGO-1 h and RGO/Ag (O2, 50 mV s−1); (d) LSV curves of RGO-1 h at different rotating speeds; (e) LSV curves of RGO/Ag at different rotating speeds. | ||
For the calculation of enhancement factor (EF), an equation of EF = (ISERS/Nsurface)/(IRS/Nbulk) was introduced and the peak at 622 cm−1 was selected for obtaining EF value. Herein, ISERS and IRS are the intensities of SERS signal and normal Raman signal at 622 cm−1, separately; Nbulk and Nsurface are RhB molecule number illuminated by the laser focus spot under normal Raman and SERS condition. The RhB solution with a concentration of 0.01 M was chosen as the reference sample and related Raman curves were exhibited in Fig. S1.† Finally, the EF was calculated to be 5.6 × 106.
In addition to SERS applications, RGO/Ag can also find its application in electrochemistry. For advanced energy storages or energy conversion devices, ORR is not only a basic electrochemical procedure but also a key process for the energy development. Therefore, the development of materials with high ORR catalytic activity is of great significance. In this study, RGO/silver was employed as an ORR catalyst. Under inert gas atmosphere (N2), cyclic voltammetry (CV) curves of RGO-1 h and RGO/Ag were measured (Fig. 6b). Compared to Liu et al.'s report,58 the peak ascribe to the dissolution of silver and the formation of single-layer Ag2O disappeared, implying the excellent adhesive stability of silver particles on RGO substrate. When oxygen gas was applied, RGO/Ag exhibited significant catalytic activity towards ORR. A cathodic reduction peaks around −0.18 V can be observed in Fig. 6c. Moreover, contributed by the pseudo capacitance produced by silver particles, the capacitance of RGO/Ag was larger than that of RGO. Furthermore, by examining LSV curves of RGO and RGO/Ag as shown in Fig. S2,† we found the onset potential of RGO/Ag was −1.0 V, which is obviously positive than that of RGO-1 h (−1.8 V). Moreover, limiting current densities of RGO/Ag were larger than those of RGO-1 h. Thus, RGO/Ag possessed significantly higher electrochemical catalytic activity.57,58 In addition, the electron transfer numbers of RGO-1 h and RGO/Ag were calculated based on the data as show in Fig. 6d and e. According to the K–L equation, the n of RGO-1 h was calculated to be 2.2 while that of RGO/Ag was 3.6. These results indicated that the redox reaction occurred on RGO-1 h was essentially a two-electron process while that on RGO/Ag was a dominant four-electron process.33,59,60
4 Conclusions
GO reduced by DMF was obtained with different reaction time. The FTIR results indicated the emergence of primary-amine molecules in the solvothermal reduction. Thus, a possible reaction mechanism was proposed. Moreover, prolonging reaction time to more than 1 hour is confirmed to be irrelevant to the reduction degree of RGO. However, the wettability of RGO is strongly dependent on the reaction time as RGO films become more hydrophobic with the prolonged reaction time. Furthermore, RGO/Ag hybrid was prepared at the boiling point of DMF. Owing to the attachment of silver nanoparticles, Raman signals of RhB can be detected in RGO/Ag while no obvious Raman signals of RhB can be observed in RGO-1 h. For the electrochemical investigation, RGO/Ag has proved to be an effective catalytic function for ORR and the n was calculated to be 3.6. Therefore, the investigation on the RGO and RGO/Ag prepared by DMF is not only providing a better understanding the reduction of GO, but also paving the way for the preparation of hybrid materials with multifunctional applications.Acknowledgements
The authors acknowledge financial support from Ministry of Education, Singapore (Academic Research Fund TIER 1-RG128/14) and Vestas Technology R & D Singapore Ltd.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. Thakur and N. Karak, Carbon, 2015, 94, 224–242 CrossRef CAS.
- F. Li, X. Jiang, J. Zhao and S. Zhang, Nano Energy, 2015, 16, 488–515 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- J. McDonald-Wharry, M. Manley-Harris and K. Pickering, Philos. Mag., 2015, 95, 4054–4077 CrossRef CAS.
- H. Feng, R. Cheng, X. Zhao, X. Duan and J. Li, Nat. Commun., 2013, 4, 1539 CrossRef PubMed.
- A. C. Faucett and J. M. Mativetsky, Carbon, 2015, 95, 1069–1075 CrossRef CAS.
- H.-L. Gao, X.-L. Li, W. He, R.-T. Guo and B. Chai, Acta Phys.-Chim. Sin., 2015, 31, 2117–2123 CAS.
- X. H. Wang, I. Kholmanov, H. Chou and R. S. Ruoff, ACS Nano, 2015, 9, 8737–8743 CrossRef CAS PubMed.
- S. Dubin, S. Gilje, K. Wang, V. C. Tung, K. Cha, A. S. Hall, J. Farrar, R. Varshneya, Y. Yang and R. B. Kaner, ACS Nano, 2010, 4, 3845–3852 CrossRef CAS PubMed.
- K. Ai, Y. Liu, L. Lu, X. Cheng and L. Huo, J. Mater. Chem., 2011, 21, 3365–3370 RSC.
- V. H. Pham, T. V. Cuong, S. H. Hur, E. Oh, E. J. Kim, E. W. Shin and J. S. Chung, J. Mater. Chem., 2011, 21, 3371–3377 RSC.
- X.-Z. Tang, X. Li, Z. Cao, J. Yang, H. Wang, X. Pu and Z.-Z. Yu, Carbon, 2013, 59, 93–99 CrossRef CAS.
- X.-Z. Tang, X. Chen, G. Wu, X. Hu and J. Yang, RSC Adv., 2015, 5, 49257–49262 RSC.
- Y. Shen, H.-B. Zhang, H. Zhang, W. Ren, A. Dasari, G.-S. Tang and Z.-Z. Yu, Carbon, 2013, 56, 132–138 CrossRef CAS.
- Z. He, B. Zhang, H.-B. Zhang, X. Zhi, Q. Hu, C.-X. Gui and Z.-Z. Yu, Compos. Sci. Technol., 2014, 102, 176–182 CrossRef CAS.
- X.-Y. Qi, D. Yan, Z. Jiang, Y.-K. Cao, Z.-Z. Yu, F. Yavari and N. Koratkar, ACS Appl. Mater. Interfaces, 2011, 3, 3130–3133 CAS.
- Y. Shen, T. Jing, W. Ren, J. Zhang, Z.-G. Jiang, Z.-Z. Yu and A. Dasari, Compos. Sci. Technol., 2012, 72, 1430–1435 CrossRef CAS.
- Z. Lin, Y. Liu, Y. Yao, O. J. Hildreth, Z. Li, K. Moon and C.-P. Wong, J. Phys. Chem. C, 2011, 115, 7120–7125 CAS.
- D. Zhou, Q.-Y. Cheng and B.-H. Han, Carbon, 2011, 49, 3920–3927 CrossRef CAS.
- I. Pastoriza-Santos and L. M. Liz-Marzan, Nano Lett., 2002, 2, 903–905 CrossRef CAS.
- I. Pastoriza-Santos and L. M. Liz-Marzan, Adv. Funct. Mater., 2009, 19, 679–688 CrossRef CAS.
- J. Wang, X. Zhao, J. Li, X. Kuang, Y. Fan, G. Wei and Z. Su, ACS Macro Lett., 2014, 3, 529–533 CrossRef CAS.
- Y. Li, P. Zhang, Z. Ouyang, M. Zhang, Z. Lin, J. Li, Z. Su and G. Wei, Adv. Funct. Mater., 2016, 26, 2122–2134 CrossRef CAS.
- P. Zhang, H. Wang, X. Zhang, W. Xu, Y. Li, Q. Li, G. Wei and Z. Su, Biomater. Sci., 2015, 3, 852–860 RSC.
- Y. Li, X. Zhao, P. Zhang, J. Ning, J. Li, Z. Su and G. Wei, J. Mater. Chem. C, 2015, 3, 4126–4133 RSC.
- X. Zhao, P. Zhang, Y. Chen, Z. Su and G. Wei, Nanoscale, 2015, 7, 5080–5093 RSC.
- S. C. Joshi and A. A. Sheikh, Virtual Phys. Prototyp., 2015, 10, 175–185 CrossRef.
- S. Dutta, C. Ray, S. Sarkar, M. Pradhan, Y. Negishi and T. Pal, ACS Appl. Mater. Interfaces, 2013, 5, 8724–8732 CAS.
- Y.-K. Yang, C.-E. He, W.-J. He, L.-J. Yu, R.-G. Peng, X.-L. Xie, X.-B. Wang and Y.-W. Mai, J. Nanopart. Res., 2011, 13, 5571–5581 CrossRef CAS.
- X.-Z. Tang, W. Li, Z.-Z. Yu, M. A. Rafiee, J. Rafiee, F. Yavari and N. Koratkar, Carbon, 2011, 49, 1258–1265 CrossRef CAS.
- X.-Z. Tang, Z. Cao, H.-B. Zhang, J. Liu and Z.-Z. Yu, Chem. Commun., 2011, 47, 3084–3086 RSC.
- S. J. Guo, S. Zhang, L. H. Wu and S. H. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770–11773 CrossRef CAS PubMed.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- D. Konios, M. M. Stylianakis, E. Stratakis and E. Kymakis, J. Colloid Interface Sci., 2014, 430, 108–112 CrossRef CAS PubMed.
- J. Morlieras, J.-M. Chezal, E. Miot-Noirault, A. Roux, L. Heinrich-Balard, R. Cohen, S. Tarrit, C. Truillet, A. Mignot, R. Hachani, D. Kryza, R. Antoine, P. Dugourd, P. Perriat, M. Janier, L. Sancey, F. Lux and O. Tillement, Nanoscale, 2013, 5, 1603–1615 RSC.
- Y. Veeranagouda, P. V. Emmanuel Paul, P. Gorla, D. Siddavattam and T. B. Karegoudar, Appl. Microbiol. Biotechnol., 2006, 71, 369–375 CrossRef CAS PubMed.
- N. Colebourne, E. Collinson and F. S. Dainton, Trans. Faraday Soc., 1963, 59, 886 RSC.
- N. Staelens, M.-F. Reyniers and G. B. Marin, Ind. Eng. Chem. Res., 2004, 43, 5123–5132 CrossRef CAS.
- J. Z. Vilseck, J. Kostal, J. Tirado-Rives and W. L. Jorgensen, J. Comput. Chem., 2015, 36, 2064–2074 CrossRef CAS PubMed.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 Search PubMed.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. Ye, Chem. Mater., 2009, 21, 3514–3520 CrossRef CAS.
- G. Wu, J. An, X.-Z. Tang, Y. Xiang and J. Yang, Adv. Funct. Mater., 2014, 24, 6751–6761 CrossRef CAS.
- L. Feng, S. Li, Y. Li, H. Li, L. Zhang, J. Zhai, Y. Song, B. Liu, L. Jiang and D. Zhu, Adv. Mater., 2002, 14, 1857–1860 CrossRef CAS.
- T. Sun, L. Feng, X. Gao and L. Jiang, Acc. Chem. Res., 2005, 38, 644–652 CrossRef CAS PubMed.
- Y. Kok, X. Tan, B. T. Shu and C. K. Chua, Virtual Phys. Prototyp., 2015, 10, 13–21 CrossRef.
- W. Wu, S. B. Tor, C. K. Chua, K. F. Leong and A. Merchant, Virtual Phys. Prototyp., 2015, 10, 187–193 CrossRef.
- Z. X. Khoo, J. E. M. Teoh, Y. Liu, C. K. Chua, S. Yang, J. An, K. F. Leong and W. Y. Yeong, Virtual Phys. Prototyp., 2015, 10, 103–122 CrossRef.
- K. C. Chee and F. L. Kah, 3D Printing and Additive Manufacturing, World Scientific Publishing, Singapore, 2014 Search PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS PubMed.
- J. R. G. Navarro and M. H. V. Werts, Analyst, 2013, 138, 583–592 RSC.
- X. Z. Tang, Z. W. Cao, H. B. Zhang, J. Liu and Z. Z. Yu, Chem. Commun., 2011, 47, 3084–3086 RSC.
- Z. Zhang, F. G. Xu, W. S. Yang, M. Y. Guo, X. D. Wang, B. L. Zhanga and J. L. Tang, Chem. Commun., 2011, 47, 6440–6442 RSC.
- X. Yu, H. Cai, W. Zhang, X. Li, N. Pan, Y. Luo, X. Wang and J. G. Hou, ACS Nano, 2011, 5, 952–958 CrossRef CAS PubMed.
- X. Ling, L. Xie, Y. Fang, H. Xu, H. Zhang, J. Kong, M. S. Dresselhaus, J. Zhang and Z. Liu, Nano Lett., 2010, 10, 553–561 CrossRef CAS PubMed.
- R. Liu, S. Li, X. Yu, G. Zhang, Y. Ma and J. Yao, J. Mater. Chem., 2011, 21, 14917–14924 RSC.
- S. J. Guo and S. H. Sun, J. Am. Chem. Soc., 2012, 134, 2492–2495 CrossRef CAS PubMed.
- S. B. Yang, X. L. Feng, X. C. Wang and K. Mullen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: DOI: 10.1039/c6ra24322c |
| This journal is © The Royal Society of Chemistry 2016 |