
Mesoporous carbons: recent advances in synthesis and typical applications
Wang Xinab and
Yonghui Song*ab
aCollege of Water Science, Beijing Normal University, Xinjiekou Wai Street 19, Beijing 100875, China
bState Key Laboratory of Environmental Criteria and Risk Assessment, Chinese Research Academy of Environmental Sciences, Dayangfang 8, Anwai Beiyuan, Beijing 100012, China. E-mail: songyh@craes.org.cn; Fax: +86 10 84915194; Tel: +86 10 84915308
First published on 14th September 2015
Abstract
Mesoporous carbon materials have been extensively studied because of their vast potential applications ranging from separation and adsorption, catalysis, and electrochemistry to energy storage. Their controllable and excellent properties distinguish mesoporous carbons from traditional carbon materials in their synthetic route, adjustable specific surface area and channels, and even interfacial properties. The template carbonization method has been widely used for the synthesis of mesoporous carbons and corresponding composites and endows mesoporous carbon materials with an ordered pore arrangement, developed pore structure and mesoporosity. Novel mesoporous carbon materials with unprecedented control over their morphology, framework and rich composition have been obtained by employing various nanotechnologies. The present manuscript mainly reviews the recent synthesis of mesoporous carbons regarding the synthetic routes and elements, special morphologies, improvements of the synthesis route, synthesis from biomass or waste, and magnetic or nitrogen-containing mesoporous carbons, plus their typical applications including adsorption, electrode materials and catalysts, with a brief introduction to the functionalization and modification of mesoporous carbons. Furthermore, some foreseeable challenges and directions of future research are proposed for the better development of mesoporous carbon materials.
1. Introduction
Highlighted by some of the highest scientific awards and the importance and potential of carbon-based materials including fullerenes, carbon nanotubes, and graphene, which are nowadays studied in depth and have achieved wide attention, another family of carbon nanomaterials, mesoporous carbons (MCs), are now emerging and have promising applications in the following: adsorption, catalysts and their supports, electrochemistry, the synthesis of inorganic nanometer-scale structures, energy conversion and storage.1–5 Actually, they not only retain the original properties of carbons but are also attractive because of a widely open and highly developed pore structure in addition to a large specific surface area.In the context of this review, the term “mesoporous carbons” refers to solids that are based on either ordered or disordered networks with a broad or narrow distribution of pores in the range between 2 and 50 nm according to the definition of the International Union of Pure and Applied Chemistry (IUPAC).6 Historically, ordered mesoporous carbon (OMC) was synthesized by Ryoo et al. via the use of mesoporous silica as a hard template for the first time;7 since then, much attention has been paid to the preparation of MC materials and research on their application. The early work was primarily focused on their synthesis via the use of mesoporous silica as a scaffold and their applications.8–12 The real stage of rapid development has started since a new way for their preparation was found by self-assembly of copolymer molecular arrays and carbon precursors from the contributions of Dai et al. and Zhao et al.13–16 As the modification and improvement of the physical and chemical properties of MCs have taken place via the incorporation of inorganic components that are either embedded in the pore walls or trapped within the channels, the emergence of so-called modified MCs has largely driven the development of carbon-based electrodes or catalysts in their potential applications.17–21 On the other hand, the significant growth of clean energy technologies has inspired research on energy storage recently.22–26 Notably, the emergence of OMCs makes up for the deficiencies of traditional microporous materials such as activated carbon and zeolite in mass transfer and specific surface area and also provides motives for the development of porous materials and other aspects such as the synthesis of new nanomaterials.27–30
Because of their uniform and tunable pore sizes, large specific surface area, thermostability, chemical inertness, biocompatibility, and periodically arranged monodisperse mesopore space, this type of porous material has attracted global interest for research and applications. The literature has grown rapidly in recent years (Fig. 1a): over 3000 publications can be found in Web of Science with the keywords “mesoporous carbon” and the great majority of the references cited in this review are dated between the years 2007 and 2014. Meanwhile, China has occupied the dominant position in this field and contributed nearly half of the papers (Fig. 1b). The published articles focus mainly on the preparation and functionalization of MCs by a soft-template approach, adsorption and magnetic separation, catalysts, high-performance electrochemistry materials, energy storage, etc.31–34 Of course, the mechanism of synthesis and critical factors of preparation have also been studied, including different carbon sources and block copolymers, various types of catalyst, synthesis conditions and different preparation pathways.21,35–39 Besides research into surface properties, pore and morphology control is also necessary for potential applications and further modification.40–44
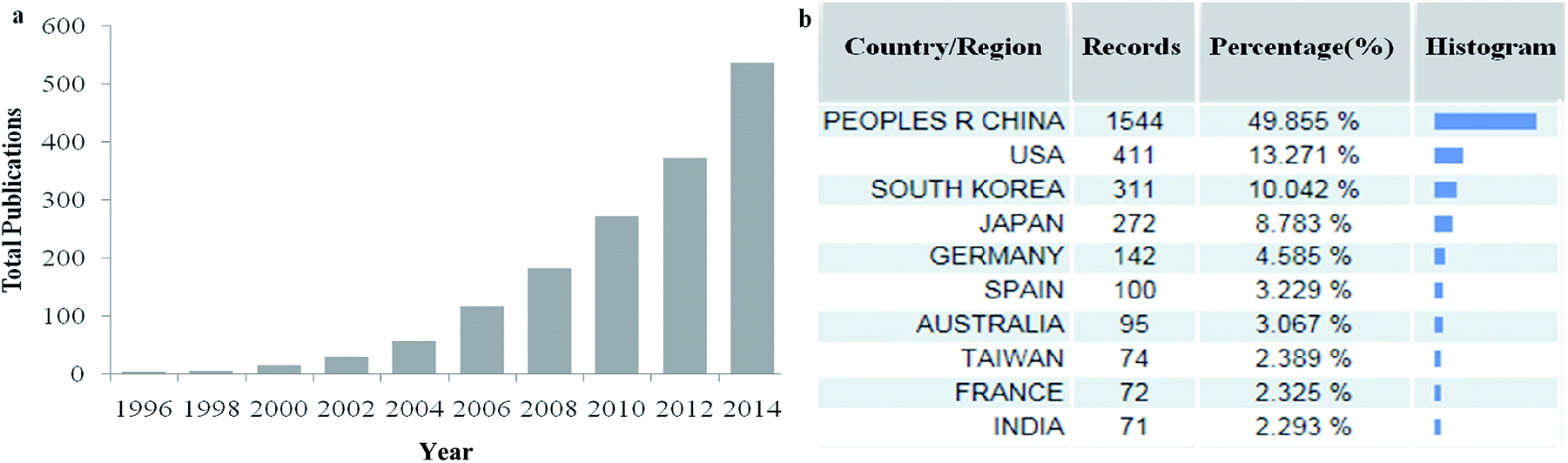 | ||
| Fig. 1 Total number of works of published literature (a) and the distribution of the main study area (b) with the words ‘mesoporous carbon’ in the title, from Web of Science. | ||
A number of reviews and perspective articles have reported about this material and its applications over the last decade. Dai et al. systematically described the synthesis of MC via both hard-template and soft-template routes, with a special emphasis on developments in methodology.45 Zhao et al. summarized the synthesis of OMCs based on supramolecular aggregates as templates and a mechanism with supramolecular aggregates as a directing agent was clarified; in addition, the morphological control and functionalization of ordered mesoporous carbonaceous materials were introduced.46 Kraehnert and Smarsly reported progress and examples in the synthesis and application of MC materials based on soft-templating approaches, reiterated the fundamental principles of self-aggregation, and furthermore proposed a mechanism of synthesis and means of controlling pore size or morphology also in hierarchical meso- or macroporous carbon materials.47 Yuan et al. reviewed a direct synthesis strategy for OMCs via organic–organic self-assembly, with a focus on their controllable preparation, modification and potential applications.48 In terms of specific applications, Guo et al. gave a description of applications as electrochemical sensors and biosensors and revealed the corresponding mechanism between ordered mesoporous carbons and the detected substances.49 Ma and Li reviewed the development of metal oxide nanomaterials that incorporated MC for high-performance supercapacitor applications and discussed the relationship between porous carbon structure and metal oxides for good performance.50 Wang et al. summarized the electrochemical properties of MC-based materials when applied to fuel cells and lithium battery electrode materials, as well as supercapacitors, and proposed future challenges for further development.51
This review aims to summarize and give an overview of some focuses of research, including the preparation and control of MCs, loading and doping, as well as typical applications such as magnetic separation and adsorption, solid catalysts and electrochemical electrode materials, and moreover describes highlights by collecting excellent work from researchers all over the world. Moreover, some challenges and schemes of preparation are presented in order to attract more attention and better improve the properties of MCs. In this review, we will first present an overview of popular synthetic strategies and conditions, also including recent advances that have been made in morphological control, utilization of waste materials for preparation, and magnetic or nitrogen-containing MCs. Secondly, we will discuss the unique physical properties of individual MCs after functionalization or modification. This area has advanced significantly in the understanding of the physical properties of MCs. Thirdly, we will look at the potential applications of MCs. Key examples are also given of how these materials have already displayed improved performance in different applications. These sections will serve as a framework for an extended discussion of the future of MCs. Finally, the latest trends in the synthesis, characterization and applications of these novel carbon materials are summarized to stimulate more innovative solutions from global networks for a sustainable and clean future in this rapidly expanding field.
2. Synthesis
These materials are usually prepared according to a template carbonization route: hard-template as well as soft-template methods have been employed. The preparation of OMCs via a hard template involves several fixed steps (also known as the nanocasting strategy), which include: (i) the preparation of a mesoporous silica matrix with controlled architecture; (ii) the introduction of a suitable carbon precursor into the mesopores by either wet impregnation or chemical vapour deposition (CVD); (iii) the polymerization of the resulting organic–inorganic composite and its carbonization at high temperature; and (iv) the removal of the silica mould by etching in HF or alkaline dissolution (shown in Fig. 2A). As the space that was once occupied by the host hard template is converted into the pores of the carbon material, the resulting carbon structure is a replica of the mesoporous silica. Inspired by the successful replication of OMCs from mesoporous silica, some new “hard templates” with similar function were also developed later.52,53 Meanwhile, the predominant soft-template route for the direct synthesis of OMCs from amphiphilic block copolymers has emerged, which follows the organic–organic self-assembly method via mainly using phenolic resin or its derivatives as the carbon precursor and amphiphilic surfactants as structure-directing agents and is a simpler and more scalable method.54 In this self-assembly method, cooperative assembly between organic surfactants and carbon precursors usually occurs during evaporation (so-called evaporation-induced self-assembly, EISA)55,56 or takes place in solution driven by hydrogen bonding (hydrothermal or phase separation).57 However, EISA is not suitable for the industrial production of carbon powders due to engineering difficulties, such as the evaporation of a large amount of organic solvents, sample collection, reactor design, etc. On the other hand, this time-consuming and costly synthesis procedure is not suitable for scalable production for almost the same reasons as exist in the hard-template route. In contrast, the hydrothermal method may be more suitable for large-scale processes.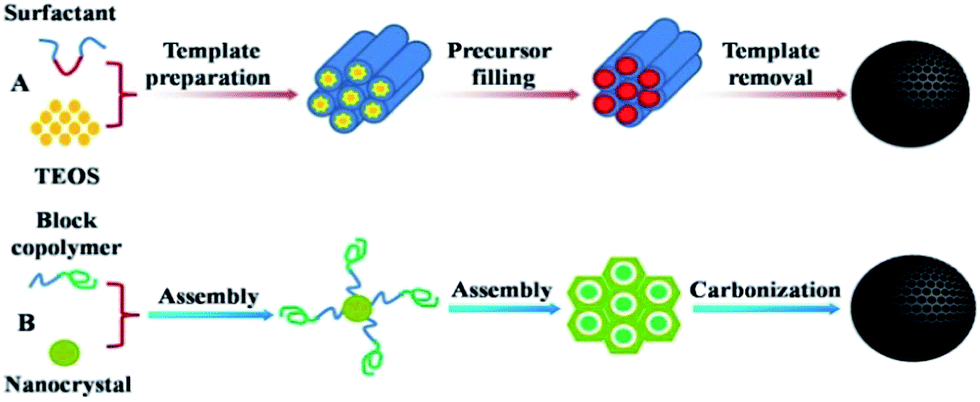 | ||
| Fig. 2 Schematic of a hard-template method (A) and a soft-template method (B). | ||
In the soft-template route, it is the case that MCs could not be obtained only by the direct pyrolysis of self-assembled block copolymers, because of their very poor carbon yields in carbonization reactions, the unsuitably low melting temperature of the linearly structured copolymers and the massive loss of carbon in carbonization, which all result in an inability to preserve pre-organized ordered nanostructures. Therefore, adding suitable carbon precursors to the supramolecular aggregates of block copolymers is necessary. Due to the driving force from hydrogen-bonding interactions, or even other weak interactions like coulombic and van der Waals forces, between the aggregated template and the thermosetting carbon precursor, the supramolecular self-assembly of both can be completed. Another important point is that the template can be removed via calcination in an inert gas or an extraction method, relying on the difference in chemical and thermal stability between the block copolymers and the carbon precursor58–60 and causing the formation of a pore structure or interconnected pore network. This usually involves the steps: (i) formation of a supramolecular arrangement of molecules; (ii) thermopolymerization to give a highly cross-linked composite; (iii) removal of templates; and (iv) carbonization (shown in Fig. 2B). Moreover, a variety of disordered MC materials with wide pore size distributions have been synthesized by the traditional chemical activation or pyrolysis of appropriate carbon precursors (organic aerogels or polymer/polymer blends)61–64 or by employing amphiphilic cationic surfactants that exhibit extremely weak interactions with organic polymer frameworks via a soft-templating route.65–67 Whether using the nanocasting strategy or the soft-template route, MCs from these two strategies exhibit nearly the same chemical compositions and surface functional groups. However, the pore size distribution from the soft-template route is more uniform than that from the hard-template method and the mesophase in the former can be rationally adjusted via different block copolymers.68,69 Moreover, the stability under thermal and oxidative treatments and mechanical properties of MCs from the soft-template route are much higher owing to their thick pore walls and continuous framework.70,71 In any case, although the hard-template method needs more procedures, it also allows more various and less restrictive conditions for the fabrication of MC materials than the soft-template route. The above methods are the main routes for the synthesis of MCs. Nevertheless, as research proceeds, some more convenient, more economical and environmentally friendly methods are also introduced.
2.1 Synthesis and necessary conditions
In this field, in either the hard-template or soft-template method, carbon sources, templates and catalysts are the three main constituents that are added to a solvent to prepare MCs via nanocasting, EISA, an aqueous route, phase separation or hydrothermal autoclaving processes. The carbon precursors and templates have a great impact on the structure and pore size of MC materials. The choice of an appropriate set of thermosetting carbon sources and thermally decomposable surfactants is the most important factor in controlling the structure of these ordered mesostructures. So far, the majority of carbon precursors that are employed to synthesize MC are phenolic types of thermosetting resins such as formaldehyde-cross-linked phenol, resorcinol, phloroglucinol, and hexaphenol.72 The main reason for incorporating phenolic resins is that the large number of phenolic hydroxyl groups form very strong hydrogen bonds with the miscible segment of block copolymer surfactants, which form micelles. The micelle domains are responsible for producing mesoporosity in the resulting carbon. Among these resins, resorcinol formaldehyde (RF) resin is the most frequently used organic monomer for the synthesis of carbon gels and OMC via an organic sol–gel process. Although mostly organic–organic self-assembly has been applied for phenolic resins so far, there have still been some attempts to use other precursors for the synthesis of MCs. Various organic compounds including sucrose,73 furfuryl alcohol,74 aromatic hydrocarbons,75 ethylene,76 acenaphthene,77 polyacrylonitrile (PAN)78 and 1,5-dihydroxynaphthalene (DHN) are suitable as carbon sources. Some researchers synthesized a kind of mesoporous carbon spheres containing in-frame incorporated nitrogen via a facile polymerization-induced colloid aggregation method using melamine–formaldehyde resin (MF)79 as the carbon precursor. Even biomass or waste has been employed as the carbon-yielding component,61–63 as well as using triethyl orthoacetate (EOA) or tetraethoxysilane (TEOS) as carbon co-precursors for pore size control.80–82 Until now, amphiphilic block copolymers (e.g. CTAB, F127, and P123) are the main choices as pore-forming components. However, other inorganics such as CaCO3,83–85 CaCl2,86 and ZnCl2 (ref. 87) have also been adapted as templates via their removal by diluted hydrochloric acid or deionized water. High-molecular-weight amphiphilic block copolymers like poly(ethylene oxide)-block-poly(styrene) (PEO-b-PS),88 poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL),89,90 and poly(isoprene)-block-poly(styrene)-block-poly(ethylene oxide) (ISO)91 have been used as templates for the preparation of MCs with large pore sizes and a tunable porous structure. Most reported syntheses resort to a solvent EISA pathway in non-aqueous media,92–96 using N,N′-dimethylformamide media,97 THF solution,98 an ethanol/H2O mixture or another solution of low polarity as the volatile solvent.34,99 With regard to hydrothermal or dilute aqueous processes, usually water is the unique medium.82,100 In summary, MCs can be successfully prepared via different routes and some typical examples are listed in Table 1.| Precursor | Catalyst | Solvent | Template | Reaction pH | Pathway | Character | Ref. |
|---|---|---|---|---|---|---|---|
| Fe3O4/phenolic resin | Not used | Water | Not used | Basic | Hydrothermal | Magnetic mesoporous Fe/carbon aerogel | 101 |
| Water hyacinth | Not used | Phosphoric acid solution | Not used | Acidic | Chemical activation | Mesoporous activated carbons | 102 |
| Phenolic resin; TEOS | HCl | Ethanol | F127 | Acidic | EISA | — | 103 and 104 |
| MF resin/silica composite | Not used | Water | Silica colloid | Neutral | Nanocasting | N-enriched MC | 105 |
| Urea–phenol–formaldehyde (UPF) resin | NaOH | Water | F127 | Basic | Dilute aqueous; hydrothermal | N-doped OMC | 106 |
| Phenolic resin; Ca(NO3)2·4H2O | Not used | Ethanol | F127 | Neutral | EISA | MC-supported nanocrystalline calcium oxides | 107 |
| Chitosan | H2SO4 | Acetic acid; hydrochloric acid | SBA-15 | Acidic | Nanocasting | — | 108 |
| P123; TEOS | H2SO4 | Water | P123 | Acidic | Phase separation | — | 109 |
| RF resin | HCl | Ethanol | F127 | Acidic | Phase separation | — | 110 and 111 |
| Phenolic resin | NaOH | Ethanol | F127; AAO membrane | Basic | EISA; nanocasting | MC nanofibers | 112 |
| Ferrocene | Not used | Not used | Mesoporous silica spheres | Not used | Chemical vapor deposition | Monodisperse MC spheres | 113 |
| Phenolic resin; dicyandiamide; TEOS | HCl | Water; ethanol | F127 | Acidic | EISA | N-doped OMC | 114 |
| Fructose; urea | Not used | Not used | Mesoporous KIT-6 silica | Neutral | Nanocasting | N-/O-modified MC | 115 |
| Phenolic resin | NaOH | Ethanol | F127; cordierite | Basic | EISA; nanocasting | Honeycomb monolithic OMC | 116 |
| Phloroglucinol–terephthalaldehyde (PT) resin | HCl | Ethanol | F127 | Acidic | EISA | Interconnected hierarchically ordered micro/mesoporous carbon | 117 |
| MF resin | HCl | Water | Commercial fumed silica (Aerosil-200) | Acidic | Nanocasting | Monodisperse N-containing MC spheres | 79 |
| Sucrose | H2SO4 | Water | SBA-15 silica | Neutral | Nanocasting | — | 118–121 |
| Furfuryl alcohol | Not used | TMB or ethanol | SBA-15 monolith | Neutral | Nanocasting | Hierarchical macro/mesoporous carbon monoliths | 122 |
| RF resin | Na2CO3 | CTAB | CaCO3 | Basic | Hydrothermal | MC aerogels | 83 |
| Gelatin | Not used | Water | CaCO3 | Neutral | Hydrothermal | N-doped MC | 85 |
| Phenolic resin | Not used | THF | Poly(ethylene oxide)-block-poly(styrene) (PEO-b-PS) | Neutral | EISA | Ordered large-pore MCs | 88 |
The table mentioned above indicates that some “no template” methods for the synthesis of MC can not be avoided chemical catalysis under conditions of heat while removing limits regarding the solvent. Acidic or basic catalysts can both play an effective role in the soft-template method, but interactions between the aggregated template and the carbon precursor such as hydrogen bonding, which is necessary for the formation of a supramolecular arrangement of molecules, require a weakly polar solvent environment for the soft-template method in some cases. In the hard-template method, although there are a variety of carbon precursors, some monomeric compounds still need to be polymerized under the action of a catalyst (sucrose and sulfuric acid, for example) to cover the surface or fill the pores of the sacrificial template via nanocasting or chemical vapor deposition as other polymers do. Most importantly, the nature and elemental composition of the precursors largely determine the properties of the obtained materials; some special morphologies cannot be achieved without the influence of a hard template.
As well as the synthesis of basic elements, many researchers have also made efforts in the improvement of the synthetic route. Generally speaking, OMCs can be synthesized using aqueous phase-induced self-assembly.123,124 However, it usually takes a long aging time (about 3–7 days) to complete the reaction. Therefore, a method in which the self-assembly and condensation process takes place rapidly at room temperature is highly desirable. Although some studies have made progress in both the polymerization of RF resins125 and the reaction time,100,126,127 strong acid conditions or relatively high temperatures are needed. In this field, the polymerization of resorcinol with formaldehyde can be catalyzed by both an acid and a base. The whole process can be divided into the production of hydroxymethyl compounds (the first step), which react rapidly at low temperatures in the presence of a basic catalyst,128 and the condensation reaction between the hydroxymethyl compounds (the second step), which has been considered as the rate-determining step under acidic conditions.129 Based on the hypothetical assumption that the reaction of resorcinol and formaldehyde is catalyzed by a base in the first step and then the condensation reaction is catalyzed by an acid in the second step, it is expected that highly cross-linked polymers can be obtained rapidly. Thus, Zhang et al. designed a new two-step method (Fig. 3A) for the rapid synthesis of ordered mesoporous RF polymers and carbons.130 Compared with the initially reported EISA method as well as the one-step liquid-phase self-assembly method, in the present method the production of the hydroxymethyl compounds and the condensation process can be carried out rapidly using low amounts of basic and acidic catalysts at room temperature. Even after activation by CO2, the carbon material still retained its ordered mesostructure (Fig. 3B), whereas the BET surface area and total pore volume were remarkably increased.
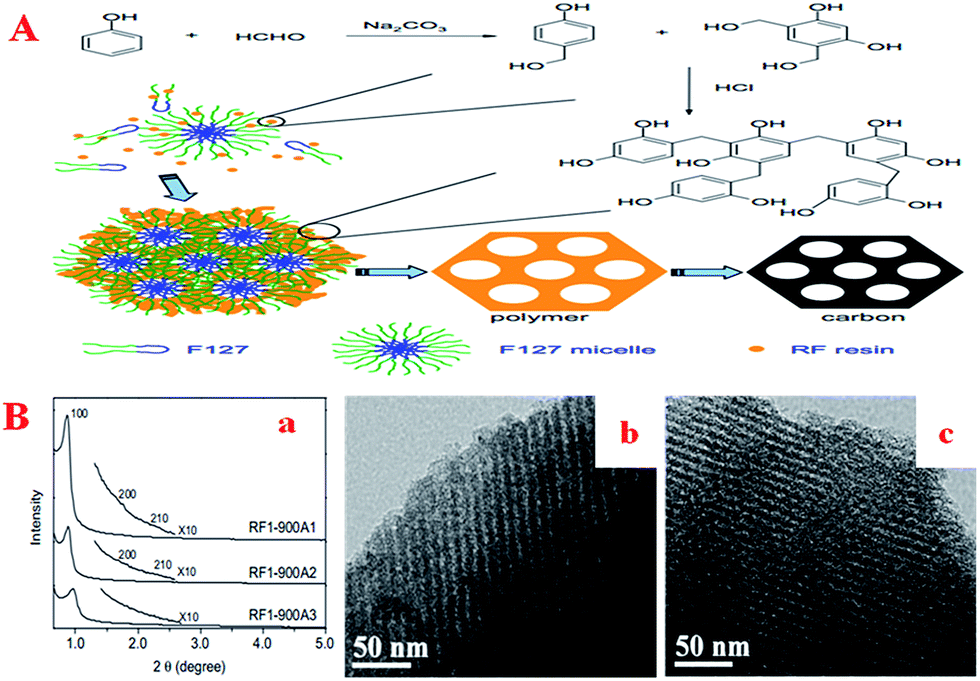 | ||
| Fig. 3 (A) Schematic of the two-step synthesis of ordered mesoporous RF polymer and carbon; (B) ordered mesostructure activated by CO2: (a) low-angle XRD patterns of OMCs after activation by CO2 at 900 °C for different times; TEM images of RF1-900A2 (b) and RF1-900A3 (c). Reprinted with permission from ref. 130. | ||
Here is a similar problem: because the condensation kinetics of phenol with formaldehyde is difficult to control in the directed assembly process, pre-polymerized PF oligomers with low reactivity instead of molecular precursors are always used as starting materials. Moreover, the hydrothermal pathway is faster and more energy-efficient than the non-aqueous EISA method and the dilute aqueous route. Therefore, a simple method of preparing OMCs under hydrothermal conditions using hexamethylenetetramine (HMT) as a source of formaldehyde during the synthesis was introduced by Lei et al. (Fig. 4A).100 In this process, HMT could be subjected to hydrolysis into formaldehyde and ammonia at elevated temperature or acidity, resulting in well-controlled organic–organic self-assembly. It is worth noting that this method omits the additional pre-polymerization step in the present process and less acid or base as a catalyst is added to the reaction system compared with the previous method, which possibly results in the formation of ordered mesoporous polymers and carbons with large surface areas by a one-pot procedure in an aqueous system. Furthermore, as research continued, Lei et al. modified the above method and made it simpler.131 In this new method, neither pre-polymerization nor thermal solidification was needed and hexamine still served as a source of formaldehyde. Consequently, a mesophase transformation from a body-centered cubic (Im3m) to a 2D hexagonal (p6m) structure could be achieved by simply adding 1,3,5-trimethylbenzene. Especially, this synthesis can be performed not only under weakly basic conditions but also under highly acidic conditions. After all, optimum control of the polymerization reaction of precursors is the basis of increasing the reaction rate, thus reducing the catalyst dosage and shortening the synthesis time.
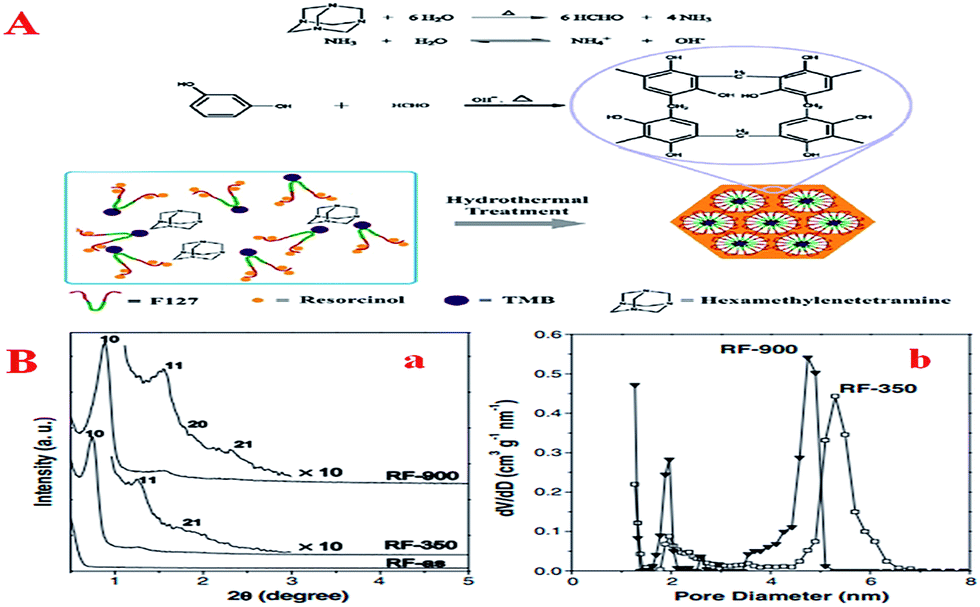 | ||
| Fig. 4 (A) Schematic of the formation of ordered mesostructured RF/F127 composites from resorcinol and hexamine; (B) characterization of RF MC: (a) XRD patterns of as-made RF (RF-as), RF calcined at 350 °C (RF-350) and RF calcined at 900 °C (RF-900), (b) pore size distributions of RF-350 and RF-900. Reprinted with permission from ref. 100. | ||
Another aspect that attracts considerable attention is the use of inorganic templates instead of a triblock copolymer; as reported, MCs with large surface areas have been prepared via soluble or hydrolysable carbohydrates with ZnCl2 as a template.87 This method has the advantages that the template is cheap and could be recycled and the MCs thus prepared are of high quality. The same method was also successful in the preparation of nitrogen-containing MCs with melamine–formaldehyde resin as a carbon source and CaCl2 as a suitable template.86 The resulting MCs containing 26% nitrogen that were prepared exhibited amphipathic surfaces (both hydrophilic and lipophilic) and adsorbed large amounts of water and benzene. In addition, the incorporated N atoms exhibited quite strong basicity for the adsorption of a large amount of SO2.
In general, CMK-8 is synthesized using a KIT-6 silica matrix, which belongs to the cubic system and possesses the Ia3d space group. This 3D structure facilitates better mass transfer kinetics in adsorption-based applications and is beneficial for obtaining carbon replicas. In order to optimize the synthesis conditions and composition of CMK-8, a simple one-pot route for the synthesis of MC with three-dimensional cubic symmetry (Ia3d) has been presented via organic–inorganic self-assembly of TEOS, Pluronic P123, n-butanol and sucrose in acidic conditions.132 In this process, the preparation of carbon could be completed with the simultaneous generation of a KIT-6 silica composite, which not only simplified the preparation process but also eliminated the complex pre- or post-treatment techniques that were widely reported.
In summary, the most impressive fact is that a stable resol precursor enables the co-assembly of multiple components to fabricate mesoporous composite materials and avoids the aggregation of nanoparticles in the final carbon framework with good dispersion.107,133–135 Again, this versatility in the synthetic procedure also provides a facile route to a variety of ordered mesoporous metal–carbon and metal oxide–carbon materials.98,136,137
2.2 The synthesis of special morphologies
With a narrow distribution of pore size and regular arrangement, MCs are available with various morphologies, such as rods,138 spheres,139 single-crystal-like forms,140,141 fibers,142 films,143,144 monoliths or foams,145 etc. This section mainly focuses on the special cases.Typically, MC films are fabricated on rigid substrates that act to provide mechanical stability. Instead of utilizing rigid substrates, a thin sheet of polyimide (Kapton), which is thermally stable, could also serve as a substrate to enable the continuous production of sheets of soft-templated MC films.135 More importantly, these materials were quite robust and could be cut to shape after carbonization, which provides a method of shaping mesoporous carbon films for applications after carbonization. Furthermore, to obtain large-pore (>10 nm) MC films, solvent vapor annealing with a soft shear method (SVA-SS) as a facile method of macroscopic alignment of a block copolymer (BCP) has been presented.151,152 Two different BCP templates, PEO-b-PBA and PS-b-PSS-DMODA, that were used in combination with SVA-SS to align MC films with a relatively large pore size (>10 nm) with both cylindrical and spherical nanostructures from each template, achieved broad applicability.153 It is noteworthy that highly aligned cylindrical MC films exhibited a narrower pore size distribution and anisotropic electrical conductivity, with a 20% increase in conductivity parallel to the alignment direction when compared to an analogous MC film without alignment.
Very recently, OMC membranes have attracted considerable attention due to their advantages such as high thermal stability, chemical stability and wide applications such as size exclusion separation of molecules. Especially, the hollow fiber morphology is a preferred geometry in industrial-scale applications because of its high packing density and easy modular assembly. With the assistance of commercialized polymeric hollow fiber ultrafiltration membranes, OMC hollow fiber membranes could be obtained via a confined soft-templating route in an ethanol solution that contained a phenolic resin and a Pluronic triblock copolymer (Fig. 5).154 After the processes of solvent evaporation, drying and pyrolysis, the OMC hollow fiber membranes possessed continuous membrane walls with an average thickness of 113 μm. Surprisingly, the membrane wall had a hierarchical pore structure: one coming from hexagonal OMC with a pore diameter of ∼4.3 nm and the other being disordered defect holes with a size of 8–50 nm randomly distributed inside the OMC matrix.
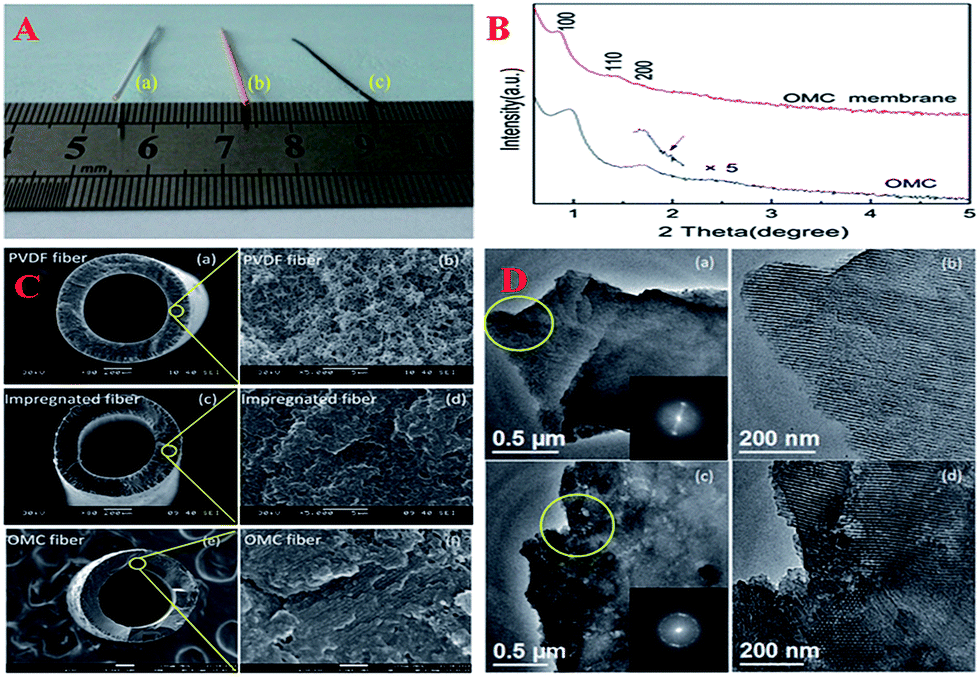 | ||
| Fig. 5 (A) Digital image of a PVDF hollow fiber membrane (a) impregnated hollow fiber membrane (b) and OMC hollow fiber membrane (c); (B) small-angle XRD patterns of unsupported OMC membrane and OMC hollow fiber membrane; (C) SEM images of PVDF hollow fiber membranes (a and b), impregnated hollow fiber membranes (c and d) and FESEM images of OMC hollow fiber membranes (e and f); (D) TEM images of unsupported OMC membrane (a and b) and OMC hollow fiber membrane (c and d). The insets are the corresponding FFT diffractograms. Reprinted with permission from ref. 154. | ||
Unlike most OMC membranes that are prepared from synthetic phenolic resins using a soft-template strategy, the liquefaction of widely available and renewable biomass such as waste wood as a substitute for phenol in the synthesis of novel carbon materials with specific morphology and porous structure has proved promising. Liu et al. prepared OMC membranes via liquefaction, resinification, assembly and carbonization steps using natural renewable larch sawdust as the starting material and triblock copolymer F127 as the template.155 The mesostructure that was formed by the assembly of larch-based resin with F127 was controllable from disordered to ordered by varying the carbonization temperature. Moreover, some MC composite membranes have also emerged in this context: two good examples of ordered mesoporous silica/carbon composite membranes and mesoporous carbon–graphitic carbon nanocomposite membranes both exhibited outstanding permeability and selectivity for the separation of mixed gases.156,157 Additive components play an important role in developing and optimizing the pore structure for special applications. In addition, some free-standing MC membranes were synthesized by casting a homogeneous polymeric composite solution on a hydrophilic Mylar sheet via a tape-casting technique.158,159 After drying and curing, the free-standing polymer film could be detached from the Mylar support and cut to the right size. Furthermore, an MC aerogel membrane with a thickness and average pore size of approximately 10 μm and 10 nm, respectively, that was made via a sol–gel process has the advantages of low cost, high mechanical strength and reusability compared to polymeric and ceramic membranes.160
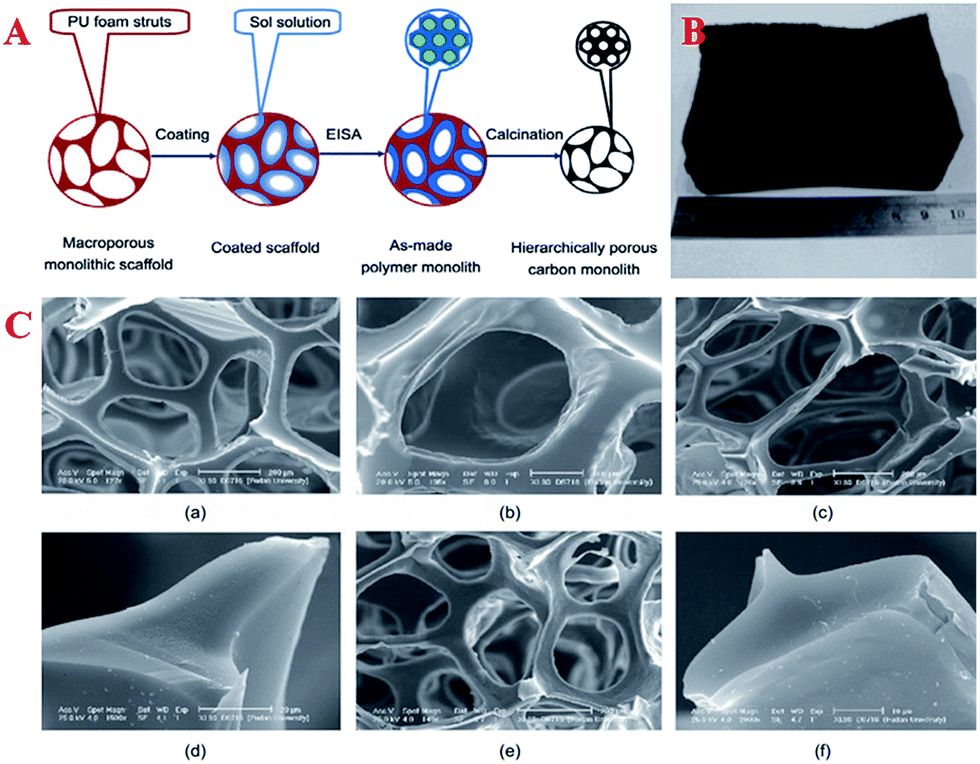 | ||
| Fig. 6 (A) Fabrication of hierarchically porous carbonaceous monoliths templated by PU foams via an EISA method; (B) image of the final hierarchically porous carbonaceous product; (C) SEM images of (a) PU foam scaffold; (b) hierarchically porous carbonaceous monolith HPCM-2-130 prepared by an EISA method on a PU foam scaffold; (c) hierarchically porous monolith HPCM-2-600 obtained by calcination at 600 °C; (d) cross-section of struts of HPCM-2-600; (e) hierarchically porous monolith HPCM-2-900 obtained by calcination at 900 °C; (f) cross-section of struts of HPCM-2-900. Reprinted with permission from ref. 52. | ||
Usually, nanocasting methods using mesoporous silicas as a “hard template” have been the traditional way of preparing these monolith materials. Hierarchically macro/mesoporous carbon monoliths with an extraordinarily high cumulative macropore volume (48.6 cm3 g−1), large specific surface area (1354 m2 g−1), ordered hexagonal mesoporous structure and regular cylindrical shape have been successfully synthesized using silica monoliths as a template followed by a nanocasting pathway via a simple and time-saving hydrothermal process.122 However, this method makes the undesirable extra step of the removal of silica necessary. Therefore, Wiesner et al. first presented a silica-free method of producing well-defined, ordered, pure MC monoliths with tunable macroscopic dimensions via heat treatment of cured organic–organic hybrid monoliths.91 On the other hand, another novel way of forming carbon monoliths is the “soft-template” method. This method is more flexible and has achieved success by using a hydrothermal approach in a conventional oven or the EISA method.165,166 The macropores and micrometer-sized mesoporous walls in hierarchical carbons provide sufficient pathways for the release of decomposition gases and good strain relaxation upon carbonization. However, the long processing time and high cost of the synthetic protocol make it difficult for this method to be employed for the large-scale manufacture of these materials. Compared with typical aqueous chemical processes, microwave heating is faster, cheaper and more energy-efficient. Therefore, Elaigwu et al. developed a new microwave-assisted hydrothermal synthesis method for the preparation of MC monoliths via a soft-template approach.167 This method appears to be simpler and much faster compared to hydrothermal methods that use a conventional oven, as it effectively reduces the synthesis time from hours to a few minutes, which could be an advantage in the large-scale production of the material. A common tool that is used to synthesize inorganic materials by transferring the energy of sound waves to the reaction system and consequently generating bubbles is ultrasound, not only because its intrinsic properties are consistent with the requirements of the principles of green chemistry, but also due to its surprising synergetic effects, which result in an improvement in reaction efficiency in terms of product quality, reaction rate and time. In order to develop a simple and scalable synthesis strategy for the preparation of hierarchical carbon monoliths with intrinsic macropores and mesopores, Lu et al. adopted an ultrasound-assisted air bubbling method for the fabrication of carbon monoliths with a foam-like macropore system and mesopore structure without the aid of an inorganic foaming agent and solid templates.168 The synthesis involved ultrasound-assisted bubbling of air through a poly(benzoxazine-co-resol)-based solution to form emulsions that were stable in air, followed by a fast polymerization step at 90 °C (Fig. 7A). During the process, the ultrasound treatment initiated the rearrangement of the surfactant F127 and induced the generation of a defective mesostructure. With the assistance of ultrasound, the mesopore sizes increased from 4.8 to 5.9 nm (inset in Fig. 7C). A weak and broad peak in the low-angle XRD pattern represents the local mesostructure of crack-free foam-like carbon monolith (FMCM-U), which corresponds to the short-range order of a mesostructure. On the contrary, there is no reflection observed in the XRD pattern of foam-like carbon monolith (FMCM), which indicates a worm-like pore arrangement (Fig. 7D). Polymerization-induced phase separation (PIPS) is also an important manufacturing route for the production of multiphase materials. Many types of MCs have been prepared by combining PIPS at constant temperature with subsequent pyrolysis and the morphological type of the obtained MC is largely dependent on the phase separation. These presented results have given rise to the production of MC monolith by a facile route based on polymerization-induced phase separation under a temperature gradient (TG).169 First, a graded biphasic structure of phenolic resin-rich and ethylene glycol-rich phases is formed in precursor form under a pre-curing temperature gradient and then the precursor form is pyrolyzed to obtain the MC monolith. The pore size, apparent porosity and specific surface area of the monolith change gradually along the TG and the phase separation kinetics at varying temperatures may further reveal the gradient porous structure of the monolith.
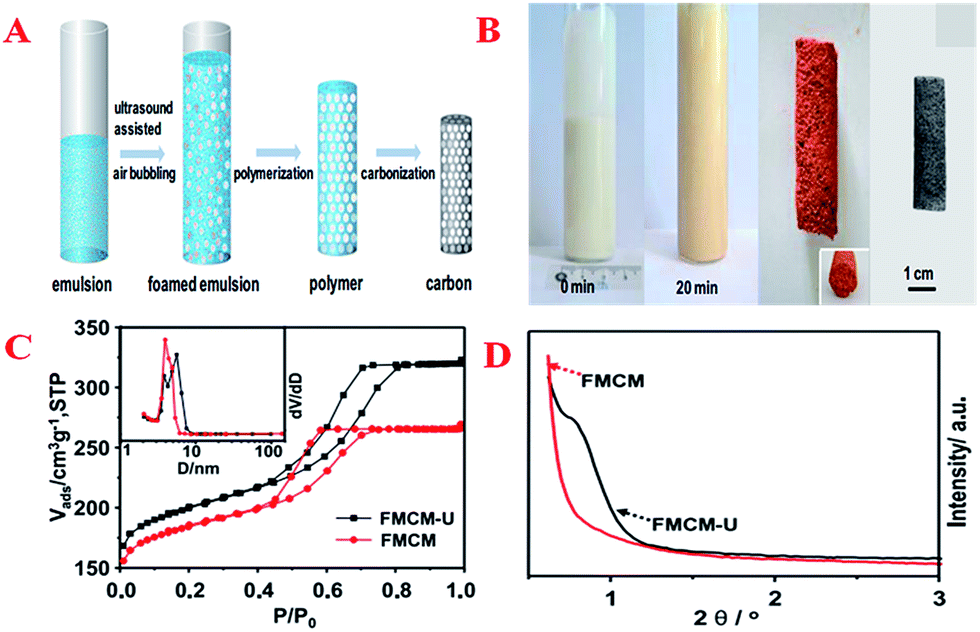 | ||
| Fig. 7 (A) Illustration of the preparation of foam-like mesoporous carbon monolith via an ultrasound-assisted air bubbling strategy; (B) images of the obtained foam-like polymer and the corresponding carbon monoliths; (C) N2 sorption isotherms of FMCM and FMCM-U (the inset is the corresponding PSDs); (D) low-angle powder XRD patterns of FMCM and FMCM-U. Reprinted with permission from ref. 168. | ||
The production of porous carbon materials from cheap natural precursors by environmentally friendly processes is a hot topic in modern materials science research. To reduce the cost of precursors, Elaigwu et al. employed a readily available and sustainable organic waste material (Prosopis africana shell) as a precursor for the synthesis of crack-free MC monolith by the EISA method.170 They provided a cost-effective strategy for the production of an advanced material via a green process that scales down and minimizes the volume of chemicals used by converting waste material into a useful product. The MC monolith with good mechanical stability had a surface area of 219 m2 g−1 and a narrow pore size distribution of 6.5 nm.
Carbon foam is a sponge-like material with advantageous features such as low density (0.2–0.8 g cm−3), a large external surface area and an open cell structure. Related reports indicated that carbon foams with a developed pore structure and consisting of textural and framework pores are superior to those with unimodal framework pores. In recent years, carbon foams with particular properties have been developed for use as catalyst supports, adsorbents for liquid or gas purification, porous electrodes and other battery components. In general, the most commonly produced carbon foams can be made by carbonization of polymeric foams. The polymeric foams that are typically used include polyurethane, furfural resin, phenol–formaldehyde, polyvinylidene chloride, and polyacrylonitrile. Carbohydrates, as a replacement for synthetic materials, can be adopted as potential suitable carbon precursors for the preparation of carbon foam as they easily undergo dehydration to produce carbon. For example, carbon foam with a bimodal micro/mesopore distribution was prepared by transforming larch sawdust by liquefaction, resinification, foaming, and carbonization at different temperatures.171 After activation by KOH, a well-connected 3D network and a developed ligament/pore structure (surface area of 554–1918 m2 g−1) containing bimodal pores of 2.1 and 3.9 nm in diameter were obtained. A carbon foam with a bimodal micro/mesoporous structure that was prepared at 700 °C exhibited much higher removal of toluene in the gas phase than commercial activated carbon fiber. Moreover, the synthesis of carbon foam can also be achieved by a templating method that may allow the formation of foam-shaped carbon monolith using a foam-shaped template and a suitable precursor. In this way, commonly available polyurethane foam and zeolite foam have been used as templates for the preparation of foam-shaped carbon monolith.172 This template-coating approach mainly involved polymerization of sucrose on the template surface with the aid of sulphuric acid. The obtained carbon foams with hierarchical porosity (involving micro- and mesopores) exhibited a characteristic cellular structure of foams as a faithful replica of their respective templates and displayed an enhancement in compressive strength from that of their respective templates. In addition, a Pechini method is based on intensive blending of positive ions in a solution, controlled transformation of the solution into a polymer gel, removal of the polymer matrix and development of an oxide precursor with a high degree of homogeneity. Zhang et al. developed a modified Pechini method and successfully synthesized MC foam with a large specific surface area and suitable pore size distribution via a facile, cost-effective and template-free Pechini method.173 This modified Pechini method can be extended to fabricate other types of MC foam by changing metal salts and organic reagents. To improve the texture of materials, Gan et al. used a cobalt-oxo cluster 2[Co3O(Ac)6(H2O)3]·H2O (Co-OXO) as a precursor to prepare Co-containing MC foams for the first time, which exhibit a highly ordered mesostructure with a specific surface area of 614 m2 g−1 and a uniform pore size of 2.7 nm.174 During the carbonization process, Co-OXO was transformed into metallic cobalt nanoparticles embedded in the carbon matrix. In this way, the method provided an effective strategy for the incorporation of cobalt nanoparticles into MC foams.
 | ||
| Fig. 8 (A) SEM images (a–d) and TEM images (e and f) of ordered MC-MSs prepared by a confined self-assembly method; (B) SEM image of the obtained MC-MS matrices; (C) TEM image of a Co–MC-MS sample. Reprinted with permission from ref. 176. | ||
Moreover, carbon nanoparticles with ordered mesostructures and controlled morphologies have great advantages due to their remarkable and complementary properties of mesochannels and quantum effects on the nanoscale. Many efforts have been made to fabricate mesoporous carbon nanospheres (MCNs). However, the synthesis of MCNs, especially with diameters below 200 nm, remains a great challenge due to weak interactions between the carbon precursors and soft templates, as well as the uncontrollable cross-linking rate of carbon precursors. Zhao et al. developed a low-concentration hydrothermal route to synthesize highly ordered body-centered cubic (Im3m) MCNs with a uniform particle size (20–140 nm) for the first time,178 initiating the controlling synthesis of MCNs using a soft-template method. This method used a very low concentration of templating agent to decrease the cross-linking rate so as to match the weak interactions between the carbon precursor and template. However, this method is a multi-step procedure and needs a large volume of water to dilute the precursor solution, which limits large-scale synthesis. By extending the synthesis method of mesoporous silica nanoparticles,179 Zhao et al. put forward an efficient, facile, general and environmentally friendly method for the synthesis of mesoporous polymer nanospheres (MPNs) and MCNs with ordered mesopores, a controllable particle size from 80 to 400 nm and morphologies (Fig. 9A).180 By finely tuning the synthesis parameters, hollow multi-layered mesoporous resorcinol–formaldehyde nanospheres could be successfully synthesized. On the same lines, MCNs and hollow nanospheres with a large surface area were further obtained via carbonization of polymer spheres. In many common cases, the synthesis of MCNs takes place under alkaline conditions, whereas this method operates under acidic conditions, which enhances the hydrogen-bonding interaction between the template molecules and the carbon precursor. In another special case, which is similar to the “I+X–S+” mechanism for the synthesis of mesoporous metal oxides under highly acidic conditions via an enhanced hydrogen-bonding interaction presented by Dai et al., as illustrated in Fig. 9B, Wang et al. developed another simple acid-assisted organic–organic self-assembly approach to synthesize uniform MCNs via direct hydrothermal treatment of phenol–formaldehyde resol and the triblock copolymer Pluronic F127 under highly acidic conditions (2 M HCl).181 Because this method can be used to prepare MCNs at much higher concentrations of F127 (almost 104 times higher than those reported before),178 the enhanced hydrogen-bonding interaction via coulombic interactions with Cl− (as a mediator) due to the increased mass ratio of the template to resol and a more highly acidic environment can further reduce the cross-linking rate of resol molecules that are trapped in the triblock copolymer micelles and thus avoid macroscopic phase separation during the hydrothermal synthesis. In this way, the MCNs displayed a large surface area (596 m2 g−1), a large pore volume (0.77 cm3 g−1) and a highly controllable diameter ranging from 20 to 150 nm, simply by changing the mass ratio of the template to resol. Unexceptionally, with regard to the hard-template method, mesoporous silica nanospheres that were employed as a template have also achieved success in the synthesis of MCNs with a particle size of about 65 nm and primary mesopores of about 2.7 nm in diameter by carbonizing a silica/ferrocene composite.113 With respect to morphological control, as the morphology of the solidified composite polymer particles is determined by the respective original morphology of the F127 micelles, highly ordered MCNs with well-controlled morphology from spherical and worm-like to rod-like structures could be obtained by controlling the concentration of F127 templates.182
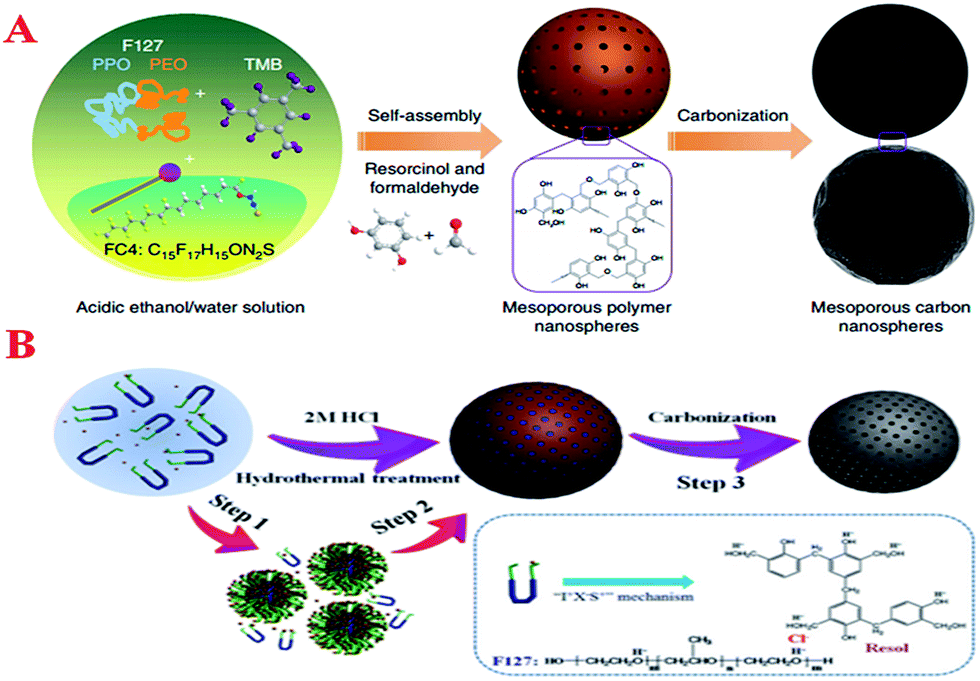 | ||
| Fig. 9 (A) Schematic of the formation of MCNs and self-assembly of mesoporous polymer spheres followed by carbonization for the synthesis of MCNs. Reprinted with permission from ref. 180; (B) schematic of the formation of uniform MCNs via an acid-assisted organic–organic self-assembly process. Reprinted with permission from ref. 181. | ||
Hollow MC spheres are generally fabricated using sacrificial templates because this method allows control of the pore structure and morphology of the resulting carbon materials after infiltration of hard mesoporous templates with carbon precursors and then carbonization under non-oxidizing conditions to etch the templates and generate porous carbon. Here, the carbon precursors are usually sucrose, furfuryl alcohol, and phenol–formaldehyde resin. Early in 2002, Yoon et al. reported the fabrication of hollow carbon spheres with a mesoporous wall and aluminum must be incorporated into a silicate framework before introduction of the carbon source.183 In a similar way, Shi et al. reported the direct synthesis of hollow MC spheres with highly ordered, three- dimensional cubic mesostructured channels in their shells by employing simple incipient-wetness impregnation with furfuryl alcohol as the carbon precursor and cubic hollow mesoporous aluminosilicate spheres as a hard template.184 In this way, aluminum that is present in the wall of hollow mesoporous aluminosilicate spheres could in situ catalyze the polymerization of furfuryl alcohol in the mesopore channels, allowing a direct and faithful replica of the hollow mesoporous structure to be made and avoiding the conventional catalyst-loading step or acidic catalyst. This team also achieved the modulation of the size (between 80 and 470 nm) of hollow MC spheres by simply changing the diameters of a solid silica core/mesoporous silica shell aluminosilicate template.185 The adjustment of the template is an effective way to control the diameter of hollow MC spheres. Yu and Zhang synthesized nitrogen-doped hollow MC spheres with uniform and tunable morphology by a simple method of impregnation of a silica template with carbonization, using an ionic liquid as the precursor and monodisperse silica spheres as a hard template.186 The size and shell thickness of the obtained hollow MC spheres are uniform and can be adjusted by controlling the molar ratio of ionic liquid: silica spheres. By twin polymerization on hard templates from spherical SiO2 particles, Spange et al. presented a universal synthetic approach that allows the design of hollow MC spheres with adjustable size and micro- or mesoporous shells using different monomers.187 The size of the produced hollow MC spheres is determined by the template particles, whereas the carbon content and shell thickness of the resulting hollow MC spheres can be adjusted by varying the amount of monomer. A second way is the nanocasting chemical vapor deposition (CVD) method, in which the carbon source is usually styrene, acetonitrile, or benzene. Recently, Xia and Mokaya reported the synthesis of hollow MC spheres with one-dimensional pore channels using conventional particulate mesoporous silica as hard templates by a CVD method.188 With the aid of the hollow mesoporous silica spheres, the synthesis of hollow MC spheres became easy.189 Chen et al. fabricated hollow MC spheres from core/shell-structured mesoporous silica sphere templates using chemical vapor deposition.190,191 Differently from the previously reported methods, this method only needs one CVD process to produce carbon-filled silica spheres and does not need the surfactant to be removed or replaced with carbon sources. More importantly, CTAB that is trapped in the silica channels could accelerate the deposition of carbon during CVD, resulting in more carbon filling the silica channels and controllable diameters. Another important way is the hydrothermal method. Zhang et al. fabricated hollow MC spheres with a hierarchical pore structure using phenolic polymer-coated polystyrene spheres as templates by a hydrothermal reaction.192 By one-step thermal treatment with sulfuric acid, SO3H-functionalized hollow MC spheres with uniform hollow morphology and developed mesostructure were successfully synthesized via a facile strategy using inexpensive resins as precursor in combination with a metal oxide template.193
Porous carbon nanofibers have received much attention owing to their thermal and mechanical stability, many surface-active groups, high surface-to-volume ratio and low ohmic resistance. Despite their excellent properties, it is difficult to precisely and simultaneously reproduce their pore texture and morphology on a mesoporous scale, which has greatly encouraged many studies to focus on producing MC nanofibers. Originally, a free-standing MC nanofiber array on a silicon wafer was prepared by a confined self-assembly process within the pores of AAO membranes.194 However, this typical procedure involves the preparation of ordered mesoporous silica templates, impregnation of carbon precursors, carbonization, and removal of the templates with hydrofluoric acid (HF) solution, which are very complex and entail high cost. Compared with artificial templates, biological templates are generally abundant, renewable, inexpensive and environmentally benign. For example, crab shell, which has a well-aligned porous mesostructure, is a perfect candidate as a template for generating hierarchical structures with uniform morphology. Xia et al. prepared highly ordered OMC nanofiber arrays by combining the surfactant-templated self-assembly of resol with a crab shell templating process, in which crab shell was adopted as a hard template for the formation of nanofiber arrays and P123 was used as a soft template for mesopores.195 The obtained MC nanofibers (70 nm in mean diameter and 11 nm in mesopore diameter) retain an interpenetrated ordered array, an interspacing void of 70 nm between nanofibers, and 1 micrometre of pores between nanofiber arrays. This team also found that this unique hierarchical porous structure contributes to attractive capabilities as a promising material in energy storage and conversion; for example, this type of OMC nanofiber arrays coated with ruthenium oxide displayed outstanding performance as a catalyst for lithium–oxygen batteries.196 After that, this crab shell templating method has also been pursued and developed by other researchers.197,198 To develop a facile template-free method for the preparation of MC nanofibers, Li et al. reported a novel self-templating method for the preparation of MC nanofibers with a 3D interconnected mesoporous structure and large surface area using ethylene glycol as the carbon precursor and Zn(CH3COO)2 as a structure-building agent and porogen.199 Electrospinning is a mature technology used in the production of fiber materials, which has been successfully employed to fabricate MC nanofibers. The typical precursor is a homogeneous solution of polyacrylonitrile (PAN) in N,N-dimethylformamide (DMF).200 Because PAN and poly(methyl methacrylate) (PMMA) exhibit stable, emulsion-like phase separation in DMF and have different thermal stabilities, Yang et al. fabricated hierarchical porous MC nanofibers with embedded graphene using PAN fibers containing graphene as the carbon precursor and PMMA as a pore-forming agent by an electrospinning method.201 The obtained MC nanofibers have good morphology and superior material properties, such as large surface area, the presence of micro- and mesopores, and increased electrical conductivity due to the dispersion of graphene. Also using electrospinning technology, Song et al. prepared 1D hierarchical macroporous/mesoporous carbon nanofibers using PAN as the carbon precursor and commercially available nano-CaCO3 as a dual-purpose template.202 During the carbonization process, the nano-CaCO3 template decomposed and released CO2 to develop mesopores, then macropores were generated by the subsequent removal using acid of the as-formed CaO nanoparticles. This type of carbon nanofibers combines the advantages of interconnected macro- and mesopores and has displayed excellent performance as a catalyst support for Pd nanoparticles in liquid-phase catalysis. Han et al. fabricated MC nanofibers with large cage-like pores via thermal treatment of electrospun fibers of polyvinyl alcohol containing a tin compound.203 When the fibers were heated over 400 °C, mixtures of Sn and SnO with rod-like shapes appeared in the matrix, then SnO and SnO2 were reduced to Sn along with the consumption of carbon and the melting Sn migrated out of the carbon fibers forming pores in the fibers at higher temperatures. The specific surface area of these MCFs can reach 800 m2 g−1 with the average diameter of the interior pores being about 10.3 nm.
The tubular morphology and high aspect ratio of carbon nanotubes could induce a confinement effect in gases or liquids that are trapped inside the tube, leading to completely different physical behavior when compared to the bulk material. In addition, the small size of carbon nanostructured materials significantly contributes to the final catalytic performance of systems, as catalytic reactions are governed by the phenomena of mass and heat transfer between the catalyst particles and the reactants. Therefore, MC nanotubes have begun to receive more attention in recent years and have been fabricated via templating approaches involving SiO2 nanoparticles,204 anodised aluminum oxide,205 polysulfone,206 ZnO nanorods,207,208 triethylamine hydrochloride nanocrystals,209 et al. Most of these were decorated with other metal nanoparticles and exhibited enhanced performance in energy storage and conversion applications.
An aerogel is a kind of solid material form with the lowest solid density in the world and different kinds of matrix such as silicone, carbon, sulfur, metal oxide, metal, etc. Considering the avoidance of the traditional supercritical drying step and solvent-exchange step, a low-cost approach for the synthesis of MC aerogel using calcium carbonate particles as templates was presented, which provided a low-cost and easy-to-scale-up method for producing MC aerogels with large pore volumes and tailored pore structures in conditions of ambient pressure drying.83
Overall, although some progress in the control of morphology has been made, there is still some elaborate work that needs to be done. The fabrication of an MC film or membrane cannot be separated from the support, so finding a suitable support or free-standing method to ensure high mechanical strength of an MC membrane will be an important trend in the future. On one hand, the sacrificial template is still a common method for the preparation of MC monoliths or foams; on the other hand, the soft-template method has the problems of a lengthy synthesis with high cost, whereas some green synthesis methods, such as microwave-assisted hydrothermal synthesis and ultrasound-assisted air bubbling, could enhance the synthetic efficiency. In addition, template-free methods including PIPS for MC monolith and the Pechini method for MC foam will produce a residual product that is mixed with large micropores and a wide pore size distribution. Gaining a clear idea of the kinetics of phase separation is the key point for the better utilization of PIPS. The synthetic mechanism of MC nanospheres is still unclear; maybe effective control of the cross-linking rate between the carbon precursor and template to match the interaction between them is an effective method. The hard-template method, including typical hollow mesoporous aluminosilicate spheres and core/shell-structured mesoporous silica spheres, is still the main route for the synthesis of hollow MC spheres, whereas the soft-template method has rarely been reported. Among these morphologies, MC nanofibers have implemented a real template-free method in the form of electrospinning, but its prospects for industrialization are not optimistic because of the discontinuous fibre products and low yield. After all, cheaper and environmentally friendly expendable templates will be pursued intensively for better development of the control of special morphologies via the hard-template method.
2.3 Synthesis from biomass or waste
The production of porous carbon materials from cheap naturally occurring precursors via environmentally friendly processes is a hot topic in modern materials science research. Waste management is an issue of global dimensions that is intensified by population growth and the pattern of resource utilization. The utilization of horticultural waste for producing new resources to apply in various fields such as environmental remediation is attractive because it offers solutions for the management of solid waste, reduces the cost of raw materials and addresses environmental issues. In principle, like the preparation of conventional porous carbon materials, MCs can also be synthesized by pyrolysis and physical or chemical activation of organic precursors at elevated temperatures.The unique physicochemical properties of hollow carbon spheres (HCSs) such as low density, large inner spaces and specific surface area make them widely used as a catalyst support, adsorbent, storage medium and template for the synthesis of other useful hollow materials. Hitherto, great efforts have been made to convert virgin or waste plastics including polypropylene (PP), polyethylene (PE) and polystyrene (PS) into high-value-added carbon nanomaterials.216–219 In this regard, uniform mesoporous HCSs with controllable diameter and large surface area were first prepared by the carbonization of mixed plastics consisting of PP, PE and PS under the catalysis of organically modified montmorillonite (OMMT)/Co3O4 at 700 °C (Fig. 10A).62 During this process, OMMT not only promoted the dispersion of Co3O4 in the mixed plastics (Fig. 10B(c) and (d)), which favored control of the diameter of HCSs, but also promoted the degradation of mixed plastics into light hydrocarbons and aromatics, which facilitated the growth of HCSs.
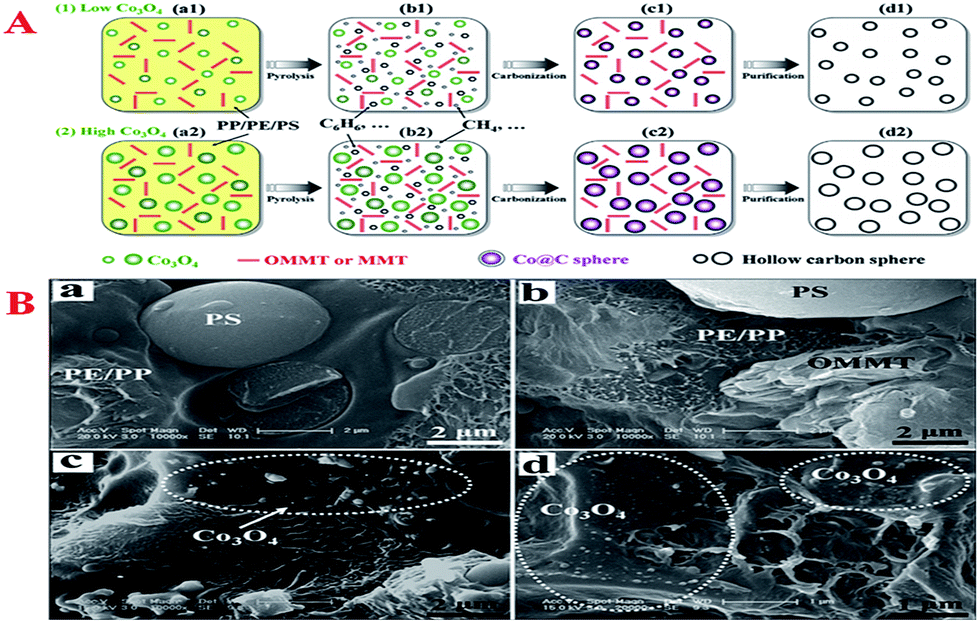 | ||
| Fig. 10 (A) Possible mechanism of the conversion of mixed plastics (PP, PE and PS) into uniform HCSs with controllable diameter under the combined catalysis of OMMT/Co3O4 at 700 °C; (B) Typical SEM images of polymer (mixed plastics) (a), polymer/OMMT (b), polymer/Co3O4-10 (c), and polymer/OMMT-Co3O4-10 (d). Reprinted with permission from ref. 62. | ||
Besides ZnCl2, H3PO4 and KOH are also commonly used as effective activators. The renewable and economical biomass material Lemna minor, known as duckweed, can be used as a potential low-cost precursor to fabricate activated carbons due to its lignin-cellulose structure and to improve the economic viability of the adsorption process. Huang et al. fabricated mesoporous activated carbon with abundant hydroxyl, carboxyl, amide and phosphate surface functional groups, high mesoporosity (92.2%) and a large surface area of 531.9 m2 g−1 from Lemna minor using one-step H3PO4 activation.220
Lignin is the third most abundant natural polymer, next to cellulose and chitin, and ranks as one of the most abundant phenolic natural polymers. The availability of plentiful hydroxyl groups in lignin macromolecules and their low cost make them preferred precursors for the synthesis of sustainable MC. Naskar et al. utilized a solvent-swollen gel of acid-treated, pre-cross-linked hardwood lignin as a carbon source and further modified its morphology by employing the templating agent Pluronic F127 to introduce micelles of a surfactant in the lignin matrix.221 After removal of the solvent and further cross-linking of the matrix, nearly 80% mesoporosity was achieved by following a slow heating process through pyrolysis and carbonization. Furthermore, the resulting MC was further activated by treatment with either CO2 or KOH at high temperatures to enhance its porosity.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles).
000 cycles).With regard to continued efforts toward the environmentally friendly disposal of biomass and resource recovery, Yu et al. proposed a facile and sustainable approach to synthesize MgO nanoparticles (mPC–MgO) that were stabilized by MC via fast pyrolysis of waste biomass loaded with MgCl2 (Fig. 11A).222 As shown in Fig. 11B, biomass loaded with MgCl2 can be easily obtained by adsorption of MgCl2 from seawater using biomass as a sorbent. In the fast pyrolysis process, MgCl2 was hydrolyzed and decomposed to form MgO nanoparticles (Fig. 11C), while the biomass was decomposed and carbonized to form MC, which could serve as a support to stabilize the MgO nanoparticles. Furthermore, another pyrolysis product, bio-oil, which is a renewable liquid that can be used as a fuel or source of chemicals, was also produced. More importantly, mPC–MgO displayed excellent performance in the process of CO2 capture with a maximum capacity of 5.45 mol kg−1. Its capacity for CO2 capture remained almost unchanged in 19 cycles of reuse and it could be regenerated at low temperatures.
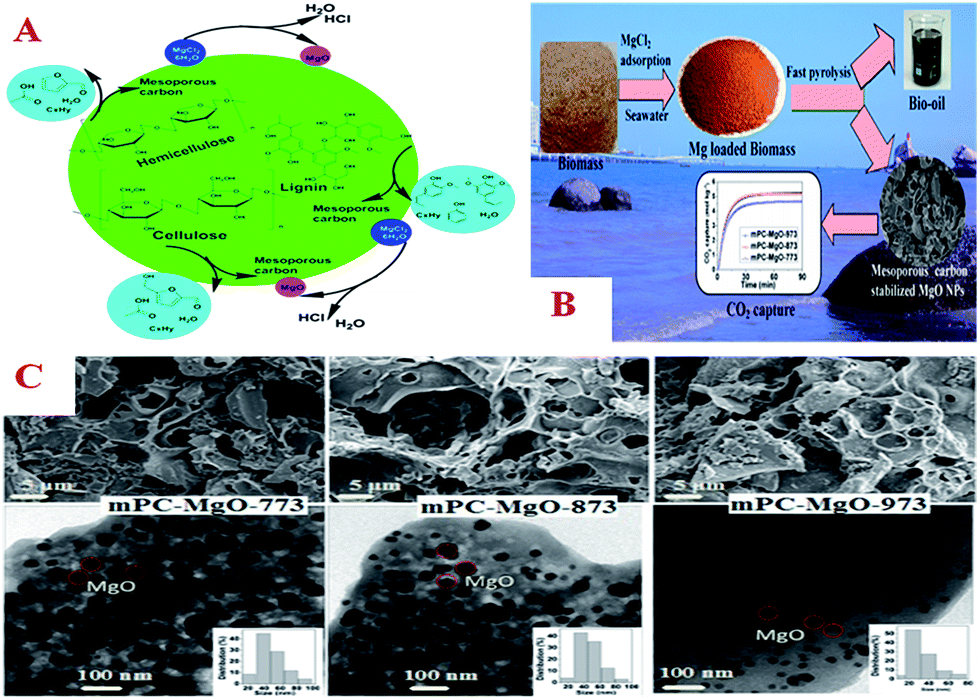 | ||
| Fig. 11 (A) Schematic of the mechanism of the decomposition of biomass and formation of MgO NPs stabilized by mesoporous carbon in a fast pyrolysis process; (B) process of the synthesis of MgO NPs stabilized by mesoporous carbon and their CO2 capture; (C) SEM (upper), TEM (lower) images, and particle size distributions (insets) of MgO NPs stabilized by carbon and prepared at different temperatures. Reprinted with permission from ref. 222. | ||
In addition, numerous MC materials can be prepared by the method of pyrolytic decomposition of organic precursors to obtain large surface areas and high pore volumes. In traditional pyrolysis methods, two polymers of appropriate mutual affinity but with different degrees of thermal stability (one of them tends to carbonize at high temperatures, whereas the other decomposes into gaseous products) are first intimately mixed. Then the as-obtained nanostructured precursor phase serves as a template for the target MC material, which is obtained by pyrolysis accompanied by volatilization of the sacrificial polymer. Although Poizot et al. devised a variation of this approach, in their case both the carbon source and the gas precursors (sacrificial atoms) are directly included at a molecular level within a single compound (squaric acid H2C4O4).64 H2C4O4 crystals at Tc = 121 °C exhibit a particular thermal behaviour that is related to the phase transition; when T > Tc, all C–O bonds of squaric acid become statistically equivalent in a perfect square, making a discrete thermal decomposition reaction possible. Based on this phenomenon, a template-free approach was presented for the synthesis of expanded foams of MC that exhibit large surface areas ranging from 550 to 1100 m2 g−1 by the exceptional carbonization of squaric acid (H2C4O4). The pyrolysis reaction proceeded at just above 300 °C via a surprising single-step massive release of gas that behaved as a “fluid” template during the production of carbon and thereby promoted the formation of a porous structure or expanded foams.
In brief, chemical activating agents such as ZnCl2, H3PO4 and KOH have been shown to play an important role in pore formation during the process of carbonization, whereas a high mass ratio of activating agent to biomass is essential to obtain a high proportion of mesopores and high pore volume. Therefore, it is necessary to reduce the dosage and recycle effectively; moreover, maybe the use of a natural mineral containing alkaline-earth elements that can replace previous activating agents is a relatively cheap way. Although fast pyrolysis has been found to be superior in the synthesis of MC from biomass via the pyrolysis route, the inevitable by-product of coal tar is dangerous to the environment. Compared with other methods, obtaining MC material by the volatilization of a sacrificial polymer or atoms is a simple and convenient method, which should be encouraged.
2.4 Magnetic mesoporous carbons
In this field, polymerization using Co, Ni, and Fe compounds is particularly attractive because these metal elements can be incorporated in the final MCs, thus affording the possibility of preparing MC materials that contain magnetisable metal nanoparticles. On one hand, the carbon coating could protect the magnetic nanoparticles; on the other hand, the magnetization parameters can be adjusted via the content of the magnetic source and the carbonization temperature. These materials that contain such magnetic nanoparticles have suitable applications in the development of heterogeneous catalysts and adsorbents that can be separated by a magnetic field after use in the liquid phase. So far, there are generally two routes for inserting metal nanoparticles into MCs. One route is the incorporation of metal nanoparticles into pre-synthesized MCs using an incipient-wetness impregnation procedure. Another route is the infiltration of an appropriate carbon precursor and a metal source (Fe, Co, Ni compounds) into the mesopores of a silica template, followed by thermal polymerization, carbonization and subsequent removal of the silica framework with HF or NaOH solution. However, these traditional methods are time-consuming and high-cost multi-step synthesis procedures, including repeated impregnation with carbon and metal precursors and the removal of hard templates, which severely hamper the wide application of magnetic composite materials. In order to simplify the synthetic process, the direct synthesis of magnetic Ni/OMC composites has been proposed in an alkaline non-aqueous solution medium using a solvent EISA approach (Fig. 12A).223 The composites were fabricated by self-assembly of F127, RF polymer and [Ni(H2O)6](NO3)2, where RF was used as a carbon precursor, the triblock copolymer Pluronic F127 was used as a templating agent and [Ni(H2O)6](NO3)2 as a nickel source. Ni2+ was captured by the network of F127/RF and further reduced to metallic Ni nanoparticles during carbonization. Direct evidence of good dispersion in the OMC walls was observed, as displayed in Fig. 12B. Most of all, the Ni/OMC composites exhibited soft ferromagnetic behavior and excellent acid-resistant properties.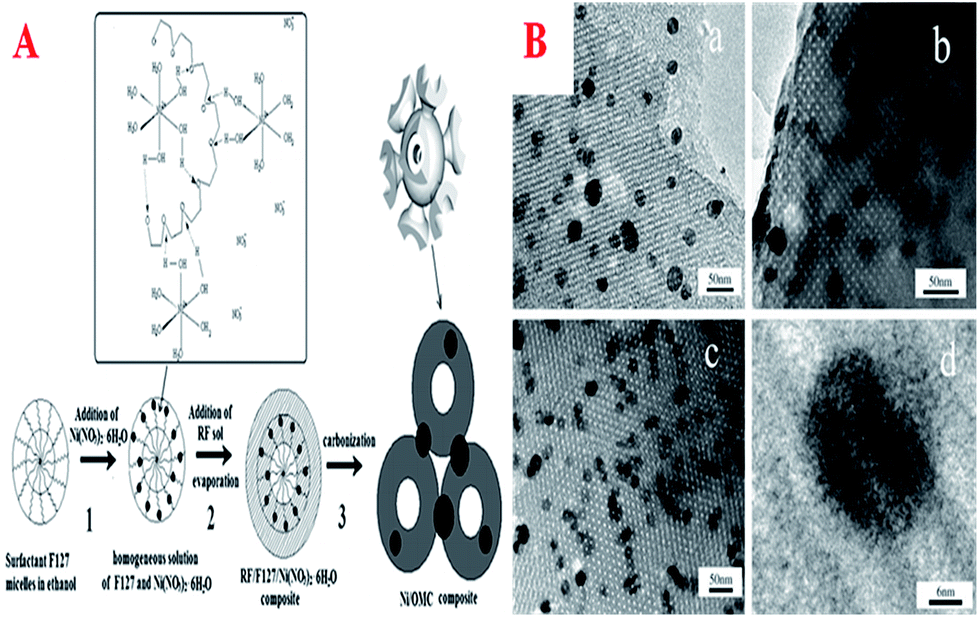 | ||
| Fig. 12 (A) Schematic of the preparation of Ni/OMC; (B) high-resolution TEM images of sample Ni/OMC(12)-700, (a) along the [110] direction, (b) along the [100] direction, (c) along the [111] direction, and (d) nickel nanoparticle embedded in the carbon wall. Reprinted with permission from ref. 223. | ||
Similarly, OMC materials with magnetic frameworks were prepared by co-assembly of resols, ferric citrate and the triblock copolymer F127 via a solvent EISA method in an ethanol/water solution.224 This one-pot synthesis route is related to the interaction between the iron complex and the phenolic resin matrix, which is important for avoiding the aggregation of iron ions and makes magnetic γ-Fe2O3 nanoparticles embedded in the carbon framework with high stability during the process of oxidation by H2O2. However, serious shrinkage of the skeleton during the high-temperature carbonization procedure led to a small surface area, small pore size, and low pore volume of the products, which limit their adsorption performance in processes that involve large molecules. Luckily, the presence of rigid silicates in the composites can greatly reduce structural shrinkage during carbonization, creating large mesopores. Therefore, a fast and simple synthesis of magnetic MCs with a large surface area, large mesopores and magnetic frameworks has been proposed using resol, nickel nitrate and TEOS as precursors and Pluronic F127 as a template.137 In this research, many complementary small pores that were caused by the removal of silica were observed in the carbon pore walls, which contributed to the large surface area. Nickel species were spontaneously reduced to metallic nickel nanoparticles during the carbonization process and were well dispersed in the framework.
Although Fe, Fe3O4, and Co have improved magnetic properties compared to Ni, acidic aqueous solutions can leach the transition metal into solution, thereby decreasing the potential for reuse of such materials.225,226 However, the stability of embedded magnetic nanoparticles in MC can be significantly improved by secondary treatment and subsequent carbonization to yield a graphitic shell.225 It is known that some organometallic cobalt precursors yield a graphene shell around Co nanoparticles by simple heating in an inert atmosphere,227,228 so MCs that contained cobalt nanoparticles were synthesized by the three- or four-component self-assembly of Pluronic F127, phenol–formaldehyde oligomer as a carbon source, cobalt acetylacetonate as a magnetic source and TEOS as a pore-expanding agent.98 In addition, the in situ synthesis of Co nanoparticles yielded a carbon shell that could partially protect Co from leaching into acidic media; after 96 h in 2 M HCl, the powders remained magnetic and enabled magnetic separation from an aqueous suspension. Coincidentally, the same protective effect of a coated carbon shell on magnetic nanoparticles in acid was also suggested by other researchers.229
Usually, magnetic iron oxide nanoparticles can be selectively deposited into the intratubular pores of CMK-5 with an iron content of up to ∼12 wt%,230 which is enough for some catalytic reactions but not so attractive when adopted as an active component for adsorption and separation. In general, at a high metal loading level and/or a high temperature of conversion, the metal species normally aggregate severely into large particles and block the pores to a greater or lesser extent, especially in the loading process. It is therefore extremely challenging to fix highly concentrated (e.g., >20 wt%) and uniformly dispersed crystalline nanoparticles into predefined mesopores of carbons without them aggregating and blocking the open pore networks. Such disadvantages are largely associated with a lack of efficient control in avoiding substantial diffusion/aggregation of the metal precursors during their conversion into oxides, which is especially challenging in the case of high loading levels. On the other hand, the synthesis of MC-based nanocomposites that encapsulate high-content but uniformly dispersed and spatially separated nanoparticles that retain an open mesopore system is highly favoured, because such features are highly desirable for adsorption and catalysis, which depend on molecular diffusion and transportation for their effectiveness. Interestingly, Zhao et al. proposed a general post-synthetic route using ammonia-atmosphere pre-hydrolysis for the construction of OMC that encapsulated a wide range of iron oxide nanoparticles with high concentrations (>40 wt%) that were homogeneously dispersed in predefined mesopores.231 The materials that were obtained possessed uniformly dispersed, spatially separated nanoparticles that were exclusively confined in mesopores even at a very high metal oxide content of up to 52 wt%. As depicted in Fig. 13, the primary mesopores (5.6 nm) of a surfactant-templated ordered bimodal MC matrix played an important role in selectively loading nanoparticles with a very high concentration, while its connected mesopores (2.3 nm) were left empty in order to retain an open pore network so that fast molecular diffusion/transportation could be achieved.
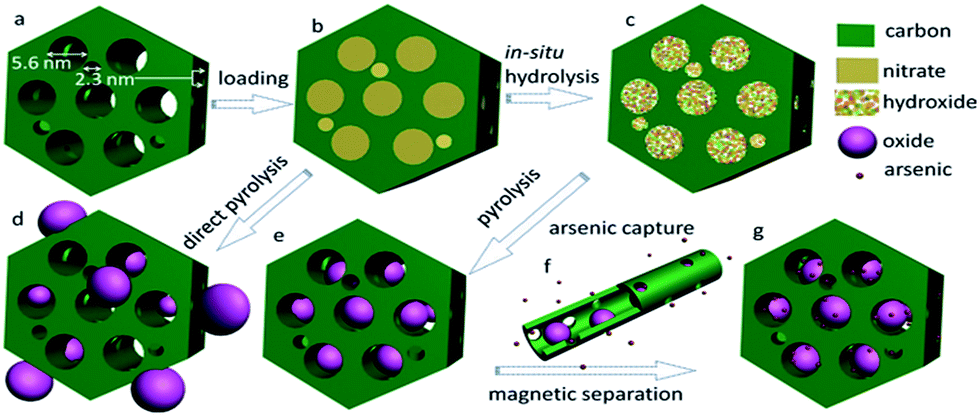 | ||
| Fig. 13 Illustration of the processes of synthesis and arsenic capture for ordered encapsulated mesoporous Fe2O3@C: (a) bimodal MC, (b) carbon loaded with hydrated iron nitrate precursor, (c) carbon loaded with iron hydroxide obtained by in situ hydrolysis under an ammonia atmosphere, (d) iron oxide@carbon composites obtained by direct pyrolysis, (e) encapsulated Fe2O3@C obtained by pyrolysis following pre-hydrolysis, (f) arsenic capture, and (g) arsenic-enriched encapsulated composite. Reprinted with permission from ref. 231. | ||
In general, methods of preparing carbon-based nanocomposites, including thermal decomposition, self-assembly, electrochemical deposition, and the layer-by-layer technique, can effectively control nanostructures; however, they either have low efficiency for scaled-up production or require multiple complex processes for successful synthesis. As the supplementary role that was discussed in the synthesis of MC monoliths, microwave-assisted heating synthesis is a promising technology for manufacturing nanomaterials with its extremely fast heating and cooling rates, which could not be achieved in a conventional heating process. In addition, microwave heating can reduce the overall thermal gradients in the reaction and thus yield more uniform products that exhibit enhanced electrochemical energy storage compared to their nanocomposite counterparts from conventional heating in a tubular furnace.232 With the help of microwave heating and the better protective effect of a coated carbon shell on magnetic nanoparticles in acid, mesoporous magnetic carbon nanocomposite fabrics could be manufactured via conventional and microwave energy-assisted annealing processes. Commercial T-shirt cotton fabric and iron nitrate served as carbon and iron precursors, respectively, and were further converted into carbon nanocomposite fabrics with doped metal/metal oxide nanostructures.233
3. Functionalization and modification
Two approaches, namely, direct synthesis and post-synthetic treatment, have been used to functionalize OMCs. The former can work under a wide range of conditions and produce carbons with high loadings and a relatively uniform distribution of functional groups, although the structural ordering that is achieved seems to be worse; the latter exhibits high variability in the introduction of functional groups. However, it is still a major challenge to prepare multifunctionalized MCs by a single method. Therefore, the combination of direct synthesis with post-synthetic treatment may be an efficient approach for achieving this goal.234 Nevertheless, all MCs exhibit rather unreactive surfaces regarding chemical functionalisation, with a reduced number of oxygen-containing groups after high-temperature carbonization; therefore, their further chemical modification is not straightforward. However, the amount of these functional groups can be increased using subsequent oxidation reactions with acids or ozone235,236 or substitution of these groups by different functionalities containing heteroatoms such as N and S.237,2383.1 Surface treatment
Great effort has been devoted to the surface functionalization of porous carbon materials in past years. A series of functional groups can be attached onto a carbon surface via surface oxidation and/or activation,239–241 halogenation,242,243 sulfonation,244,245 and grafting via diazonium chemistry,246,247 etc. Among these, surface oxidation is one of the most convenient and simplest methods for modifying a carbon surface, which not only attaches oxygen-containing groups but also alters the surface hydrophobic/hydrophilic balance. It generally includes dry and wet oxidation based on different treatment media, such as plasma treatment,248 electrochemical modification249 and reactions with oxidizing gases at high temperatures (>700 °C).250,251 Wet oxidation is widely adopted and involves reactions between a carbon surface and oxidizing solutions, such as HNO3, H2O2, NaClO and (NH4)2S2O8, under relatively mild reaction conditions (20–150 °C). Among these oxidants, ammonium persulfate (APS, (NH4)2S2O8) solution is a gentle and less toxic oxidant with a high capability of generating surface oxides without obvious damage to the porous structure,252 whereas other oxidants are not widely acceptable because of their reduction of the specific surface area and porosity, damage to the mesostructure, or liberation of toxic gas into the environment to a greater or lesser extent.253–255 Moreover, given their great potential and ease of industrial-scale production, it is desirable to generate surface functional groups, especially carboxylic groups (COOH), in such novel carbon materials for specific applications, such as highly efficient adsorbents for the immobilization of heavy metal ions as well as functional organic compounds and biomolecules and solid and stable supports for water treatment and the immobilization of organic molecules/biomolecules, with high capacities and excellent binding capabilities.256 For example, MC foams that were treated with acid using HNO3 with different molarities and reaction temperatures were tested as adsorbents of Pb(II) ions from aqueous solutions.257 Unluckily, an increase in the concentration of acid and the temperature was found to be highly destructive to the pore structure of the carbon foams. However, this aggressive acid treatment also created a wealth of oxygen-containing functional groups (–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O) on the surface of the material, which acted as binding sites for Pb(II) ions with extremely high capacities of up to 188 mg g−1. Moreover, OMCs that were functionalized with SO3H with large specific areas and large pore sizes exhibited excellent performance in many reactions.258–262 Concentrated sulfuric acid is an efficient sulfonating agent263–265 and generally the sulfonation of carbon materials in concentrated sulfuric acid has to be carried out at high temperatures. For the purpose of retaining the ordered mesostructure of OMCs, synthesized OMCs have been sulfonated in concentrated sulfuric acid (98%) at elevated temperatures.266 The results suggested that the mesostructural stability and content of surface sulfonic acid (SO3H) groups depended mainly on the pyrolysis temperature of the OMCs and the sulfonation temperature.
O, C–O) on the surface of the material, which acted as binding sites for Pb(II) ions with extremely high capacities of up to 188 mg g−1. Moreover, OMCs that were functionalized with SO3H with large specific areas and large pore sizes exhibited excellent performance in many reactions.258–262 Concentrated sulfuric acid is an efficient sulfonating agent263–265 and generally the sulfonation of carbon materials in concentrated sulfuric acid has to be carried out at high temperatures. For the purpose of retaining the ordered mesostructure of OMCs, synthesized OMCs have been sulfonated in concentrated sulfuric acid (98%) at elevated temperatures.266 The results suggested that the mesostructural stability and content of surface sulfonic acid (SO3H) groups depended mainly on the pyrolysis temperature of the OMCs and the sulfonation temperature.
As well as modification by acid, modification of MC materials by alkali might provide them with other special properties. For example, MgO is of special importance in catalysis and medicine and ordered mesoporous MgO/carbon materials have strong surface basic properties, which make them promising as candidates for selective adsorption and catalysis involving strongly basic materials.267 The disadvantages of these chemical modifications, however, lie in the low degree of final functionalization and corrosion of the carbon surface during oxidative treatment. Another pathway towards mesoporous functional carbons can be achieved using hydrothermal carbonisation, which is a well-established method for creating hydrophilic carbon materials starting from water-soluble carbohydrates under mild conditions.268 These hydrothermal carbons have a hydrophilic shell that contains a large number of functional groups such as COOH, OH, and CO, which remain from the carbohydrate precursor.269 The generation of highly ordered mesoporous materials with oxygen-containing groups on the surface can be regarded as a versatile method for the easy production of a wide variety of functionalised ordered carbon materials.270 In summary, the choice of suitable operating conditions and parameters is necessary to obtain a certain density of functional groups on the surface of MC materials by surface treatment.
3.2 Heteroatom doping
The properties of porous carbon materials depend to a large extent not only on the raw material and its surface structure and porosity but also on the heteroatoms that are built into its structure. The surface chemical nature of OMCs can be modified by the introduction of heteroatoms such as oxygen, nitrogen, phosphorus, sulfur, boron, etc. using heteroatom-containing precursors. The improvement in the properties of doped carbons is well known: for example, oxygen functional groups enhance the hydrophilic character of the carbon surface, act as active sites in catalytic reactions or in the selective adsorption of cationic species, provide specific sites for anchoring other functionalities and facilitate the dispersion of metals on the carbon surface;271,272 nitrogen functional groups provide basic character to the surface, increase the adsorption of acidic molecules, improve catalytic activity and pseudocapacitance by means of redox reactions and enhance the anion-exchange properties of carbon;273,274 and phosphorus-containing surface groups increase the resistance of the carbon material to oxidation, provide strongly acidic character to the carbon surface, improve the cation-exchange properties and increase the energy density of supercapacitors.275,276 The doping process requires not only the introduction of large amounts of heteroatom-containing groups onto the carbon surface, but also as accurate a characterization as possible of the type and density of the surface functionalities in order to adapt their chemical nature to a specific application. This issue is complex and becomes quite a challenge especially when several types of heteroatom are present on the same carbon surface. To shed light on the surface chemical characteristics of doped carbons, OMCs containing nitrogen, oxygen and phosphorus surface functionalities were synthesized using SBA-15 as a solid template and 3-aminobenzoic acid as the carbon, nitrogen and oxygen precursor and phosphoric acid was selected to achieve doping with phosphorus.277 During this process of doping, selecting a certain phosphoric acid concentration and carbonization temperature is an effective way to design ordered carbons that exhibit a specific surface composition. For catalytic applications where a transition metal is necessary, especially some expensive metals, transition metal-doped carbon xerogels with mesoporous structure can be prepared by solubilization of metal salts in an aqueous solution of resorcinol–formaldehyde; moreover, rather large metal particles can be observed in the carbon framework.278Very recently, the ability of graphene layers to covalently attach heteroatoms (e.g., O, N, S) has been exploited in order to incorporate oxygen, nitrogen or sulphur-based functional groups into carbonaceous materials.279 Similarly, this approach was adopted in order to incorporate nitrogen and sulphur functionalities into a layer of carbon that was deposited inside the pores of silica with the purpose of enhancing the range of potential applications of silica–carbon composites.280 The prepared silica–carbon composites contained ∼25 wt% of carbonaceous matter, a large number of nitrogen functional groups that were incorporated into the aromatic rings in pyridine (N-6) and quaternary (NQ) positions, and sulphur functional groups, which mainly formed sulphide bridges (–C–S–C–) connecting the aromatic rings. After the removal of silica from the composites, the resulting carbons exhibited a large surface area and pore volume, in addition to a large amount of nitrogen in the case of N-doped templated carbons (6.7 wt% N) and sulphur in the case of S-doped templated carbons (3.9 wt% S). Highly ordered and nitrogen- and oxygen-rich MCs were also synthesized by applying the innovative methods of solid-state thermal polymerization and organic-phase synthesis.281 In both methods, 3-aminobenzoic acid and SBA-15 were used as precursor and template, respectively. After carbonization at 900 °C and removal of the template, OMCs with very narrow pore size distributions and nitrogen and oxygen contents as high as ∼6 and 6.4–11.5 wt%, respectively, were obtained. Moreover, this was an attractive pathway for the preparation of N- and O-doped OMCs in comparison with the chemical vapor deposition technique or liquid infiltration processes using other carbon precursors. Notably, some research has found that boric acid instead of other strong acids can control the formation and assembly of silica nanotubes.282 Based on this mechanism, a strategy was proposed to prepare boron-doped MCs by a sol–gel method using D-fructose as the carbon source and boric acid as a multifunctional reagent: a catalyst for the hydrolysis/condensation of a small amount of silica precursor, a dopant during subsequent thermal reactions and an important agent for the formation of micropores by the removal of boron oxide.283 This short-cut route can simplify the synthesis process and obtain a homogeneous dispersion of boron in carbon in both energy- and time-saving fashions, which provides a new strategy for making doped carbon materials.
Hitherto, it has been demonstrated that the modification of the framework and/or surface of mesocarbons with heteroatoms such as boron,284,285 phosphorus,286,287 sulfur288 and nitrogen289 can significantly improve their physicochemical properties. Especially, the incorporation of nitrogen into MCs can alter the electronic and crystalline structures of carbon, increasing its chemical stability, surface polarity, electrical conductivity, and electron-donor properties. As a well-ordered mesoporous carbon nitride material (Fig. 14B) has been prepared by inorganic templating via a polymerization reaction between ethylenediamine and carbon tetrachloride (Fig. 14A),291,292 nitrogen-containing MCs have attracted wide interest. Nitrogen functionalities generally provide basic properties, which can enhance interactions between the carbon surface and acidic molecules such as dipole–dipole, hydrogen bonding, covalent bonding, etc. Possible structures of nitrogen functionalities include amides, imides, lactams, pyrrolic and pyridinic groups, as shown in Fig. 15. In general, nitrogen-containing functional groups are generated on the surface of partially oxidized porous carbons via post-treatment in ammonia, whilst the treatment temperature can markedly affect the type of nitrogen-containing groups: for example, nitrile, lactam, imide or amine-type functional groups are generated below 300 °C; pyridine-type nitrogen groups are formed between 300 °C and 500 °C; when the temperature reached is higher than 500 °C, pyridinic, pyrrolic or quaternary-type nitrogen groups are the dominant species. Moreover, the types of nitrogen-containing groups could be adjusted by adopting different precursors via self-assembly.
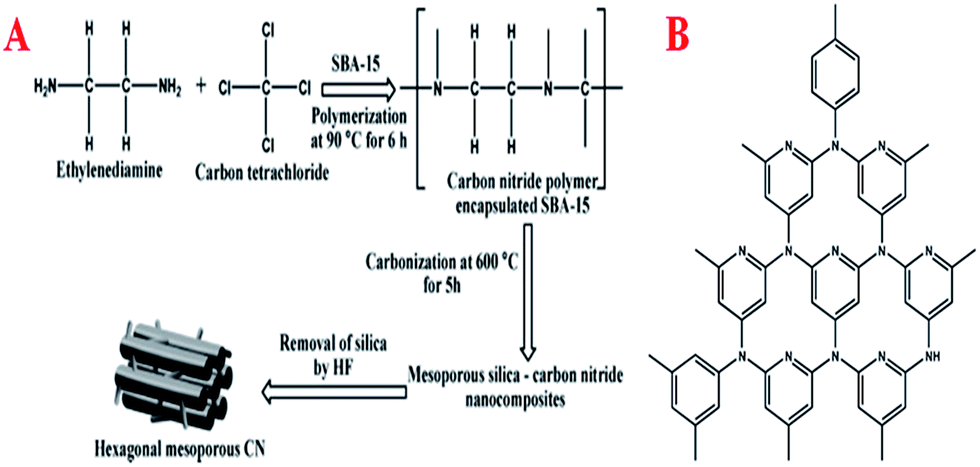 | ||
| Fig. 14 (A) Preparation of mesoporous CN using SBA-15; (B) schematic of wall structure of mesoporous CN. Reprinted with permission from ref. 292. | ||
 | ||
| Fig. 15 Types of nitrogen-containing surface functional groups: (a) pyrrole, (b) primary amine, (c) secondary amine, (d) pyridine, (e) imine, (f) tertiary amine, (g) nitro, (h) nitroso, (i) amide, (j) pyridone, (k) pyridine N-oxide, (l) quaternary nitrogen. Reprinted with permission from ref. 290. | ||
From a large number of studies,84,118,293,294 it should be noted that the enhancement of the properties of N-doped MCs should be dependent on the amount of nitrogen atoms and their chemical state when incorporated into the carbon backbone. In systematic research on this subject, Long et al. synthesized N-doped MCs with controllable nitrogen contents (Fig. 16B(a) and (c)) in the range of 0–12 wt% by incorporating melamine, which has a high nitrogen content, into phenolic precursors that can control the amount of nitrogen atoms that are doped into the carbon framework and maintain similar mesoporous structures for N-doped MCs (Fig. 16A).134 The N 1s spectra (Fig. 16B(c)) are fitted to three peaks with binding energies of 398.7 ± 0.3, 400.3 ± 0.3, and 401.4 ± 0.5 eV, which correspond to pyridinic N (N1), pyrrolic N (N2), and graphitic N (N3), respectively. Reliable relationships between nitrogen doping or content and the physicochemical properties of MCs such as pore structure, stability to oxidation, conductivity, adsorption and catalytic activity were clarified.
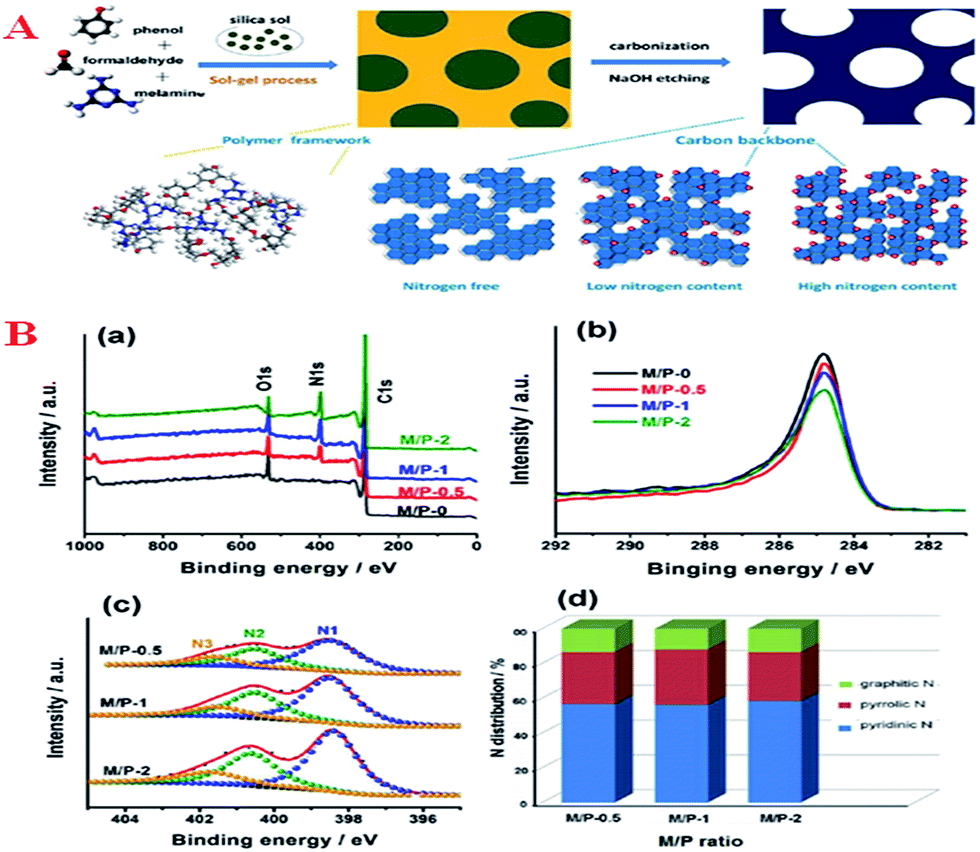 | ||
| Fig. 16 (A) Schematic of the preparation of N-doped MCs; (B) (a) XPS survey and (b) high-resolution C 1s and (c) high-resolution N 1s spectra of N-doped MCs with different M/P ratios; (d) distribution of N species in N-doped MCs from resolution of the peaks of the N 1s spectra. Reprinted with permission from ref. 134. | ||
MC materials that are doped with N-based functionalities have commonly used acetonitrile or pyrrole as the basic feedstock for the synthesis.295,296 Similarly, a chelating diamine functionality can also be integrated into N-doped MCs using 1,10-phenanthroline as the starting material.297 However, these N-doped materials have been limited to no more than 8% N content, which is presumably due to loss of N at the elevated temperatures required for the carbonization stage.296,297 In addition, linear polymers suffer from the potential drawback of thermal depolymerization during the carbonization stage of the synthesis. Therefore, developing a precursor that would be compatible with heteroaromatic functionalities and provide the greatest degree of cross-linking at low temperatures would be useful for the carbonization process to allow as much as possible of the N functionality to be retained in the final product. For this purpose, a dendritic polymer built from alternating benzene and pyridine rings that was prepared by utilizing the cyclotrimerization of 2,6-diacetylpyridine induced by SiCl4 is suitable.298 Cyclotrimerization allows a high degree of cross-linking to take place at low temperatures, which stabilizes the mesostructure and allows carbonization to be carried out at only 600 °C; the functional MC that was formed in this way was found to have a surface area of 1275 m2 g−1 and contain 6.8% N with the predominant species being pyridine N.
An important nitrogen-containing organic carbon precursor, resorcinol–urea–formaldehyde (RUF) resin, which can be formed from RF and urea–formaldehyde (UF) joined either by methylene or ether bridges, can also interact with Pluronic F127 by hydrogen bonding, leading to the formation of ordered mesostructures during polymerization. To better mediate the formation of a nitrogen-enriched organic carbon precursor, HMT is a good choice, which is usually susceptible to hydrolysis and results in the generation of formaldehyde and ammonia at elevated temperatures for the self-assembly of resorcinol–urea–formaldehyde resin. In a similar way, a one-pot cooperative assembly approach to prepare a highly ordered nitrogen-containing MC using RUF resin as an organic precursor without pre-polymerization or hydrothermal treatment was successful.299 As expected, a similar synthetic strategy was adopted to prepare an ordered nitrogen-doped MC via direct self-assembly of resorcinol–melamine–formaldehyde (RMF) resin and amphiphilic triblock copolymer (Pluronic F127); additional pre-polymerization and hydrothermal solidification steps were also omitted.300 In any case, HMT as a pH buffer or curing agent plays an important role in controlling the kinetics of polymerization reactions to prepare nitrogen-doped OMCs during self-assembly without a pre-polymerization step.
Owing to the low nitrogen content of most precursors and the loss of N at high temperatures during the carbonization process, the N content of the resulting carbon materials is not considerable. Therefore, finding how to obtain MCs with a high N content is highly desirable. However, most mesoporous nitrogen-enriched carbons, especially those with ordered mesostructures, are synthesized by a hard-templating method using appropriate nitrogen sources such as acrylonitrile,301 acetonitrile,302 pyrrole,303 aniline,304 and melamine–formaldehyde resin.305 Carbon and nitrogen sources are filled into the pores of the hard template, followed by polymerization, carbonization, and removal of the silica template. This process is obviously time-consuming and the nitrogen content of the resulting carbon materials is highly dependent on the precursors and pyrolysis conditions. Alternatively, a nitrided silica template can also transfer nitrogen into templated carbon during carbonization.306 The above methods normally deliver a nitrogen content of <10 wt%. Although the incorporation of nitrogen into OMC by a hard-templating approach is fairly well developed, there is still a high demand for new types of mesoporous nitrogen-enriched carbon materials for large-scale production. With this aim, OMC materials prepared by an organic–organic soft-templating approach bring new opportunities for development, whereas finding a suitable nitrogen-containing precursor that can not only copolymerize with phenolic resin and co-assemble with surfactants into an ordered mesostructure by a soft-templating approach but also retain nitrogen atoms to a greater or lesser extent after the carbonization process is still a great challenge. Learning from the success in the enrichment of nitrogen in activated carbons,307,308 post-treatment may more easily introduce nitrogen into soft-templated carbon materials. OMCs that were treated with ammonia with a variable nitrogen content of 3.6–6.0 wt% are a successful example.309 With a simple wet-impregnation step, melamine as a precursor can be loaded into the mesopore channels of a pristine carbon matrix. The basic principle relies on the confinement of melamine molecules in the mesochannels so that they self-condense into carbon nitride, which is uniformly dispersed under heat treatment at about 500 °C and subsequently leads to the formation of mesoporous nitrogen-enriched carbon materials at 700–900 °C with well-retained ordered mesostructure and large surface area.310 Like melamine or urea, another kind of nitrogen-rich compound, dicyandiamide (DCDA), which possesses one nitrile group and two amine groups, has a high nitrogen content (66.7%) and can be converted into graphitic carbon nitride (g-C3N4) at high temperatures. The direct assembly of DCDA and Pluronic PEO–PPO–PEO copolymers will lead to porous carbon nitrides with high N content but poor mesostructure and small surface areas, which is mainly due to the weak interaction between DCDA and PEO–PPO–PEO and the collapse of the framework during pyrolysis for the removal of templates.311 However, a one-pot controllable method to synthesize N-doped OMCs with a high N content (up to 13.1 wt%) using a low-molecular-weight soluble resol and DCDA as carbon and nitrogen sources, respectively, and ethanol and water as a mixed solvent via an EISA process is quite different133 because resol molecules can bridge both the Pluronic F127 template and DCDA via hydrogen-bonding and electrostatic interactions during the EISA process (Fig. 17). Notably, this synthesis concept can also be applied to the design of MCs that are doped with other elements by choosing appropriate precursors that can interact with resols. Pyrolyzing some nitrogen-containing chemicals such as polyacrylonitrile, polypyrrole and polyaniline seems to be the most common way to obtain nitrogen-rich porous carbon materials. Gelatin is an amphoteric polyelectrolyte, which consists of three polymer chains in a triple helix with a molecular weight of approximately 300000; moreover, it is cheap, environmentally friendly and commercially available. The above characteristics enable gelatin to be pyrolyzed between 700 and 900 °C using a nano-CaCO3 template method to obtain nitrogen-rich MC materials. FTIR and XPS studies indicate that nitrogen in the material exists in the form of pyridinic, pyrrolic/pyridonic and graphitic nitrogen with a nitrogen content of 10.72 wt%.312
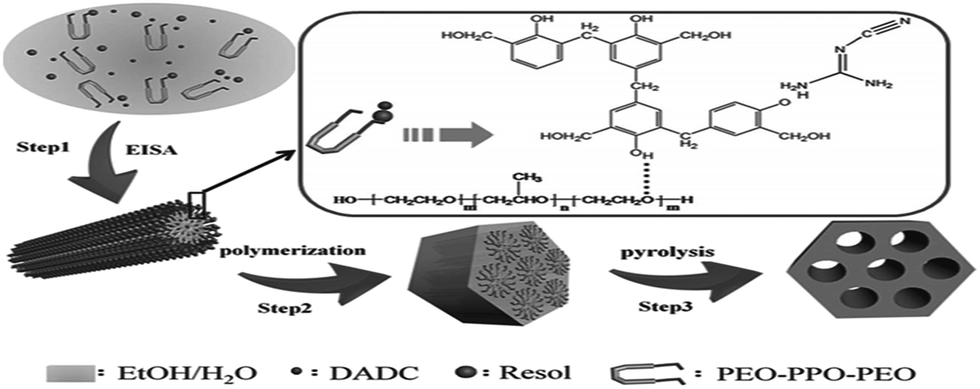 | ||
| Fig. 17 Process of formation of ordered N-doped MC by a one-pot assembly method using dicyandiamide (DCDA) as a nitrogen source. Reprinted with permission from ref. 133. | ||
Although post-treatment of OMCs with nitrogen compounds such as melamine313 is a feasible way of preparing nitrogen-containing OMCs and represents an alternative way to incorporate nitrogen-containing functionalities, the direct synthesis of OMCs with a high nitrogen content is highly desirable. A simple preparation of highly nitrogen-enriched OMCs by the direct pyrolysis of a soft-templated phenolic resin-F127 nanocomposite in NH3 has achieved success without using any nitrogen-containing precursor or post-treatment.314 This approach not only takes advantage of the preferential reaction and/or replacement of oxygen with nitrogen species,315 which are generated by the decomposition of NH3 at elevated temperatures, in oxygen-rich carbon precursors during pyrolysis, but also combines carbonization, nitrogen-functionalization, and activation into one simple process. The obtained N-doped OMCs exhibited uniform mesopore size, large surface area (up to 1400 m2 g−1), and high nitrogen content (9.0 wt%). Unlike traditional precursors, for example, the assembly of phenolic resin with nitrogen compounds or nitrogen-containing chemicals, biopolymers are attractive as renewable resources for preparing nitrogen-doped carbon-based materials. Chitin is the main structural component in the shells of crabs, shrimps, and insects and the cell walls of fungi and contains ∼6.9 wt% nitrogen from N-acetyl groups. Because of this, nanocrystalline chitin has been used both as a soft template and as a carbon and nitrogen source for preparing mesoporous nitrogen-doped carbon materials with a layered structure.316 As another cheap, environmentally friendly and commercially available biopolymer, gelatin is an animal derivative composed of various proteins with an average nitrogen content of 16 wt%, which is produced by the partial hydrolysis of collagen extracted from the skin, bones and connective tissue of animals. Again, a simple preparation of nitrogen-doped MC was developed using gelatin as a precursor in a nano-CaCO3 template approach.85 Biomass derivatives such as carbohydrates are more sustainable and available than nitrogen-containing precursors like acetonitrile, pyrrole or polyacrylonitrile and in addition, it is also a cheap and sustainable way to obtain chemicals and carbons from raw materials other than crude oil or natural gas lead to a re-exploration of this field. Thus Titirici et al. represent a green and sustainable alternative way to produce nitrogen-rich carbonaceous materials, which is based on the hydrothermal carbonization of biomass that includes nitrogen-containing carbohydrates such as chitosan or glucosamine.317
In contrast to direct assembly, pyrolysis or post-treatment for the synthesis of N-doped OMCs, solid-state grinding is a method of introducing guest species into the channels of ordered mesoporous materials.318,319 Grinding can efficiently elevate the surface temperature among particles and change their tensile force to improve the state of dispersion of the guest chemicals, which is possible to realize on a large scale. Chen et al. employed a simple solid–solid grinding/templating route to fabricate N-doped OMC with a 2D hexagonal symmetric structure using the ionic liquid (IL) 1-cyanoethyl-3-methylimidazolium chloride as the precursor and SBA-15 as the template.320 The as-synthesized N-doped OMC exhibited a uniform mesopore size (3.5 nm), rope-like morphology (0.4–1 m in length) and large surface area (803 m2 g−1). Quantitative analysis revealed that the nitrogen content on the surface of N-doped OMC was 5.5 at%. The further introduction of nitrogen-containing functional groups into the graphitic matrix would provide nitrogen-doped carbon nanofibers with an improved capacity to capture some acidic gases. Such nitrogen-doped MC nanofibers with an aligned mesoporous structure were synthesized by a co-confined carbonization method using anodic aluminum oxide (AAO) membrane and TEOS as co-confined templates and ionic liquids as precursors.321 The as-synthesized nitrogen-doped carbon nanofibers with a diameter of 80–120 nm possessed a bulk nitrogen content of 5.3 wt% and a bimodal mesoporous structure. The nitrogen atoms were mostly bound to the graphitic network in two forms, i.e., pyridinic and pyrrolic nitrogen, which provided adsorption sites for acidic gases like SO2 and CO2. Cycling experiments revealed considerable stability over 20 runs for adsorption of SO2 and 15 runs for adsorption of CO2. The nitrogen-doped carbon nanofibers also displayed preferable adsorption performance for Cd2+.
Carbon nitride is a well-known fascinating material that has attracted worldwide attention because the incorporation of abundant nitrogen atoms in the carbon nanostructure can effectively enhance its mechanical, conducting, field emission, and energy storage properties.322–325 Moreover, carbon nitride is a potentially useful substitute for amorphous and graphitic carbon in a variety of applications such as catalysis, gas storage, and purification of contaminated water. A soft-template method generally produces mesoporous carbon nitride with a smaller surface area, whereas the nanocasting technique using mesoporous silica as a hard template is an advisable strategy for synthesizing mesoporous carbon nitride with a larger surface area and controlled mesoporous structures. In any case, further improvements in surface area and pore volume are highly desirable. Although by the post-treatment of MC well-ordered mesoporous carbon nitride with a large surface area can be obtained, the N content is commonly less than 5 wt% and this method generally requires a nitrogen-containing atmosphere such as highly toxic NH3 at high temperatures. In contrast, in situ doping of carbon using various nitrogen-containing precursors (melamine, cyanamide, dicyandiamide, gelatin, etc.) can achieve the homogeneous incorporation of nitrogen into the carbon material with controlled chemistry; however, the synthesis of mesoporous carbons with an ultra-large surface area of up to 1000 m2 g−1 and a large pore volume of up to 1.0 cm3 g−1 still remains an insurmountable challenge. In order to enhance the performance of this material in various applications, Zhao et al. developed a facile and efficient method of synthesizing ordered mesoporous carbon nitride materials with an ultra-large surface area (971–1124 m2 g−1), ultra-large pore volume (1.31–1.79 cm3 g−1) and higher N content (9.2–23%) by a self-polymerization reaction of HMT using SBA-15 as a hard template via a facile and efficient nanocasting approach (Fig. 18).326 The specific surface area and pore volume as well as N content were strongly dependent on the chosen precursor and pyrolysis temperature. Furthermore, the current synthetic strategy can be extended to the preparation of various mesoporous carbon nitrides with different textural characteristics using diverse templates under changeable preparation conditions.
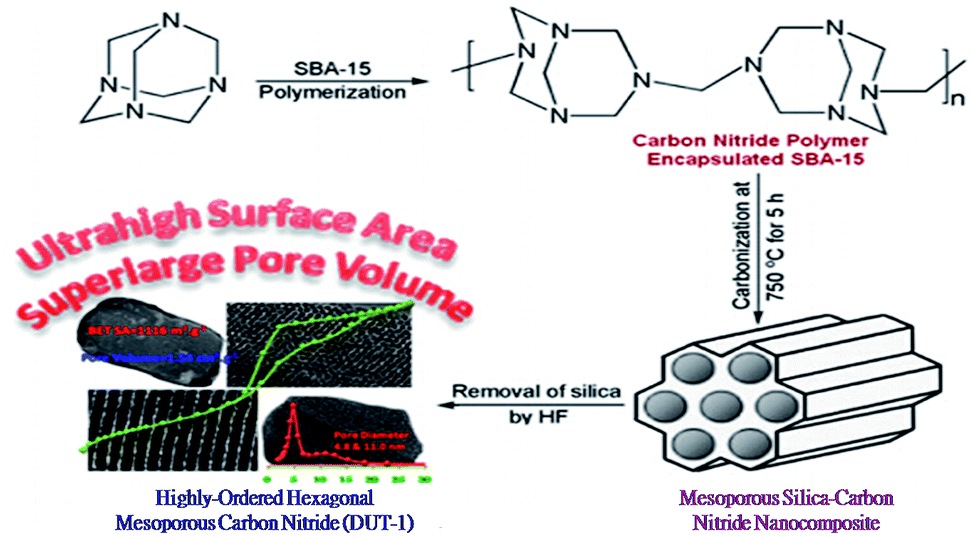 | ||
| Fig. 18 Illustration of synthetic procedure of highly ordered mesoporous carbon nitride material using SBA-15 as a hard template. Reprinted with permission from ref. 326. | ||
In addition to the above methods, choosing a highly functional precursor may also be a good choice for broadening the functionality of MCs. The synthesis of highly ordered fluorinated MCs by a direct triblock copolymer templating approach is a typical case.327 The organic precursors were phenol, formaldehyde, and p-fluorophenol, which served as functional monomers in the modification of carbon products, similarly to alkoxysilanes containing organic groups in the synthesis of functionalized mesoporous silicates. The triblock copolymer F127 was selected as a structure-directing agent. After high-temperature carbonization at 900 °C, highly ordered OMC with covalent C–F bonds could be obtained. In addition, the fluorinated carbon products had large surface areas and narrow pore sizes. Preliminary application in electron transfer kinetics indicated that the fluorinated MCs possessed high redox ability. By analogy, m-aminophenol was employed as a carbon and nitrogen co-precursor to synthesize nitrogen-containing OMC via a co-assembly process with F127 in the aqueous phase.328
As is widely known, carbonization conditions can lead to a loss of functionality in those systems in which depolymerization or fragmentation can readily occur. Therefore, finding a functional precursor with high thermal stability is important. Cyclotrimerization of acetylenes forms trisubstituted benzene rings and is therefore not easily reversible or prone to depolymerization.329 Cyclotrimerization can inherently produce a high degree of cross-linking, which results in a carbon backbone that is thermally quite robust. Based on this point, the synthesis of inherently functional MCs has been devised using the cyclotrimerization of heteroaryl acetylenes to create carbon frameworks that are built around a specific heteroaromatic functionality (a pyridine ring).330 In general, compared with surface treatment, doping with a heteroatom could provide more types and higher contents of functional groups with the advantages of covering the whole framework and no destruction of the surface and porous structure of materials. More importantly, maybe some lattice defects that are produced by the removal of heteroatoms will bring about superior performance such as catalytic effects.
3.3 Carbon–inorganic nanocomposites
Adsorption, catalysis, separation and energy-related applications often require MCs with incorporated inorganic species or carbon–inorganic nanocomposites. Furthermore, the addition of silicates to phenolic resins is widely used in industry to enhance the toughness of polymers and carbons and reduce thermal shrinkage. Inorganic species can be incorporated into MCs during their synthesis or by post-synthetic infiltration.331,332 The latter strategy, although it is often used, has several shortcomings due to the limited loading of inorganic species, difficulties in controlling their size and distribution and possible pore blocking. However, the above limitations do not occur when inorganic species are incorporated during the synthesis of carbon. Therefore, several attempts have been made to incorporate inorganic species during hard-templating and soft-templating syntheses of MCs.333–336 In comparison to hard templating, many studies show that soft-templating synthesis is better suited for the incorporation of inorganic nanoparticles as well as their formation from suitable inorganic precursors.223,337,338 Moreover, colloidal templating is well suited for the incorporation of inorganic nanoparticles into carbons because these particles can be easily co-assembled with silica colloids to form a hard template.339 Therefore, a combination of colloidal hard templating with soft templating has been shown to afford mesoporous inorganic–carbon composites with a high loading of inorganic species.340 This approach can be used in the self-assembly of block copolymer templates and carbon precursors together with inorganic nanoparticles and TEOS (Fig. 19A) in order to obtain a mesoporous silica–carbon composite that contains nanoparticles. The resulting mesoporous silica–carbon composites (Fig. 19C (left)) contained spherical silica colloids in addition to uniformly distributed silica, which originated from TEOS. Dissolution of silica led to carbons with a large surface area (Fig. 19C (right)), in addition to primary mesopores and an enlarged pore size that were formed by thermal degradation of the block copolymer template and the accumulation of TEOS on the PEO copolymer blocks and colloidal silica nanoparticles (Fig. 19B), respectively. Moreover, this approach can also be used to incorporate other inorganic nanoparticles into MCs with extra microporosity that was generated after dissolving silica species generated from TEOS. | ||
| Fig. 19 (A) Illustration of soft-templating synthesis of MCs in the presence of colloidal silica without (left side) and with (right side) tetraethyl orthosilicate (TEOS); (B) proposed enlargement of pore size due to the accumulation of TEOS on PEO copolymer blocks and colloidal silica nanoparticles ((a) shows the self-assembly process without TEOS and colloidal silica; (b), the system with TEOS; (c), the system with TEOS and colloidal silica); (C) comparison of nitrogen adsorption isotherms and the corresponding pore size distributions for the silica–carbon composite obtained using 50 nm silica colloids (left) and the corresponding carbon after dissolution of silica samples (right). Reprinted with permission from ref. 340. | ||
Besides, ordered mesoporous organic–silica nanocomposites can also be obtained by surface functionalization,341 encapsulation of organic moieties in the channels of mesoporous silica materials,342 and the direct synthesis of periodic mesoporous organosilicas (PMOs).343 However, these processes have their drawbacks: one is that organosilanes and functional surfactants are expensive and difficult to obtain. The other drawback is that organic functional groups may block the pores and their random distributions limit further applications. Subsequently, a tri-constituent co-assembly approach for preparing well-ordered mesoporous polymer–silica and carbon–silica nanocomposites using resols as polymer precursors, silicate oligomers as inorganic precursors, and the triblock copolymer F127 as a template via EISA has been put forward (Fig. 20).81 By adjusting the aging time of TEOS and the amount added, highly ordered OMC could be synthesized with tunable bimodal pores and large surface areas. More importantly, the presence of silicates in nanocomposites dramatically inhibited shrinkage of the framework during calcination, resulting in highly ordered large-pore mesoporous carbon–silica nanocomposites. It happens that there is a similar case. Another simple one-pot route for the synthesis of highly ordered OMCs and silicas with two-dimensional hexagonal symmetries (p6mm) via the organic–inorganic self-assembly of tetraethoxysilane (TEOS), the triblock copolymer Pluronic P123 and sucrose under highly acidic conditions has been presented by Kao et al.82 Using a P123/sucrose/silica composite that was directly catalyzed by H2SO4 in the synthesis mixture, MCs or silicas were generated after carbonization followed by removal of silica or calcination of the aforementioned as-synthesized composite in air, respectively. Otherwise, this synthetic procedure had the advantage of being a single-step self-assembly approach and only involved the use of simple organic precursors and water.
 | ||
| Fig. 20 Schematic of tri-constituent co-assembly of ordered mesoporous polymer–silica and carbon–silica nanocomposites and the corresponding ordered mesoporous silica and carbon frameworks. Reprinted with permission from ref. 81. | ||
Commonly, mesoporous crystalline metal oxides undergo structural collapse when crystallizing, resulting in the formation of large grains,344 whereas a carbon component has been successful when applied to support a mesoporous crystalline titania framework.345 The synthesis of mesoporous carbon–titania (MCT) composites with crystalline titania and amorphous carbon components has been proposed using phenolic resins and acid–base pairs [acidic TiCl4 and its basic counterpart Ti(OC4H7)4] as carbon and titanium sources, respectively.346 The composites possessed highly crystalline anatase pore walls, ordered mesostructure, large surface areas (∼200 m2 g−1), large pore volumes (∼0.15 cm3 g−1) and a high content (∼87 wt%) of TiO2 nanocrystals. The MCT composite favored the immobilization of proteins and exhibited enhanced electrocatalytic properties in relation to the reduction of hydrogen peroxide. In any case, carbon–silica nanocomposites hold a dominant position in the world of carbon–inorganic nanocomposites. The incorporation of silica has brought about more desired properties than those of pristine carbon such as toughness and thermostability. Looking back over past studies in this field, TEOS was frequently chosen as the main source of silicon in laboratory research because of its good fusion and hydrolytic properties, but some cheaper silicon precursors (e.g. sodium silicate) will be more popular for industry.
3.4 The design of carbon texture
How to design and construct the pore structure, microcrystalline structure, and surface chemistry of porous carbons is an important task for a specific application. Methods such as activation treatment by ZnCl2, KOH, CO2, and NH3 have proved to be very effective in extending their potential for applications by providing microporosity to nanoporous carbons, especially those with highly developed mesoporous structures. Nevertheless, in many cases, these activation treatments often cause the skeleton to collapse or even destroy the mesoporous structure, thus damaging the uniqueness of the original nanostructure as well as limiting its application to certain fields. Wu et al. presented a novel and simple strategy for introducing micropores into the skeleton of carbon aerogel (CA) without damaging its unique 3D mesoporous nanonetwork by employing NH3 as a porogen and surface modifier.347 During the NH3-assisted semi-carbonization process (with a partial pressure of ammonia in an inert atmosphere), nitrogen functional groups that are highly thermally decomposable like pyrrole/pyridone (N-5) and pyridine (N-6) could be introduced into the semi-carbonized aerogel framework by replacing oxygen functional groups that are less thermally decomposable, like CO quinone-type groups, which then escape from the resulting CA framework during the subsequent carbonization, forming abundant micropores inside the carbon framework while keeping the remarkable stability of the mesoporous nanonetwork structure. Compared with traditional activation methods including post-synthetic activation by NH3, the present strategy provides an easy and effective route to create micropores and a much higher activation ability of nitrogen functional groups (the surface area to burnoff percentage (S/B) relative to the unactivated precursor is up to 60 m2 g−1 wt) than in other carbon materials like OMCs, while maintaining their original nanostructure.
Hydrophilization of a carbon surface is demanded in various application fields because it makes it possible to employ aqueous/polar solvents as reaction and/or dispersion media. Among surface modifications, oxidative treatment is one of the most convenient and frequently used methods. For example, carboxylic functional groups can be introduced by oxidizing the surface of carbon materials using various oxidizing agents such as concentrated nitric or sulfuric acids, ozone, and ammonium persulfate solution ((NH4)2S2O8, APS), which could help change the inert and hydrophobic nature of carbon materials, enhance their wettability for polar solvents and make the surface active for the immobilization of organic compounds via adsorption.348 Although chemical modifications of a carbon surface have been applied in previous hydrophilization techniques, an alternative hydrophilization pathway that is free from surface modifications is still expected to provide considerable advantages because the carbons that are obtained retain their inherent surface properties. Structure-directed hydrophilicity is an alternative promising approach for achieving surface hydrophilization. Theoretically, a rough surface with a texture on micron or nanometer scales can increase its hydrophilicity when the corresponding flat surface exhibits θCA < 90°. If θCA of flat carbon is 80°, hydrophilization is in principle possible by designing the mesotexture of MC films. Tokudome et al. developed a pathway to flexibly design the mesostructure of MC films with a hydrophilic surface via soft templating by a dewetting-free coating process.349 First, a precursor polymer film was coated using a mixture of poly(styrene)-b-poly(4-vinylpyridine) (PS-b-P4VP), resorcinol, and pore-swelling agents (polystyrene (PS) or 1,3,5-trimethylbenzene (TMB)) dissolved in a volatile solvent. Then the film was treated with paraformaldehyde to develop RF networks. Subsequent calcination in an inert atmosphere selectively removed the PS domain to produce a MC film. After that, the obtained MC films exhibited a water contact angle below 30° when 1,4-dioxane and toluene were employed as coating solvents. The present hydrophilization method was free from chemical modifications and therefore the obtained carbons possessed a pristine surface. Again, special functional groups on the carbon surface can also alter the surface hydrophobic/hydrophilic balance.
Although the mesophase symmetry and morphology of OMCs can be transformed by tuning the PEO/PPO ratios of amphiphilic triblock copolymers350 or changing the HCl concentration,351 other characteristics such as pore geometry or pore size distributions are also needed for special requirements. Very recently, in order to meet the need for rapid mass transport in many applications such as separation, catalysis and energy storage, many types of hierarchical porous carbon materials have been successfully fabricated.352–354 For example, bimodal MCs have been prepared using a one-step templating method.355 This technique consists of the direct synthesis of sucrose/silica composites by a sol–gel process using TEOS with colloidal silica particles in the presence of sucrose, followed by carbonization of sucrose and then the removal of silica templates. In this method, small and large mesopore systems are obtained using silica gel and colloidal silica particles, respectively. Similarly, some researchers found that many silica gel frameworks in CD/silica wet gels would collapse and then become stacked into many large silica nanoparticles in composites with low CD/TMOS ratios. At the same time, the silica component in dried CD/silica composites has two structures: one is a gel skeletal structure and the other is a nanoparticle structure. Based on this point, bimodal MC has been prepared by a simple one-step nanocasting method. The overall synthetic procedures are depicted in Fig. 21.53 Most notably, this synthesis route avoids the need to use colloidal silica particles as a template for large mesopores.
 | ||
| Fig. 21 Schematic of proposed mechanism of formation of bimodal MC. Reprinted with permission from ref. 53. | ||
Hierarchical materials represent a great breakthrough in materials science, as they should favor the fast transport of molecules while retaining good storage capacity. In order to obtain hierarchical micro/mesoporous carbons with highly controllable porosity in the micro/mesopore range, Suárez-García et al. prepared ordered micro/mesoporous carbons by a simple chemical vapor deposition process using acetylene as the carbon source and a core/shell-type aluminosilicate as a template.356 In addition, the direct replication of the hierarchical templates enabled the production of micro/mesoporous carbons in one step, eliminating the activation post-treatments that are necessary for preparing micro/mesoporous carbons from OMCs.
Normally, the direct synthesis of highly ordered OMCs via EISA or a solution synthesis route makes it possible to conveniently synthesize MCs with pore sizes of 3–7 nm and various pore structures on a large scale using Pluronic block copolymers as templates, but large-pore OMCs that are suitable for applications involving large guest objects can hardly be obtained using a hard-templating approach or a soft-templating method based on Pluronic copolymers, due to the limitation of the templates' molecular weight.357,358 However, the successful synthesis of high-molecular-weight amphiphilic block copolymers has opened the door to the preparation of OMCs with large pore sizes and a tunable porous structure via an EISA approach.359–361 Recently, Zhao et al. have succeeded in the preparation of OMC C-FDU-18s with tunable pore sizes and pore wall thicknesses using poly(ethylene oxide)-block-poly(styrene) (PEO-b-PS) diblock copolymers with various PS chain lengths as templates in an EISA process.88 After oxidative treatment by a mixed solution of HNO3 and H2O2, numerous hydrophilic groups were created in the mesopore channels without destroying the ordered mesostructure of C-FDU-18. By the in situ reduction of Ag+ or Fe3+, Ag or magnetic nanoparticles could be successfully introduced into large mesopores, resulting in functional mesoporous carbons with various applications in fields such as catalysis, chemical sensing, and magnetic separation and enrichment. Due to the low molar mass of common structure-directing molecules, the accessible pore size range is limited to less than 4 nm, which can be increased to 15–16 nm by molecular swelling agents. For example, ultra-large MCs from triblock copolymers and phloroglucinol/formaldehyde polymer have been obtained under acid conditions with the assistance of decane as a swelling agent and the pore size of carbons that were templated by P123 and carbonized at 600 °C could be increased from 11.5 to 14.7 nm. The low synthesis temperature and high reactivity of phloroglucinol were two key factors for the formation of large mesopores.362 Owing to the outstanding contributions of Zhao et al., pore sizes of up to 37 nm have been achieved using poly(styrene)-block-poly(ethylene oxide) (PS-PEO) with the addition of poly(styrene) homopolymer as a pore-expanding agent and phenolic resols as carbon precursors.363 A strange morphology, gyroidal MCs with ordered and bicontinuous networks, have been reported using Pluronics or the structure-directing block copolymer poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL).89,90 Nevertheless, these materials displayed only a small pore size of 11 nm and low porosity. Subsequently, Wiesner et al. reported the tunable synthesis of two gyroidal MC (GDMC and GAMC) morphologies via an EISA process using the structure-directing triblock terpolymer poly(isoprene)-block-poly(styrene)-block-poly(ethylene oxide) (ISO) with phenol- or resorcinol–formaldehyde resols as carbon precursors.91 Characteristics such as porosity, pore size (up to 39 nm), and mesostructure could be tunable by rational design of the soft template. Gyroidal materials are ideally suited as electrode materials in fuel cells, batteries, and supercapacitors due to their high three-dimensionally connected porosity. In conclusion, among the methods of designing MC texture, the realization of controlling the pore size and distribution of MC materials is the cornerstone for widening the applications of porous materials. By adopting either a swelling agent or high-molecular-weight amphiphilic block copolymers, in any case, the control and design of channels are changed and their original properties are optimized, making MC materials more suitable for their potential applications.
4. Typical applications
Owing to their large surface areas, well-developed porous structures, and the presence of different kinds of functional groups on the surface, MCs possess obvious advantages compared to traditional microporous materials. Applications of MCs have been widely reported in recent years.110,293,364–368 In this section, we discuss some topical applications including: (1) the removal of solids or gases from the natural environment; (2) turning salt water into fresh water; (3) use as electrodes for electrochemical capacitors, batteries or electrochemical detection; and (4) the manufacture of catalysts or catalyst supports.4.1 Adsorption
Adsorption has proved to be a simple, effective and time-saving technology for the removal of pollutants, in which the key factor is the exploitation of economical and efficient adsorbents. MCs, which possess unique physical and chemical properties such as a large specific surface area, large pore volume, chemical inertness and good mechanical stability, gradually developed enormous advantages in adsorption. The presence of mesopores (2–50 nm) plays a significant role in the adsorption process of various molecules such as heavy metals, vitamins, dyes, drugs, amino acids, etc. (Fig. 22). The efficiency of adsorption on MCs is influenced by the molecular mass of the adsorbed substance and its size, geometry, solubility, polarity and functional groups. The ordered pore systems and well-defined pore size distributions of mesoporous materials influence the adsorption process of active compounds and their controlled release. | ||
| Fig. 22 Schematic of MCs as adsorbents. | ||
Furthermore, the impregnation of metals into MCs can intensify and expand the adsorption performance. Introducing metal sources into MCs, such as Fe, Ni, Mn and Co, will provide magnetism and also the possibility of creating specific binding sites whose binding force is conducive to adsorption. In addition, this also makes them easy to separate from an aqueous solution by applying an external magnetic field instead of centrifugation or filtration.
As a new agent in the field of adsorbents, OMCs have displayed a remarkable improvement in the adsorption of bulky dyes compared with commercial activated carbons.369,370 The amount of bulky dyes that is adsorbed (methylthionine chloride, basic fuchsin, rhodamine B, brilliant yellow, methyl orange, or Sudan G) is almost twice that with activated carbon, in which mesopores contribute almost 100% of the total surface area and volume. The OMC adsorbent provides a high adsorption rate (>99.9%) for low concentrations of dyes, good performance in decoloration regardless of the nature of the dye, including basic, acidic, or azo dyes, and high stability after elution of the dye. If the surface chemical properties of OMCs could be tailored by appropriate methods, effluents of better quality might be produced; moreover, heteroatoms on the surface of the carbon material can bond to the edges of the carbon layers and to a certain degree determine the surface chemistry of carbons.371 For instance, after thermal modification by ammonia, basic nitrogen-functionalized MCs displayed an enhanced adsorption capacity for three anionic dyes (orange II, reactive red 2 and acid black 1) compared to commercial activated carbon and unmodified carbon, respectively.118–120 This significant improvement is attributed to the increased dispersive forces between the carbon surface and the dye molecules that are induced by the nitrogen-containing functional groups.372 Some research also found that mesoporous nitrogen-enriched carbon materials had a new property of removing phenol not only by physisorption but also by catalytic photodegradation.310 As well as nitrogen groups, there are other novel OMCs containing magnetic components or metal nanoparticles; these adsorbents all display excellent adsorption properties for the toxic organic compounds rhodamine B,373 basic fuchsin,137 methylene green,98 and 4-nitrophenol.348 Furthermore, the adsorption capacity of the metal-containing adsorbent is relatively high compared with that of raw OMC and careful tuning of the wall chemistry and microporosity could even enable selective separations from aqueous mixtures.176 In addition, the magnetic carbon adsorbent can be regenerated by simply washing it with ethanol to recover both the adsorbent and the adsorbates.
The inert and hydrophobic nature of MC materials results in poor wettability and dispersibility in water, which would limit their potential in adsorption, whereas surface modification or functionalization of porous carbon materials is a crucial way to change the hydrophobicity and hydrophilicity of the surface of the materials in order to make them available as good adsorbents for the selective removal of some organic contaminants and biomaterials. For instance, modification of the carbon surface with COOH groups would help enhance its wettability with polar solvents and make the surface active for the covalent immobilization of proteins with enhanced adsorption capacity compared to that of pristine MC.374 After further oxidative treatment with H2O2, nanocomposites with a hydrophilic framework as well as increased mesopores and pore volume could be obtained; the modified nanocomposites displayed markedly improved adsorption properties for the dye molecule basic fuchsin in water.224 Furthermore, the characteristic hydrophobicity as well as size exclusion (against serum proteins) properties of the mesopores made OMC materials perform highly efficiently in the extraction and recovery of peptides from serum.375 Similarly, the electrostatic attraction and hydrophobic interaction between perfluorinated compounds (PFCs) and magnetic mesoporous carbon nitride materials made them more effective adsorbents to remove PFOS and PFOA pollutants from aqueous solution than previous examples.376
The effects of polarity conditions and the pH value of the solvent on adsorption have been discussed in detail for the MC CMK for the removal of vitamin E, L-histidine (His) and L-phenylalanine.377–379 The amount of vitamin E that was adsorbed on the different CMK adsorbents depended on the solvent as well as the mesopore volume and surface area of the adsorbent, whereas the specific mesopore volume rather than the specific surface area played a predominant role in its removal. It has also been found that a non-polar solvent such as n-heptane was more suitable compared to the polar solvent n-butanol to achieve high loadings of vitamin E. CMK-3 achieved the maximum adsorption of His or L-phenylalanine near their isoelectric point, which depended on the pH, and displayed a larger amount of adsorption of His or L-phenylalanine compared to ordered silicas, which was mainly due to the stronger hydrophobic interaction between the non-polar side chains of the amino acids and the hydrophobic surface of MC compared to mesoporous silica.
As well as the characteristics of the carbon surface and solvent, the morphology or pore structure of MCs also has an effect on adsorption. For the removal of bilirubin from plasma, OMC and hollow mesoporous carbon spheres (HMCSs) were employed as bilirubin adsorbents.380 The results demonstrated that HMCSs had a much higher adsorption capacity than that of OMC and commercial activated carbon as haemoperfusion adsorbents in PBS solution. In addition, HMCSs provided good selectivity in the adsorption of bilirubin against human albumin and in a hemolysis assay. To overcome some physicochemical drawbacks associated with conventional packed-bed reactors, such as fluid dynamics, pressure drop, heat and mass transfer, low contacting efficiency, and mechanical attrition, etc., hierarchically porous carbon monoliths (HPCMs) with both ordered hexagonal mesoporosity and three-dimensionally connected macroporosity highlighted their advantages.122 These monoliths shared high macropore volumes (48.6 cm3 g−1) and large specific surface areas (1354 m2 g−1), strong hydrophobicity (water contact angle: 140 ± 3°), low densities (0.017 ± 0.002 g cm−3) and regular shapes (e.g., cylinders). Importantly, these HPCMs exhibited excellent performance not only in cleaning/recycling spilled oil or organic solvents but also in removing bilirubin. They could adsorb oil with a weight of 23–48 times their own in a few seconds. Besides, their high durability in aqueous media, low density and good recycling stability made them almost ideal adsorbents for the recovery of spilled oil. On the other hand, their satisfactory compatibility with blood and regular shape made them promising adsorbents for the removal of bilirubin in clinical haemoperfusion.
For the enhancement of adsorption capacity, a hierarchical and interconnected pore structure in OMC can contribute to increased performance. OMCs with high pore interconnectivity have displayed enhanced adsorption capacities compared with non-interconnected CMK-3 and activated carbons.381 Moreover, interconnected hierarchically ordered micro/mesoporous carbons with a specifically tailored pore structure complemented the molecular dimensions of BPA with high diffusion kinetics and an ultra-high adsorption capacity of 1106 mg g−1.117 Similarly, in the adsorption of pharmaceutical antibiotics on ordered micro- and mesoporous carbons that were synthesized via a template, the size exclusion effect and a regular-shaped, open and interconnected three-dimensional pore structure were also demonstrated to be conducive to the adsorption of macromolecules and adsorption kinetics in comparison with other porous adsorbents.382 Again, adding an activating agent to increase the mesopore surface area was also a feasible way to achieve higher adsorption saturation capacity.61
Adsorbent coatings on inorganic monoliths are of great significance for industry because of their favorable properties such as low pressure drop, large geometric surface area, short diffusion lengths, lack of attrition by vibration, resistance to thermal shock, and convenient separation from media without the assistance of an external magnetic field.383,384 To expand the range of applications, Wan et al. used a honeycomb monolith coated by OMCs as a reusable adsorbent without any further activation and achieved adsorption capacities for chlorinated organic pollutants in water of 200 mg g−1 for p-chlorophenol and 178 mg g−1 for p-chloroaniline, a high adsorption ratio for low concentrations of pollutants, and large processing volumes and the adsorbent was reusable more than 200 times without obvious loss of either adsorption capacity or weight.116
Doping heteroatoms or other metal particles into pore channels is helpful for increasing adsorption sites. Nitrogen- and oxygen-containing MC materials with high heteroatom (O, N) contents of up to 7.5 wt% and 19.1 wt%, respectively, were synthesized via the utilization of a mixture of molten fructose and urea as the precursors and porous silica as a structure matrix under solvent-free conditions.115 Porous carbon materials that were modified by heteroatoms displayed significantly higher pore wall surface polarity and higher sorption capacity for Cu2+ ions from aqueous solution than those prepared from a melt of fructose. Moreover, owing to the successful application of nanosized iron particles to remove organic contaminants,386 heavy metals,387 and dyes,388 in view of their low cost, mild reaction conditions and easy separation, Tang et al. developed a new adsorbent that was endowed with improved properties by doping iron nanoparticles (FeO, Fe3O4 and λ-Fe2O3) on the OMC CMK-3.389 This adsorbent possessed enhanced ability for the adsorption and/or simultaneous reduction of Cr(VI) and it could also be easily magnetically separated and collected after loading pollutants. Moreover, Fe/CMK-3 loaded with Cr could be regenerated by 0.01 mol L−1 NaOH solution. Nevertheless, encapsulating high-content but uniformly dispersed and spatially separated nanoparticles while retaining an open mesopore system is highly desirable for adsorption. Such a carbon material with uniformly dispersed iron oxides with high content (>40 wt%) was ideal for the removal of arsenic with large capacities, fast adsorption rates, easy magnetic separation, and long cycling stability.231 Lin et al. successfully developed mesoporous Fe/carbon aerogel (CA) structures with large specific surface areas of 487 m2 g−1 via the carbonization of composite Fe3O4/phenol–formaldehyde resin.101 The mesoporous Fe/CA materials were further used for the adsorption of arsenic ions with a maximum uptake of arsenic ions calculated to be 216.9 mg g−1, which was higher than that observed for other adsorbents of arsenic. Moreover, the adsorbent Fe/CA could be easily separated from the solution using an external magnetic field. Furthermore, FeCl3·6H2O was introduced into the synthesis process of N-doped OMC to embed magnetic α-Fe nanoparticles in the framework of N-doped OMC, which was proved to provide enhanced uptake of Cu2+ and the ability to be separated from the solution under an external magnetic field, as well as reusability.320
Recently, the use of low-cost and ubiquitous lignocellulosic materials (natural materials or agricultural wastes) to synthesize adsorbing materials has attracted growing attention. Mesoporous activated carbons that used water hyacinth as an efficient raw-material precursor via activation with H3PO4 exhibited high mesoporosity (93.9%) with a surface area of 423.6 m2 g−1 and abundant oxygen-containing functional groups including hydroxyl, carbonyl, carboxyl and phosphate groups, which allowed the diffusion of Pb(II) into the pores with a maximum monolayer capacity (qm) of 118.8 mg g−1.102 Moreover, adsorption–desorption results showed that the adsorbent could be readily regenerated using 0.1 M HCl (pH = 1.0). The desorbed carbons could be reused at least six times without significant reduction in adsorption capacity. The surface of obtained duckweed-based mesoporous activated carbons could easily bind Pb(II) via the formation of strong chemisorptive bonds or ion exchange, with a monolayer adsorption capacity of 170.9 mg g−1 at 25 °C.220 In another case, mesoporous magnetic carbon nanocomposite fabrics that were prepared from precursors of commercial T-shirt cotton fabric and iron nitrate exhibited a much higher removal capacity for Cr(VI) than those of cotton and carbon fabrics and a much faster adsorption rate than other reported materials such as carbon, waste biomass and lignocellulosic substrates.233
| CO2 + 2RNH2 ↔ NH4+ + R2NCOO− | (1) |
| CO2 + 2R2NH ↔ R2NH2+ + R2NCOO− | (2) |
| CO2 + 2R3N ↔ R4N+ + R2NCOO− | (3) |
Because nitrogenous functional groups in MCs can increase their affinities for acidic carbon dioxide, mesoporous carbon materials incorporating nitrogen functional groups in their framework were prepared by carbonizing MF/silica composite precursors.105 The prepared MCs have large specific surface areas (974 m2 g−1) with well-developed mesopores. The highest adsorption capacity for carbon dioxide of 106 mg g−1 at 25 °C was achieved with the MCs-800 sample (carbonization temperature of 800 °C). Furthermore, N-doped OMCs that were based on two important nitrogen-containing precursors, resorcinol–melamine–formaldehyde (RMF) and resorcinol–urea–formaldehyde (RUF), achieved almost the same performance in the capture of CO2.299,300 Other synthesized nitrogen-doped OMC nanospheres with a mean diameter of around 240 nm possessed a variable surface nitrogen content of 0.38–1.4 wt% and a uniform pore size of around 2.8 nm.106 The nitrogen atoms were mostly bound to the graphitic network in two forms, i.e., pyridinic and pyrrolic nitrogen, which provided adsorption sites for acidic gases. N-doped OMC materials exhibited good performance in the capture of typical acidic gases such as CO2 (2.43 mmol g−1) and SO2 (119.1 mg g−1). In addition, the uptake of acid gases was similar under the same conditions in at least five repeated runs without noticeable reduction. In another case, which is ascribed to the unique features of large surface area and high N content (up to 13.1 wt%) in two dominant forms, tertiary nitrogen and pyridine N-oxide, the prepared materials exhibited high ability to capture CO2 (2.8–3.2 mmol g−1 at 298 K and 1.0 bar).133 To our knowledge, alkali metal-promoted alumina and carbonates,399,400 hydrotalcites,401 layered double hydroxides402 and calcium oxides403–405 were reported to be capable of the capture of CO2 at elevated and/or high temperatures. With large and regular mesoporosity, high thermal stability and good affinity for metal species, novel mesoporous nanocrystalline calcium adsorbents supported on carbon that were capable of the capture of CO2 were synthesized via a three-constituent co-assembly pathway.107 The composite materials were of significance for the physisorption of CO2 at ambient temperatures with competitive capacities (up to 7 mmol g−1) and selectivity over N2. Moreover, nanocrystalline calcium oxides were highly active in the chemisorption of CO2 and almost complete initial conversion and fast reaction kinetics at a low temperature (450 °C) and low pressure of CO2 could be achieved within minutes.
Hydrogen is considered a promising renewable non-polluting alternative to fossil fuels. One of the key problems in promoting a hydrogen economy is obtaining efficient and safe storage materials for hydrogen. In spite of intensive research activity in hydrogen storage, several barriers remain. For instance, considering carbon-based materials as major candidates for physisorption, although adsorption processes possess several advantages, under reasonable conditions of pressure and temperature the adsorption capacity of pure carbons could not exceed a few weight percent of hydrogen.406,407 However, metal particles dispersed in the pores of active carbons can greatly enhance storage abilities; for example, the electrosorption of hydrogen could be improved by coating with nickel nanoparticles as redox sites.408 On the other hand, the incorporation of nickel in pure porous carbons is beneficial, whereas the combination of nickel and nitrogen impairs storage capacities in terms of the electrosorption of hydrogen. Therefore, OMCs that were enriched with nitrogen, then loaded with nickel nanoparticles were prepared and the amount of hydrogen that was adsorbed at temperatures above 298 K was enhanced by the presence of both nitrogen and nickel additions or substitutions.409 In addition, the activation of MCs by KOH, CO2 or steam at high temperatures has been widely used for the generation of microporous carbon structures and can greatly enhance their hydrogen storage performance.410 As one of the most abundant biopolymers in nature, chitosan is widely used to adsorb transition metals, precious metals and rare metal ions by a chelation mechanism due to the amino groups and secondary hydroxyl groups on its chain. In virtue of the above properties, a cobalt-chelated chitosan solution was used as the carbon precursor for the synthesis of OMCs.108 The prepared OMCs with embedded cobalt possessed a markedly higher hydrogen adsorption capacity than that of pure OMC. In short, the Kubas interaction along with hydrogen spillover and physisorption on the carbon support played roles in the mechanism of hydrogen adsorption in MCs with embedded cobalt.
Hitherto, various technologies have been developed for the separation/purification of gases, such as cryogenic distillation, absorption, membrane separation, and adsorption. Among these, adsorption has received intense interest due to its great advantages: high energy efficiency, ease of control, and low capital investment costs. The separation of CO2 and N2 from CH4 is highly important in the upgrading of natural gas and the capture/removal of CO2 and CH4 from air (N2) is essential for the control of greenhouse gas emissions. Deng et al. reported a versatile OMC material that displayed both high selectivities and large adsorption capacities for separating CO2/CH4, CH4/N2, and CO2/N2 mixtures.104 At 278 K and 100 kPa, the predicted selectivities for equimolar CO2/CH4, CH4/N2, and CO2/N2 were 3.4, 3.7, and 12.8, respectively, and the adsorption capacities for CH4 and CO2 were 1.3 and 3.0 mmol g−1, respectively. In this field, MC membranes occupy an important position in gas separation. Recent progress has been made in this respect: a kind of “green” synthesis method from larch sawdust resulted in an OMC membrane that was obtained at 700 °C and exhibited high efficiency (permselectivity factor = 1.97) in the separation of CO2 from a N2/CO2 mixture.155 Some works also described that post-synthetic treatment of MC membranes with ammonia at elevated temperatures could improve the permeance and selectivity of these membranes for the separation of carbon dioxide and hydrocarbons from permanent gases; this was because treatment with ammonia at high temperatures provided a controlled method of introducing both added microporosity and surface functionality to enhance the gas separation performance of porous carbon membranes.158 Moreover, some mesoporous carbon composite membranes also exhibited good performance in this field. Two typical examples could be given by Wang et al. and Dai et al.: ordered mesoporous silica/carbon composite membranes exhibited outstanding gas permeability and selectivity for CO2 in the separation of the CO2/CH4 and CO2/N2 gas pairs, owing to the lower resistance to gas diffusion through the membrane and additional gas permeation channels that were created by the incorporation of mesoporous silicas in a carbon membrane matrix;156 and free-standing mesoporous carbon–graphitic carbon black nanocomposite membranes that supported ionic liquids had a permeability for CO2 of up to 180 Barrer and good selectivity (∼36) from CO2/N2 mixed gases under a high transmembrane pressure (1000 kPa) without degrading their separation performance, which widened the application of high-pressure processes for the capture and separation of CO2.157
As a novel desalination technology, capacitive deionization (CDI) is a kind that has low energy consumption and produces no secondary waste. CDI operates at low potentials in the range of 1–1.4 V and membranes and high-pressure pumps are not required for its operation, so problems of scaling and fouling can be avoided. As illustrated in Fig. 23B, CDI employs the principle of adsorption of ions on an electrode surface due to electrostatic force. When an external electric field is applied, ions with opposite charge move to the electrical double layer (EDL) that forms near the electrode surface. When the force is removed, the ions will be desorbed and return into the bulk solution for further use without secondary waste. On the basis of this mechanism, the capacity of CDI strongly depends on the conductivity and surface properties of the materials. It is clear that MC displays much better electrochemical performance than conventional large-surface-area activated carbon in both aqueous and non-aqueous electrolytes.412–414 Graphene (GE), which is a two-dimensional carbon, is considered a promising electrode material due to its outstanding properties such as high electrical conductivity, large surface area, good mechanical properties and chemical inertia. These intriguing mechanical and electrical properties enable GE to be a free-standing electrode for energy storage as well as electrosorption.415 Therefore, GE/MC composites that are used as CDI electrodes have been demonstrated to perform better in terms of capacitance values, conductive behaviour, rate performance and cycling stability.416 Well-dispersed GE nanosheets were confirmed to be beneficial for enhanced electrical conductivity. Hence, GE/MC composite electrodes were proved to be promising electrodes for capacitive deionization, in contrast to those based on MC and activated carbon.
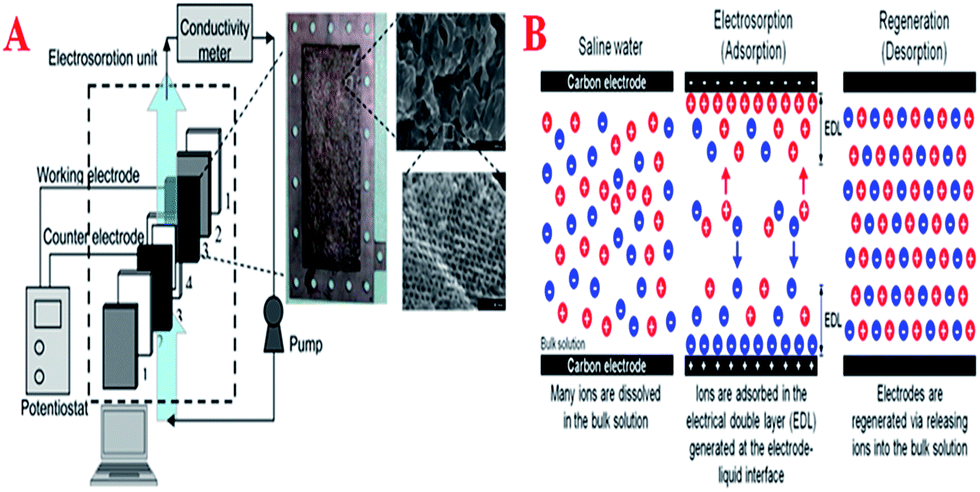 | ||
| Fig. 23 (A) Single-cell capacitive deionization reactor with two half-cells, each of which containing (1) a Plexiglas cover, (2) a current collector, (3) a carbon electrode, and (4) a middle hollow plate. Right: STEM images of phloroglucinol-based MC utilized to produce the MC-coated graphite electrode materials with uniform pores of ∼8 nm (bottom right) and combined hierarchical meso- and macroporosity (top right). Reprinted with permission from ref. 110; (B) adsorption/desorption behavior of ions in capacitive deionization of saline water. Reprinted with permission from ref. 421. | ||
Although previous works have mainly focused on carbon aerogel as an electrosorptive material,417–419 its microporosity and uncontrollable pore distribution limit the rates of ion transport, so its capacitance may not be as high as that of mesoporous materials. With regard to mesoporous materials employed for CDI, Tsouris et al. have made an in-depth study. They investigated the CDI performance of self-assembled MC (resorcinol or phloroglucinol-based) electrode materials and commercial carbon aerogel at different applied voltages and solution concentrations.110 The laboratory-scale CDI reactor consisted of a single pair of carbon electrodes and a pair of current collectors (Fig. 23A). The results that were obtained suggested that MC materials were superior to carbon aerogel with respect to capacitive deionization and phloroglucinol-based MC-coated graphite (PMCG) exhibited the highest ion removal capacity, at 21 mg salt per gram carbon for a salt concentration of 35000 ppm, which indicated that these materials were a more suitable choice for carrying out desalination of salt water by the CDI process. Another similar publication revealed that PMCG displayed a faster deionization rate than that of resorcinol-based MC-coated graphite (RMCG).420 By employing neutron imaging of lithium-6 ions to observe the electrosorption and regeneration behavior during CDI at varying electrolyte concentrations, Tsouris et al. found that the ion transport behavior during the sorption and regeneration phases in CDI could aid in making the CDI process more efficient and provided insight into why the desalination of high-salinity solutions by CDI is challenging.421 The mechanism could be explained as follows: firstly, the very fast ion transport in the discharge process produces a higher concentration of ions than in the bulk solution; later, this transport process prevents the electromigration of ions and leaves only diffusion transport, which is further slowed down by negative differences in the activity coefficients. Therefore, all these greatly decrease the efficiency of the CDI process at high saline concentrations. In combination with the theoretical model, this work could help in the design of CDI devices to improve the process for solutions with high ionic strength.
4.2 Electrode materials
A lot of research has been carried out on electrochemical applications due to the excellent properties of OMCs: (1) high conductivity to assure efficient delivery of electrons; (2) large specific surface area and uniform pore size distribution consisting of nanopores or hierarchical channels, which is useful for increasing capacitance and facilitating the diffusion of ions; and (3) a large number of surface functionalities that increase their ability to capture or immobilize active sites and speed up redox reactions to increase the capacitance via pseudo-capacitive processes.The electrochemical applications of MCs can benefit from their one-dimensional (1D) nanostructures such as nanofibers and nanotubes with well-controlled dimensions and extra surface area; besides, 1D nanostructures provide a shorter ion diffusion path than bulk forms for electron transportation.425–427 Moreover, ordered mesoporous structures can facilitate the penetration of electrolytes from a direction perpendicular to the longitudinal axis of the nanostructures. These excellent properties render MC nanofibers more desirable as electrode materials for supercapacitors than mesoporous carbon in high-rate charge/discharge operations.112 Similarly, some MC nanosphere electrodes not only exhibited a specific capacitance of 180 F g−1, but also displayed good cyclability, with 78% energy density and more than 90% power density remaining after 700 cycles at a current density of 3.0 A g−1.113 However, monodisperse MC spheres with a diameter of 1.2 μm, a large specific surface area (up to 1460 m2 g−1) and a uniform pore size of as high as 31 nm performed less effectively.79 Moreover, some nanocomposites with 40 wt% polyaniline nanowhiskers (PANI-NWs), which were grown vertically on the outer surface of CMK-3 particles via chemical oxidative polymerization, when applied in supercapacitor devices possessed a large specific capacitance of 470 F g−1 and good capacitance retention of 90.4% after 1000 cycles at a current density of 1.0 A g−1.428 Carbon microspheres are usually regular spheroids with homogeneous packing, which can decrease the resistance to diffusion of liquids and hence improve the capacity of the electrode. The space between the microspheres can provide the electrolyte with access to the electrodes, which is beneficial for the formation of double-layer capacity at the carbon/electrolyte interface. Novel MC microspheres, which were produced via a hydrothermal emulsion-activated method, as electrode materials exhibited a specific capacitance of 157 F g−1 at a high current density of 10.0 A g−1 in 6 M KOH aqueous solution.177
Surface modification under conditions of high temperature or treatment with a base or acid can generate some functional groups, such as CH, NH and COOH, which usually promote the hydrophilicity of carbons in aqueous electrolytes. In addition, these groups could introduce an amount of pseudocapacitance as a result of their redox reactions with electrolyte ions in charge–discharge processes.429,430 Some studies reported that nitrogen-enriched carbons displayed excellent electrochemical performance because of pseudocapacitive interactions between electrolyte ions and surface nitrogen active sites431,432 and improvements in the wettability of the electrode. For example, Song et al. synthesized N-doped OMC as an electrode material for supercapacitors; the obtained carbon exhibited excellent cycling stability and delivered a reversible specific capacitance of as high as 308 F g−1 in a 1 mol L−1 H2SO4 aqueous electrolyte, of which 58% of the capacity was due to pseudocapacitance.114 In another case, abundant nitrogen-containing functional groups with a nitrogen content of 10.72 wt% and a developed mesoporous structure endowed carbon with good rate capability, a relatively high capacitance of 198 F g−1 in 6 mol L−1 KOH aqueous electrolytes and excellent cycling durability.312 In order to further enhance the electrochemical performance of such N-doped MC materials, Li and Xue also developed an integrated and facile strategy to fabricate NC, using melamine resin as a nitrogen source and phenolic resin as a carbon source (Fig. 24A), and nitrogen-doped graphene (NG) that was fully covered with a layer of N-doped MC on a graphene sheet (shown in Fig. 24B).433 As compared to pure MC materials, NC and NG that were reported in this work exhibited great potential as efficient electrode materials for supercapacitors with higher specific capacitance (238 and 289 F g−1 at a current density of 0.2 A g−1) and excellent rate performance.
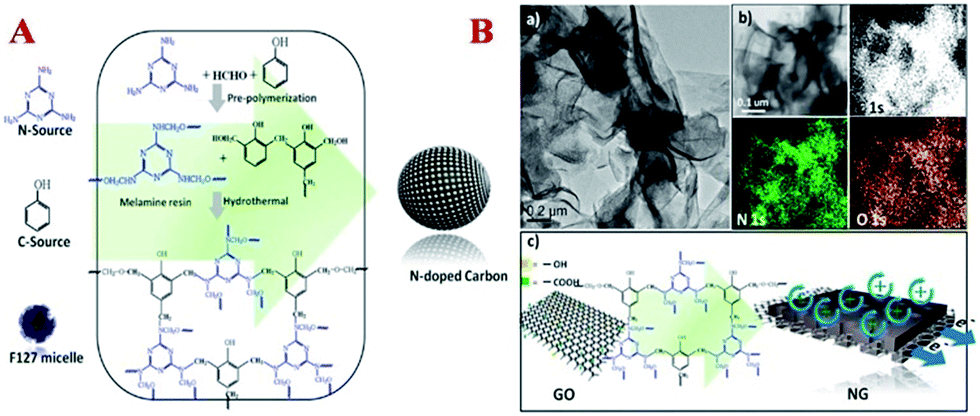 | ||
| Fig. 24 (A) Schematic of general strategies for preparing nitrogen-doped mesoporous carbon; (B) (a) TEM image of NG; (b) TEM mapping images of NG with carbon, nitrogen, and oxygen; (c) scheme illustrating the formation of N-doped graphene associated with rapid electron transport and electrolyte ions that effectively accumulated beside the N-doped porous carbon layer. Reprinted with permission from ref. 433. | ||
As reported, B-containing MC also manifested good electrochemical properties as electrodes in a supercapacitor.434 Furthermore, OMCs that incorporated B and/or P and were synthesized via a facile multi-component self-assembly method exhibited superior electrochemical performance to their non-incorporated counterparts when used as electrodes in supercapacitors.435 It was found that the specific capacitance (F m−2) of OMC increased by more than 50% via the co-incorporation of B and P. More interestingly, OMC with co-incorporated B and P exhibited significantly improved capacitance retention at a high potential scan rate, which is desirable in difficult power supply conditions. For applications of supercapacitors in a wide range of temperatures, the low-temperature operation of EDLCs was examined using MgO-templated MC or a commercially available activated carbon electrode in three kinds of ionic liquid: BMIBF4, HMIBF4, and DEMETFSA.373 MCs exhibited greatly increased capacitance in ionic liquids at 20 to −40 °C compared to conventional activated carbons. Mesopores in the carbon electrodes provided a smooth pathway for ions and minimized the influence of temperature on the diffusion resistance of the ions below 0 °C in ionic liquids.
To confirm the common assumption that the additional contribution from pseudocapacitance is due to enhanced capacitance performance originating from heteroatom-doped carbon materials,436–440 direct evidence of reversible redox peaks that originated from a faradaic reaction was observed by Zhang et al. in the synthesis of 3D cubic ordered mesoporous carbon (KNOMC) that was co-doped with nitrogen and sulfur with controlled dopant contents (4.6–10.0 atom% for nitrogen and 0.75–0.94 atom% for sulfur) and served as electrodes in supercapacitors.293 The synthesized materials exhibited excellent supercapacitive performance with a high specific capacitance of 320 F g−1 at a current density of 1 A g−1 in 2 M potassium hydroxide (KOH) by combining electrical double-layer capacitance and pseudocapacitance as well as enhanced wettability and improved conductivity. It was noticed that the pseudocapacitance, which originated from redox or faradaic charge-transfer reactions because of the chemical doping of heteroatoms into the frameworks of carbon materials, was proven by the apparent reversible redox peaks that are shown on cyclic voltammetry (CV) curves (shown in Fig. 25A, B and D). When the scan rate was increased to 100 mV s−1 all redox peaks decreased (shown in Fig. 25C), but the CV curves for all samples still retained an approximately rectangular shape with slight distortion because of a fast charge/discharge process and the high power capability of KNOMC as electrode materials in 2 M KOH aqueous solution.
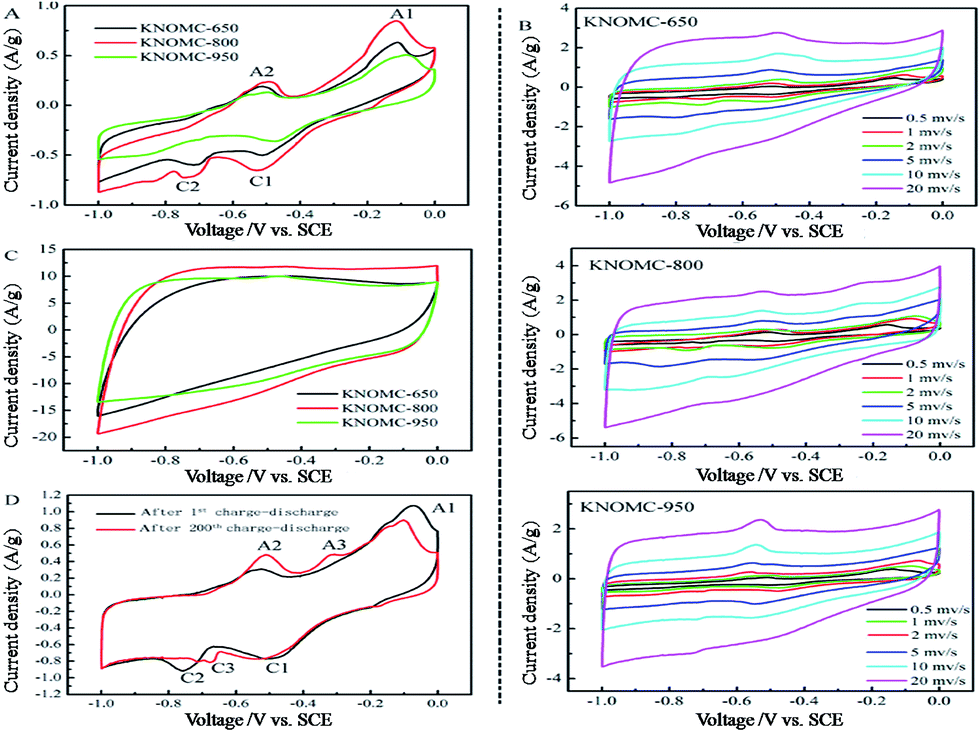 | ||
| Fig. 25 CV curves of KNOMC electrodes at a scan rate of 1 mV s−1 (A). CV curves of KNOMC electrodes at varying scan rates between 0.5 and 20 mV s−1 (B). CV curves of KNOMC electrodes at a scan rate of 100 mV s−1 (C). CV curves of KNOMC-850 electrodes at a scan rate of 1 mV s−1 after 1 and 200 cycles of charge/discharge (D). Reprinted with permission from ref. 293. | ||
The activation of MCs using an activating agent is another feasible way to improve the performance of capacitors. For example, a series of highly porous nitrogen-containing carbons have been prepared via high-temperature activation with KOH. These samples exhibited very promising potential as electrode materials for electrical double-layer capacitors with an optimized electrical capacitance of 318 F g−1.441 Similarly, Nishiyama et al. used KOH to activate OMCs that were synthesized by a soft-templating method and achieved enhanced electrochemical properties that are desirable for electrodes in electrochemical double-layer capacitors.111 Dai et al. also found that synthesized MCs when used as supercapacitor electrode materials exhibited gravimetric specific capacitances of 77.1, 102.3, and 91.7 F g−1 for pristine, CO2-activated (with 56% burn-off), and KOH-activated (200% KOH loading) carbons, respectively.221 Again, MC nanofibers with large cage-like pores were prepared by combining electrospinning and thermal treatment of the composite fibers using SnO2 as an activator.203 The capacitor that was assembled with MC nanofibers exhibited excellent rapid charge/discharge properties and its specific capacitance could reach about 105 F g−1 in 6 mol L−1 KOH aqueous solution.
Ruthenium (Ru) is known to be the best electrode material for faradaic capacitors due to its outstanding specific capacitance and long cycle life. He et al. presented a facile technique for preparing Ru/MC composites from RuCl3 and cheap MC that was derived from peanut shells for supercapacitors by a method involving microwave-assisted heating and reduction by glycol.442 The specific capacitance of the obtained Ru/MC composite increased with an increase in the mass loading of Ru and was maintained at 287 F g−1 with an energy density retention of 93.3% even after 1000 cycles at 20% loading of Ru. Moreover, the pore size distribution also has an effect on the performance. Hierarchically porous carbons with mesoporous channels and micropores have been shown to display a good electrocapacitive rate performance.443,444 Furthermore, porous carbons with a large surface area, large ion channels and nanometer-scale diffusion length are suitable for high-rate supercapacitors.445,446 Therefore, several high-capacitance OMC materials with a bimodal pore distribution were synthesized to be evaluated as electrode materials with a specific capacitance of 0.15 F m−2.447 Again, Wu et al. successfully introduced micropores into the skeleton of carbon aerogel without damaging its unique 3D mesoporous nanonetwork structure by semi-carbonization assisted by NH3. The novel carbon aerogel, which was loaded with pyrrolic/pyridone (N-5) and pyridinic (N-6) nitrogen functional groups and abundant micropores, was then used as an electrode material in supercapacitors and exhibited much higher specific capacitance (208 F g−1) and comparably high capacitance retention compared with traditional carbon aerogel.347
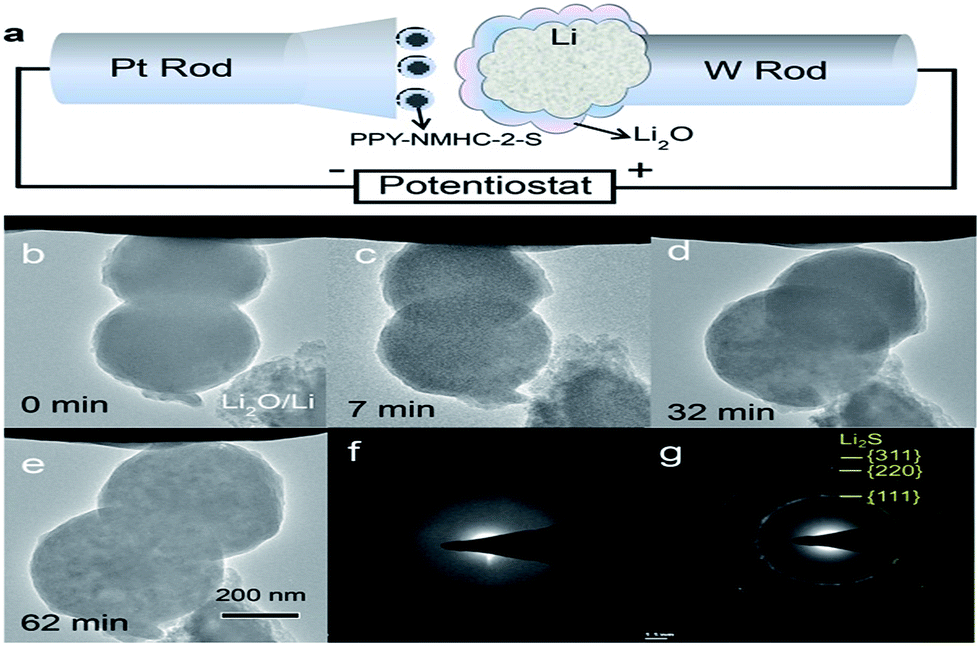 | ||
| Fig. 26 (a) Schematic of in situ TEM testing. (b–d) In situ TEM images of NMHC-2-S coated with polypyrrole during the lithiation process. (f and g) Diffraction mode of the particles in figures (b) and (e), respectively. Reprinted with permission from ref. 366. | ||
Some work also showed that a nitrogen functionality on carbon was responsible for the electrocatalytic activity of the cathode and an enhancement in the cell capacity of a lithium–oxygen battery.466 In addition, some nitrogen-rich pyrolytic carbon materials displayed reversible lithium storage capacities and stable cycling performance in lithium-ion batteries,84 because the structure of MC became more disordered during discharge and was restored during recharge. With respect to redox flow batteries, the all-vanadium redox flow battery ([VO]2+/[VO2]+ [positive]–V3+/V2+ [negative] redox couples) is limited by the redox reaction of the V(II)/V(III) couple (negative side) and the reaction kinetics between them from the electrochemical aspect. The electrocatalytic kinetics and reversibility of the redox couple [VO]2+/[VO2]+ can be significantly enhanced on a nitrogen-doped MC electrode compared with MC and graphite electrodes, as nitrogen doping facilitates the electron transfer at the electrode/electrolyte interface in both oxidation and reduction processes.467
As the core component of an electrochemical sensor, a substrate electrode (a glassy carbon electrode, for example), its modification with OMC is very simple. Usually, the prepared OMC material is first dissolved in Nafion/ethanol solution, then, after ultrasonic treatment for dispersion, the OMC-Nafion mixed solution will be cast on pre-cleaned glassy carbon electrodes (GCEs), followed by drying under ambient conditions to obtain the final product. Functionalized glassy carbon electrodes with OMC exhibited high sensitivity of 62.7 μA cm−2 per ppb towards 2,4,6-trinitrotoluene (TNT).468 Because of its large specific surface area and fast electron transfer capability, for sensing TNT OMC achieved greatly superior performance compared with other materials such as carbon nanotubes and ordered mesoporous silica. More notably, even ultra-trace nitroaromatic concentrations as low as 0.2 ppb TNT, 1 ppb 2,4-dinitrotoluene and 1 ppb 1,3-dinitrobenzene could also be detected with electrodes modified by OMC.
Selectivity is of great importance when considering biosensors for detecting phenol derivatives in compost systems; on the other hand, the appropriate immobilization of an enzyme on the electrode surface is considered a key step in the development of biosensors for the determination of phenols. By virtue of its higher affinity for the bio-activator, higher bioactivity and faster electron transfer between the bio-activator and ordered mesoporous carbon nitride (MCN) sensing sites, Zeng et al. proposed a chronoamperometric biosensor based on MCN/tyrosinase (Tyr), which exhibited high specificity toward phenol and catechol in compost samples (as shown in Fig. 27).367 In this work, Tyr could be immobilized on MCN via adsorption (Fig. 27A), cross-linking (Fig. 27B) or covalent attachment (Fig. 27C) and applied in biosensing using enzyme activity. The reduction in current for chronoamperometric measurements of phenol and catechol was proportional to their concentration in the ranges of 5 × 10−8 to 9.5 × 10−6 M and 5 × 10−8 to 1.25 × 10−5 M with correlation coefficients of 0.9991 and 0.9881, respectively. The detection limits for catechol and phenol were 10.24 nM and 15.00 nM (S/N = 3), respectively. This proposed enzyme-based biosensor provided a powerful tool for the rapid, sensitive, and especially selective monitoring of catechol and phenol simultaneously.
 | ||
| Fig. 27 Schematic of the preparation of a tyrosinase biosensor and the proposed mechanism of the electrocatalytic detection of phenol and catechol (a). Signal transduction and amplification mechanism of the biosensor (b). Reprinted with permission from ref. 367. | ||
Despite displaying high selectivity, a traditional enzymatic sensor has a lack of stability due to the intrinsic nature of the enzyme, which is especially poisoned by adsorbed intermediates, whereas carbon-based electrodes display good electrochemical behavior in the determination of some molecules even in the presence of high concentrations of poison.469,470 In virtue of the important fact that the large active surface area of carbon-based electrode materials played a key role in the electro-oxidation of glucose, a novel non-enzymatic amperometric sensor for glucose based on OMC was prepared to evaluate its electrocatalytic activity in the non-enzymatic detection of glucose in alkaline media.471 The results demonstrated that OMC displayed electrocatalytic activity in the oxidation of glucose in alkaline solution and the resulting biosensor exhibited excellent performance for the determination of glucose with high sensitivity of 10.81 μA mM−1 and a low detection limit of 0.02 mM. More importantly, the OMC-modified electrode could resist interference from common interfering substances such as ascorbic acid, dopamine and uric acid.
Removing and detecting carcinogenic compounds remains an important and major task for human health. On account of the potential adsorption of N-nitrosopyrrolidine (NPYR) and its fluorescent properties, a good fluorescence sensor for NPYR has been prepared using cyanamide as a precursor and colloidal silica as a template for the synthesis of metal-free MCN.294 Owing to its structure and nitrogen functional groups, the adsorption of NPYR on MCN has been found to occur as monolayer adsorption with the presence of heterogeneous adsorption sites. Fluorescence spectroscopy further illustrated that two excitation wavelengths, which were due to the terminal N–C and NC groups, were used to monitor the interactions between the emission sites of MCN and NPYR molecules. It was also confirmed that the intensity of the emission was quenched almost linearly with the concentration of NPYR and that the terminal N–C groups on MCN should be the favoured sites for interaction with NPYR.
Electrochemiluminescence (ECL), which is the production of light emissions from an electrochemically generated luminophore, is a noteworthy, versatile and sensitive analytical method that has emerged in various research fields such as chemical sensing, imaging, optical studies, and environmental, clinical and medical analysis. Among various ECL compounds, Ru(bpy)32+, which exhibits good electrochemical stability, high sensitivity and efficiency, has received great attention. Due to the excellent ion exchange ability of Nafion for Ru(bpy)32+, which is as high as 5.7 × 106, and the strong adsorption by OMC, Jia et al. prepared an OMC@Nafion-based composite film to immobilize Ru(bpy)32+ for modifying the surface of a glassy carbon electrode (GCE).472 The fabricated GCE, which was modified by a Ru(bpy)32+/OMC@Nafion composite film, displayed good sensitivity for the ECL determination of tripropylamine (TPA) with a wide linear range (4.75 × 10−9 to 6.25 × 10−4 M) and low detection limit (1.58 × 10−9 M). Even in the presence of TPA as co-reactant, the Ru(bpy)32+ ECL sensor could be applied in the determination of dopamine (DA) with a good response to its concentration from 5.0 × 10−9 M to 5.0 × 10−4 M. More importantly, this ECL sensor based on Ru(bpy)32+–TPA provided a convenient, fast and sensitive way to detect DA with high accuracy in real sample analysis.
A molecularly imprinted polymer (MIP) is a type of synthesized polymer material, which is termed a man-made antibody due to its high affinity for template molecules. Compared with a natural antibody, an MIP has higher resistance to environmental temperatures, pH, and salts. Therefore, MIPs have been widely utilized for separation and detection purposes as molecular recognition materials. Tan et al. developed a sensitive method for the determination of ofloxacin (OFL) based on molecularly imprinted polymer/mesoporous carbon nanoparticles (MCNs@MIP) using OFL as a template. MCNs@MIP was synthesized first by covalent grafting of the template agent onto the surface of MC nanoparticles, and then a glassy carbon electrode modified by MCNs@MIP (MCNs@MIP/GC) was developed for the selective detection of OFL.473 The imprinted sites in MCNs@MIP/GC could specifically recognize and enrich OFL molecules in aqueous solution, achieving higher sensitivity and selectivity in detection compared with MCNs@NIP/GC and bare GC electrodes. There was also a linear relationship between the peak currents in cyclic voltammetry measurements and ofloxacin concentrations in the range of 0.5–100 μM, with a limit of detection (S/N = 3) of 80 nM.
4.3 Catalyst supports or catalysts
In industrial catalysis, solid catalysts could avoid the corrosion and environmental problems associated with liquid catalysts and also lead to lower production costs because of the easy separation and recycling of the product. The development of new basic solid catalysts is a rapidly expanding field. Over the years, many types of solid catalyst supports have been developed ranging from metallic oxides, zeolites, and active carbon to ion-exchange resins. Compared with the above materials, MCs, which possess tunable and large pore sizes, large surface areas, and even periodically arranged monodisperse mesopore spaces and alternative pore shapes, are more suitable as supports for catalysts or active heterogeneous catalysts for the development of environmentally sustainable chemical processes.The MC-based catalysts that have been developed have been employed in various organic transformations such as base-catalyzed reactions, selective oxidation, dehydrogenation, photocatalysis, and electrocatalysis. To optimize the use of these catalysts in various applications, it is necessary to attach functional groups or heteroatoms to their surface or framework. At the same time, efficient methods for the modification of carbon surfaces, such as grafting via the electrochemical or chemical reduction of aryl diazoniums, reductive alkylation and arylation, have been developed and could be applied for their functionalization. The presence of an adequate number of catalytically active sites that are exposed on MC materials, which are typically characterized by a large surface area and excellent electrical conductivity, makes them attractive single-phase catalysts without any additional conductive support. For example, amorphous carbons bearing SO3H groups exhibited remarkable catalytic performance in various acid-catalyzed reactions with hydrophilic reactants, such as esterification,474 transesterification,475 hydration,476 and hydrolysis reactions.477
In heterogeneous catalysis, the introduction of nitrogen-containing groups as well as non-precious metal oxides into carbon materials alters the basic surface properties and plays an important role in catalyzing reactions or increasing the dispersion of active components/promoters and therefore the activity. The type of nitrogen functional groups and the nitrogen doping level strongly depend on the synthetic conditions that are used. Moreover, textural features (pore structure, nitrogen in a carbon skeleton, hydrophilic or hydrophobic properties) also play an important role in the activity and selectivity of a catalyst. For example, the incorporation of a transition metal oxide or N-doped carbon can further improve performance in the oxygen reduction reaction (ORR). The hydrophobic surface of carbon materials can effectively adsorb long-chain organic molecules such as free fatty acids and avoid the adsorption of the water by-product that can lead to deactivation, resulting in high catalytic performance, whereas hydrophilic functional groups inside carbon particles prevent the incorporation of hydrophobic reactants into the carbon bulk. As a result, the carbon material exhibits poor or no catalytic activity in hydrophobic reactions. Pore size is another concern with mesoporous materials in catalytic applications, such as the synthesis of biodiesel involving large organic molecules. A pore size that favors the diffusion of large organic molecules is considered to contribute to the enhancement of catalytic properties.
For the design of robust and highly efficient metal-supported carbon catalysts, it is important not only to control the size and shape of the nanoparticles, but also to take into account metal-support interactions. The most important drawback of metal nanoparticles is their tendency to aggregate, especially with high metal contents. In general, the incorporation of active metal species into MC can be accomplished by soft-templating routes, while retaining the ordered mesostructure with finite loading. The fabrication of metal-based MCs can also be achieved via a conventional hard-templating approach. The latter route is largely based on conventional techniques, such as wet impregnation followed by chemical reduction of the metal nanoparticle precursors under an inert atmosphere. This tedious post-synthetic method makes unstable catalysts with unevenly dispersed metal nanoparticles on their external surface or near the mouths of pores, which results in a significant loss of reactivity during recycling because of the weak interaction between nanoparticles and the MC support. In addition, techniques based on thermal heating are time-consuming and often lack control of the particle size and morphology. However, the use of a supramolecular organic template involving the cooperative assembly of molecules via non-covalent interactions has the prospect of becoming a candidate for the fabrication of tailored mesoporous carbons that incorporate metals, with the reduction of aggregation during thermal treatment and the course of the reaction. For example, an approach using cyclodextrin yielded a more exposed metallic surface thanks to a high dispersion of metal nanoparticles478 and the direct pyrolysis of CTAB–chitosan–nickel supramolecular aggregates resulted in an evenly dispersed nickel complex on the mesoporous carbonaceous framework, which was an economical step for the in situ construction of homogeneously dispersed metal nanoparticles that were firmly attached inside the carbonaceous matrix.368 Most importantly, nanocatalysts from well-dispersed metallic nanoparticles displayed higher catalytic activities than those obtained from conventional supported systems. Unfortunately, the resulting carbonaceous frameworks were always disordered and lacking in uniform pore size distributions, thus blocking mass transportation to some extent.
Metal-free carbon catalysis has been another hot topic recently. Current industrial processes are based on metal oxide catalysts that work at high temperatures in excess steam. Actually, carbon materials, especially OMC without the deposition of metal particles, have been demonstrated to be highly selective and active catalysts for dehydrogenation reactions under more favorable reaction conditions (lower temperatures, without steam, energy-saving) than those of industrial catalysts.479–481 Surface basic oxygen functional groups that formed during the reaction were believed to be the active sites for the reaction. For example, MC that was activated with HNO3 was demonstrated to be a robust metal-free catalyst for the direct dehydrogenation of propane without any auxiliary steam, exhibiting high selectivity and stability.482 Besides oxygen, other elements, such as N, S, P, B, Cl, I, and Se, have been doped into carbon materials to give a metal-free electrocatalyst for ORR with improved performance compared to undoped carbon. For example, boron- and fluorine-containing mesoporous carbon nitride as a metal-free catalyst also exhibited good performance in the oxidation of cyclohexane with good conversion and excellent selectivity for cyclohexanone.483 MC that was co-doped with nitrogen and sulfur displayed remarkably high electrocatalytic activity as a metal-free electrocatalyst for the ORR; in contrast to the commercially available Pt/C catalyst, it displayed much better long-term stability and tolerance toward methanol crossover in an alkaline medium.484 Phosphorus-doped OMC was demonstrated as a metal-free electrode with excellent electrocatalytic activity for ORR, coupled with greatly enhanced stability and tolerance of alcohol compared to those of platinum, via a four-electron pathway in an alkaline medium.485 Nitrogen-containing OMC as a metal-free catalyst exhibited better catalytic activity than CMK-3 for the selective oxidation of ethylbenzene486 and exhibited much better electrocatalytic activity, long-term operational stability and high tolerance of CH3OH compared to commercial Pt/C catalysts for ORR in an alkaline fuel cell.487 In recent years, metal-free graphitic carbon nitride has become a new generation of polymeric semiconductor that is relevant in the energy, photocatalysis and environmental fields.488–490 However, it was shown that the more ideal bulk carbon nitride solids perform rather poorly in some catalytic processes, whereas more disordered, polymeric versions displayed better activity, as structural defects or surface terminations seemed to play a key role in catalytic activation. The performances of some MCs as catalysts or catalyst supports reported in the literature are listed in Table 2.
| Catalyst/catalyst support | Content of active sites (wt%) | Active sites | Catalytic reaction | Properties of catalyst/catalyst support | Ref. |
|---|---|---|---|---|---|
| OMC-based titania | 40–87 | TiO2 | Photocatalytic degradation of dye | Well-dispersed and tunable titania nanoparticles confined inside carbon frameworks | 491 |
| Highly ordered mesoporous ruthenium–carbons | Not given | Ru | Hydrogenation of methyl oleate | Uniform dispersion of ruthenium nanoparticles of 1–2 nm in size | 478 |
| Nickel-based MCs | 0.33–1.47 | Ni | Reduction of 4-nitrophenol | Evenly dispersed nickel complex on a mesoporous carbonaceous framework | 368 |
| TiO2-coated MCs | Not given | TiO2 | Photocatalytic degradation of methyl orange | TiO2–C nanocomposites and improved crystallinity of TiO2 with fewer defect sites | 492 |
| Sulfonated OMCs | Not given | –SO3H | Esterification of oleic acid with ethanol | High acid density, hydrophobic surface and strong attachment of sulfonic acid group | 474 |
| Rh on OMC nanoparticles | Below 1 | Rh | Hydrogenation of carbon monoxide to higher alcohols | Higher production of C2+ alcohols with high selectivity for ethanol | 493 |
| Fe–N/MCNs | Fe: not given, N: 2.2–18.9 | Fe, Fe3C and γ-Fe2O3; pyridinic-N, pyrrolic-N, graphitic-N, oxidized-N | ORR | High catalytic activities, long-term stability and improved tolerance of methanol under alkaline conditions | 181 |
| SO3H-bearing MCs | –SO3H: 2.43–13.7; phenolic OH: 7.44–43.7 | –SO3H, phenolic OH | Selective dimerization of α-methylstyrene | Provides high accessibility of hydrophobic reactants in solution to SO3H and a high density of phenolic OH groups bonded to the carbon that prevent side reactions | 494 |
| Pt on highly stable graphitic MCs | 18 | Pt | Proton exchange | High degree of graphitization with better corrosion resistance | 495 |
| Pd on MCs | 1 | Pd | Deoxygenation of fatty acids | The decarboxylation rate of fatty acids decreased with an increase in the fatty acid to metal ratio | 496 |
| Pd on MCs | 5 | Pd | Hydrogenation of nitrocyclohexane to cyclohexanone oxime | Palladium particles are well dispersed on the surface | 497 |
| Highly ordered Fe-containing MCs | Not given | Metallic Fe, γ-Fe2O3 | Wet peroxide oxidation of phenol solution with hydrogen peroxide | Well-ordered structure is closely related to iron loading contents | 136 |
| MCs | Nothing | Nothing | Reduction of p-nitrotoluene | Serves as an adsorbent and electrical conductor to enable the reaction to occur in the presence of hydrazine hydrate as the reducing agent | 498 |
| Highly ordered mesoporous carbon nitride | 9.3–23 | Pyridinic-N, pyrrolic-N | Dehydrogenation of ethylbenzene to styrene | Nitrogen within the carbon matrix, rich CO groups and defect/edge features on the surface |
326 |
| Pt on MC nanofibers | 40–80 | Pt2+, Pt4+, Pt0 | Methanol oxidation | Textural features and high loading | 203 |
5. Summary and outlook
What makes MCs different from the synthesis of active carbons from natural precursors is the feasible control over their material features including porosity, morphology and surface chemistry. Recent progress in the synthesis of highly tunable and developed porous structures has generated a wide range of research in adsorption and magnetic separation, catalysis, electrochemistry and energy storage. Various nanotechnologies ranging from nanocasting, electrodeposition, liquid-phase impregnation, and CVD to surfactant templating have successfully yielded novel MC materials with unprecedented control over their framework and composition, plus the development of controllable morphology and mesoporosity, and the modification of the surface chemistry of porous materials via methods ranging from chemical grafting to the oxidation of the material surface has provided improved accessibility, a larger surface area and better dispersion of the active species for a further improvement in their performance.Regarding the hard-template route, few chemical interactions between hard templates and carbon precursors occur during the replication process. Although the hard-template method has its inherent drawbacks in the sacrificial use of hard templates, insufficient stabilities of replicated mesostructures and a complex operational process, it has a wider use in the preparation of MC because it simply allows a carbon source to sediment onto the surface without the need for additional forces at the laboratory level. At the same time, some “hard templates” with similar functions have played important roles in the formation of mesopores and carbon monoliths, such as rigid silicates in composites, which can greatly reduce structural shrinkage during carbonization, creating large mesopores;39 other novel templates including halloysite,499 polyurethane foam,52 cordierite116,164 and crab shell195,197,198 also improve this method on different scales. In any case, the method of using natural complex templates to synthesize MC materials has received extensive attention among materials chemists and should be promoted. Moreover, CaCl2, ZnCl2 and other inorganic templates lower the cost of the synthesis, whereas their recycling needs further study. At present, hydrogen-bonding interactions are still investigated as the main driving force of self-assembly, but other driving forces, like coulombic,500 van der Waals501 or dative forces502 need to be fully researched. Solid acids such as boric acid282 and amino acids123,503 also need to be further promoted; in particular, more environmentally friendly precursors are good candidates to replace toxic precursors such as furfuryl alcohol, which conforms to the principles of green chemistry.504 Although some progress has been achieved in the control of morphologies in recent years, some critical issues deserve attention. For example, producing MC membranes with a large continuous pore size distribution, good thermal stability and permselectivity will be an important direction in the future; although the hydrothermal process is the main approach for the fabrication of MC monoliths, new assisted approaches for introducing mesoporosity should also be advocated, such as the ultrasound-assisted air bubbling method;168 bimodal hierarchically porous MC structures show great promise in catalysis, separations, and energy storage and conversion because they display high selectivity by combining different sizes of pores and favor the rapid transport of molecules while retaining good storage capacity.117,505,506 The further development of new hierarchical materials that possess not only mesopores and macropores but also micropores will be desirable for new applications involving smaller molecules. In addition, if no consideration is given to precise control over the channel structure, synthesis from biomass or waste by activation or fast pyrolysis is an environmentally friendly and more economical route. The residual products with high mesoporosity are more attractive for environmental remediation. However, to obtain high mesoporosity, a high ratio of a chemical activating agent to the raw material is needed. Therefore, activating agents that are efficient and economical in utilization are clearly in demand for the development of this process. On the other hand, the search for two polymers with appropriate mutual affinity but different degrees of thermal stability is worthy of more research. In this field, the development of more flexible routes to produce varieties of complex nanostructured MCs derived from renewable resources can address the energy and chemical challenges of a future sustainable society.
In order to optimize the behavior of MCs in a given application, non-metallic species such as N, F, P, B, –SO3H, –COOH, and –OH groups and metallic species such as Ca, Mg, Fe, Ni, and Ti are successfully introduced into the MC matrix, giving them enhanced functionalities; also, work is increasingly focused on modification or functionalization in situ rather than subsequent chemical processing. Although supramolecular organic templates involving the cooperative assembly of molecules368,478 and stable resol precursors that enable the co-assembly of multiple components107,136 possess advantages in fabricating MC composite materials, there is still a major challenge in preparing multifunctionalized MCs by a single method. Whether by doping or loading, keeping an open and ordered mesostructure with high functional content (organic functional groups, nanoparticles, biomolecules or inorganic components) to generate high coverage of functional groups or active sites is a significant goal via direct synthesis or post-treatment, thus facilitating molecular diffusion, adsorption kinetics, etc. However, the obvious collapse or degradation of the mesostructure and a reduction in functional content are more or less inevitable at high temperatures.
MCs have displayed unprecedented adsorption kinetics, high capacity and good stability and size effect. Their active surfaces provide the possibility of creating specific binding sites for any guest. From the aspect of practical implementation, monolithic adsorbents favor practical applications in industry, whereas higher selectivity and molecular recognition would be another important issue for further research. Another important aspect of MCs in recent years is the rapid development of practical applications for electrochemical purposes. MCs play different roles in electrode materials; for example, they are used as conductive materials as well as stable substances for batteries or sensors, whereas supercapacitors mainly depend on their large surface area and pore volume, suitable distribution of pore sizes, and interconnected pore network. Extensive effects have been made to improve the performance of devices, including the improvement of their energy storage performance with the features of high rate capability, high energy density, long-term operational stability and the enhancement of sensitivity in sensors or biosensors via combination with functional agents,294 ionic liquids,507 suitable charge transfer mediators,472 prepared polymers473 or biologically active compounds.367 Actually, some foreseeable challenges still remain in developing high-quality electrochemical electrodes or devices with MC materials including the following requirements: finding strategies to improve the uniform loading and stability of active sites (nitrogen atoms, reductive nitrogen groups or metal particles) in mesoporous materials and combining materials with different or uniform pore distributions and ordered arrangements to obtain more efficient storage capacity and stability for energy applications; and higher sensitivity and selectivity in sensors and biosensors to enable analyses in real media. With regard to catalysts and catalyst supports, the presence of rich active sites and a controllable pore structure are responsible for their selectivity and catalytic activity. In these cases, the presence of mesoporosity significantly enhances the dispersion of the active phase and the accessibility of the target, which suggests that advanced properties in mesoporous materials will play a significant role in the future of industrial catalysis. With the assistance of nitrogen-containing groups and active nanoparticles, it can be anticipated that the employment of mesoporous N-containing carbons supporting well-dispersed active nanoparticles as highly efficient and stable catalysts has wide prospects for application in the chemical industry; moreover, novel photocatalytic mesoporous carbons developed for environmental decontamination can be regarded as building blocks for next-generation materials for water treatment.
In general, extending the range of properties of MCs to increase their potential applications largely requires broadening and diversifying the composition of MCs. The control of interfacial properties via tunable surface functionalities is essential in the development of functional MCs. In addition, the hierarchical porosity and structures, especially wall thickness, pore geometry and size, and pore volume of MCs, whose relationship with performance in potential applications should be well established, and interfacial problems need further study. Nevertheless, research should also focus on mesoporous multi-component and multi-layered systems, which may not only provide the anticipated properties, but could also lead to unexpected effects that are likely to be exploited with multifunctional materials in many applications. Maybe the synthesis of novel MCs via multiple templates is a good solution;508,509 as well as the pore architecture, novel multiple compositions of MCs have the promise of achievement. In summary, the synthesis and applications of MC materials have the following complementary directions: (a) the use or development of nanotechnology to fill more varieties of precursors into a templated mesostructure; (b) the electrochemically assisted generation of a special morphology (e.g. MC nanofibers or nanospheres) on electrodes; (c) the comparative analysis of the interactions between templating agents and precursors under different synthesis conditions; (d) the search for attractive, environmentally friendly and expendable template materials for fabricating MC materials with more industrial prospects; and (e) the study of the application of MC materials in soil remediation. Although some topics have already been well developed, much about these remains to be investigated and discovered.
Acknowledgements
The authors gratefully acknowledge the financial support of this work by the National Key Scientific and Technological Project for Water Pollution Control and Management (2012ZX07202-002 & 2012ZX07202-005).References
- D. P. Upare, S. Yoon and C. W. Lee, Korean J. Chem. Eng., 2011, 28, 731–743 CrossRef CAS.
- G. Walker and L. Weatherley, Water Res., 1997, 31, 2093–2101 CrossRef CAS.
- W. Ahn, K. Min, Y. Chung, H.-K. Rhee, S. Joo and R. Ryoo, Stud. Surf. Sci. Catal., 2001, 135, 313–317 CrossRef.
- M. Sevilla, S. Alvarez, T. A. Centeno, A. Fuertes and F. Stoeckli, Electrochim. Acta, 2007, 52, 3207–3215 CrossRef CAS PubMed.
- A. H. Lu, W. Schmidt, A. Taguchi, B. Spliethoff, B. Tesche and F. Schüth, Angew. Chem., Int. Ed., 2002, 41, 3489–3492 CrossRef CAS.
- J. Rouquerol, D. Avnir, C. Fairbridge, D. Everett, J. Haynes, N. Pernicone, J. Ramsay, K. Sing and K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- A. H. Lu, W. Schmidt, B. Spliethoff and F. Schüth, Adv. Mater., 2003, 15, 1602–1606 CrossRef CAS PubMed.
- T. W. Kim, I. S. Park and R. Ryoo, Angew. Chem., 2003, 115, 4511–4515 CrossRef PubMed.
- A.-H. Lu, W. Schmidt and F. Schüth, New Carbon Mater., 2003, 18, 181–185 CAS.
- A. Vinu, C. Streb, V. Murugesan and M. Hartmann, J. Phys. Chem. B, 2003, 107, 8297–8299 CrossRef CAS.
- H. Zhou, S. Zhu, M. Hibino and I. Honma, J. Power Sources, 2003, 122, 219–223 CrossRef CAS.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- C. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., 2005, 117, 7215–7221 CrossRef PubMed.
- F. Zhang, Y. Meng, D. Gu, Y. Yan, C. Yu, B. Tu and D. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CrossRef CAS PubMed.
- Y. Huang, Y.-E. Miao, W. W. Tjiu and T. Liu, RSC Adv., 2015, 5, 18952–18959 RSC.
- L. Wang, X. Dong, H. Jiang, G. Li and M. Zhang, Catal. Commun., 2014, 56, 164–167 CrossRef CAS PubMed.
- J. Xu, F. Wu, H.-T. Wu, B. Xue, Y.-X. Li and Y. Cao, Microporous Mesoporous Mater., 2014, 198, 223–229 CrossRef CAS PubMed.
- F. Li, K.-Y. Chan, H. Yung, C. Yang and S. W. Ting, Phys. Chem. Chem. Phys., 2013, 15, 13570–13577 RSC.
- S. Thieme, J. Brückner, I. Bauer, M. Oschatz, L. Borchardt, H. Althues and S. Kaskel, J. Mater. Chem. A, 2013, 1, 9225–9234 CAS.
- D. Yuan, F. Zeng, J. Yan, X. Yuan, X. Huang and W. Zou, RSC Adv., 2013, 3, 5570–5576 RSC.
- P.-Y. Chang, C.-H. Huang and R.-A. Doong, Carbon, 2012, 50, 4259–4268 CrossRef CAS PubMed.
- F. A. Viva, M. M. Bruno, E. A. Franceschini, Y. R. Thomas, G. R. Sanchez, O. Solorza-Feria and H. R. Corti, Int. J. Hydrogen Energy, 2014, 39, 8821–8826 CrossRef CAS PubMed.
- M. Xu, Y. Rong, Z. Ku, A. Mei, T. Liu, L. Zhang, X. Li and H. Han, J. Mater. Chem. A, 2014, 2, 8607–8611 CAS.
- A. Trifonov, K. Herkendell, R. Tel-Vered, O. Yehezkeli, M. Woerner and I. Willner, ACS Nano, 2013, 7, 11358–11368 CrossRef CAS PubMed.
- Y. Fang, Y. Lv, F. Gong, Z. Wu, X. Li, H. Zhu, L. Zhou, C. Yao, F. Zhang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 2808–2811 CrossRef CAS PubMed.
- J. Xu and T. Zhao, RSC Adv., 2013, 3, 16–24 RSC.
- X. Bian, J. Zhu, L. Liao, M. D. Scanlon, P. Ge, C. Ji, H. H. Girault and B. Liu, Electrochem. Commun., 2012, 22, 128–132 CrossRef CAS PubMed.
- R. Wu, G. Xia, S. Shen, F. Zhu, F. Jiang and J. Zhang, RSC Adv., 2014, 4, 21325–21331 RSC.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. R. Eckert and L. Giebeler, J. Phys. Chem. C, 2015, 119, 4580–4587 CAS.
- D. Liu, D. Zheng, L. Wang, D. Qu, Z. Xie, J. Lei, L. Guo, B. Deng, L. Xiao and D. Qu, J. Phys. Chem. C, 2014, 118, 2370–2374 CAS.
- F. Cao, J. Chen, M. Ni, H. Song, G. Xiao, W. Wu, X. Gao and K. Cen, RSC Adv., 2014, 4, 16281–16289 RSC.
- C. Marino, L. Boulet, P. Gaveau, B. Fraisse and L. Monconduit, J. Mater. Chem., 2012, 22, 22713–22720 RSC.
- J. R. Schuster, R. Köhn, M. Döblinger, A. Keilbach, H. Amenitsch and T. Bein, J. Am. Chem. Soc., 2012, 134, 11136–11145 CrossRef CAS PubMed.
- M. Wu, P. Ai, M. Tan, B. Jiang, Y. Li, J. Zheng, W. Wu, Z. Li, Q. Zhang and X. He, Chem. Eng. J., 2014, 245, 166–172 CrossRef CAS PubMed.
- Y. Zhang, Z. Qiang and B. D. Vogt, RSC Adv., 2014, 4, 44858–44867 RSC.
- X.-W. Yin and L. Quan, Trans. Nonferrous Met. Soc. China, 2013, 23, 1652–1660 CrossRef.
- K. Wenelska, K. Kierzek, R. J. Kaleńczuk, X. Chen and E. Mijowska, ACS Appl. Mater. Interfaces, 2013, 5, 3042–3047 CAS.
- T. Z. Minović, J. J. Gulicovski, M. M. Stoiljković, B. M. Jokić, L. S. Živković, B. Z. Matović and B. M. Babić, Microporous Mesoporous Mater., 2015, 201, 271–276 CrossRef PubMed.
- K. Chen, L. Kang, M. Zhu and B. Dai, Catal. Sci. Technol., 2015, 5, 1035–1040 CAS.
- S. Dutta, A. Bhaumik and K. C.-W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 CAS.
- W. Zhu, Q. Zhao, C. Sun, Z. Zhang, T. Jiang, J. Sun, Y. Li and S. Wang, Mater. Sci. Eng., C, 2014, 39, 13–20 CrossRef CAS PubMed.
- M.-J. Kim, H.-J. Chae, K. S. Ha, K.-E. Jeong, C.-U. Kim, S.-Y. Jeong and T.-W. Kim, J. Nanosci. Nanotechnol., 2013, 13, 7511–7518 CrossRef CAS PubMed.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- Y. S. Ying Wan and D. Zhao, Chem. Mater., 2008, 20, 932–945 CrossRef.
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801–10812 RSC.
- T.-Y. Ma, L. Liu and Z.-Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC.
- J. C. Ndamanisha and L.-P. Guo, Anal. Chim. Acta, 2012, 747, 19–28 CrossRef CAS PubMed.
- H. Jiang, J. Ma and C. Li, Adv. Mater., 2012, 24, 4197–4202 CrossRef CAS PubMed.
- J. Wang, H. L. Xin and D. Wang, Part. Part. Syst. Charact., 2014, 31, 515–539 CrossRef CAS PubMed.
- C. Xue, B. Tu and D. Zhao, Nano Res., 2009, 2, 242–253 CrossRef CAS.
- D. Wu, Y. Liang, X. Yang, Z. Li, C. Zou, X. Zeng, G. Lv and R. Fu, Microporous Mesoporous Mater., 2008, 116, 91–94 CrossRef CAS PubMed.
- Y. Wan, Y. Shi and D. Zhao, Chem. Mater., 2007, 20, 932–945 CrossRef.
- D. Grosso, F. Cagnol, G. D. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS PubMed.
- C. K. Tsung, J. Fan, N. Zheng, Q. Shi, A. J. Forman, J. Wang and G. D. Stucky, Angew. Chem., Int. Ed., 2008, 47, 8682–8686 CrossRef CAS PubMed.
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- X. Du, B. Shi, J. Liang, J. Bi, S. Dai and S. Z. Qiao, Adv. Mater., 2013, 25, 5981–5985 CrossRef CAS PubMed.
- J. Liang, R. F. Zhou, X. M. Chen, Y. H. Tang and S. Z. Qiao, Adv. Mater., 2014, 26, 6074–6079 CrossRef CAS PubMed.
- J. Liang, X. Du, C. Gibson, X. W. Du and S. Z. Qiao, Adv. Mater., 2013, 25, 6226–6231 CrossRef CAS PubMed.
- A. Jain, S. Jayaraman, R. Balasubramanian and M. Srinivasan, J. Mater. Chem. A, 2014, 2, 520–528 CAS.
- J. Gong, J. Liu, Z. Jiang, X. Chen, X. Wen, E. Mijowska and T. Tang, Appl. Catal., B, 2014, 152, 289–299 CrossRef PubMed.
- W.-J. Liu, K. Tian, Y.-R. He, H. Jiang and H.-Q. Yu, Environ. Sci. Technol., 2014, 48, 13951–13959 CrossRef CAS PubMed.
- M. Leclère, M. Lejeune, L. Dupont, A.-L. Barrès, S. Renault, F. Dolhem and P. Poizot, Mater. Lett., 2014, 137, 233–236 CrossRef PubMed.
- I. Moriguchi, A. Ozono, K. Mikuriya, Y. Teraoka, S. Kagawa and M. Kodama, Chem. Lett., 1999, 1171–1172 CrossRef CAS.
- Z. Li, W. Yan and S. Dai, Carbon, 2004, 42, 767–770 CrossRef CAS PubMed.
- K. T. Lee and S. M. Oh, Chem. Commun., 2002, 2722–2723 RSC.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS PubMed.
- Y. Xia, Z. Yang and R. Mokaya, Nanoscale, 2010, 2, 639–659 RSC.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan and A. Stein, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- Z. Wu, P. A. Webley and D. Zhao, Langmuir, 2010, 26, 10277–10286 CrossRef CAS PubMed.
- D. Saha, C. I. Contescu and N. C. Gallego, Microporous Mesoporous Mater., 2012, 155, 71–74 CrossRef CAS PubMed.
- J.-S. Lee, S. H. Joo and R. Ryoo, J. Am. Chem. Soc., 2002, 124, 1156–1157 CrossRef CAS PubMed.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed.
- C. H. Kim, D.-K. Lee and T. J. Pinnavaia, Langmuir, 2004, 20, 5157–5159 CrossRef CAS.
- W. H. Zhang, C. Liang, H. Sun, Z. Shen, Y. Guan, P. Ying and C. Li, Adv. Mater., 2002, 14, 1776–1778 CrossRef CAS.
- T. W. Kim, I. S. Park and R. Ryoo, Angew. Chem., 2003, 115, 4511–4515 CrossRef PubMed.
- M. Kruk, B. Dufour, E. B. Celer, T. Kowalewski, M. Jaroniec and K. Matyjaszewski, J. Phys. Chem. B, 2005, 109, 9216–9225 CrossRef CAS PubMed.
- W. Li, D. Chen, Z. Li, Y. Shi, Y. Wan, G. Wang, Z. Jiang and D. Zhao, Carbon, 2007, 45, 1757–1763 CrossRef CAS PubMed.
- S. Tanaka, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Commun., 2005, 2125–2127 RSC.
- R. Liu, Y. Shi, Y. Wan, Y. Meng, F. Zhang, D. Gu, Z. Chen, B. Tu and D. Zhao, J. Am. Chem. Soc., 2006, 128, 11652–11662 CrossRef CAS PubMed.
- C.-C. Ting, H.-Y. Wu, S. Vetrivel, D. Saikia, Y.-C. Pan, G. T. Fey and H.-M. Kao, Microporous Mesoporous Mater., 2010, 128, 1–11 CrossRef CAS PubMed.
- G.-P. Wu, J. Yang, D. Wang, R. Xu, K. Amine and C.-X. Lu, Mater. Lett., 2014, 115, 1–4 CrossRef CAS PubMed.
- Y. Mao, H. Duan, B. Xu, L. Zhang, Y. Hu, C. Zhao, Z. Wang, L. Chen and Y. Yang, Energy Environ. Sci., 2012, 5, 7950–7955 CAS.
- B. Xu, S. Hou, F. Zhang, G. Cao, M. Chu and Y. Yang, J. Electroanal. Chem., 2014, 712, 146–150 CrossRef CAS PubMed.
- Y. A. Huang, F. Yang, Z. Xu and J. Shen, J. Colloid Interface Sci., 2011, 363, 193–198 CrossRef CAS PubMed.
- Y. A. Huang, S. Hu, S. Zuo, Z. Xu, C. Han and J. Shen, J. Mater. Chem., 2009, 19, 7759–7764 RSC.
- Y. Deng, Y. Cai, Z. Sun, D. Gu, J. Wei, W. Li, X. Guo, J. Yang and D. Zhao, Adv. Funct. Mater., 2010, 20, 3658–3665 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan and A. Stein, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- J.-G. Li, Y.-D. Lin and S.-W. Kuo, Macromolecules, 2011, 44, 9295–9309 CrossRef CAS.
- J. G. Werner, M. R. Scherer, U. Steiner and U. Wiesner, Nanoscale, 2014, 6, 8736–8742 RSC.
- P. Li, Y. Song, Q. Guo, J. Shi and L. Liu, Mater. Lett., 2011, 65, 2130–2132 CrossRef CAS PubMed.
- Y. Deng, T. Yu, Y. Wan, Y. Shi, Y. Meng, D. Gu, L. Zhang, Y. Huang, C. Liu and X. Wu, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS PubMed.
- Y. Huang, H. Cai, T. Yu, F. Zhang, F. Zhang, Y. Meng, D. Gu, Y. Wan, X. Sun and B. Tu, Angew. Chem., 2007, 119, 1107–1111 CrossRef PubMed.
- C. Liu, L. Li, H. Song and X. Chen, Chem. Commun., 2007, 757–759 RSC.
- S. Tanaka, Y. Katayama, M. P. Tate, H. W. Hillhouse and Y. Miyake, J. Mater. Chem., 2007, 17, 3639–3645 RSC.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- M. Dai and B. D. Vogt, J. Colloid Interface Sci., 2012, 387, 127–134 CrossRef CAS PubMed.
- X. Wang, C. Liang and S. Dai, Langmuir, 2008, 24, 7500–7505 CrossRef CAS PubMed.
- D. Liu, J.-H. Lei, L.-P. Guo and K.-J. Deng, Carbon, 2011, 49, 2113–2119 CrossRef CAS PubMed.
- Y.-F. Lin and J.-L. Chen, J. Colloid Interface Sci., 2014, 420, 74–79 CrossRef CAS PubMed.
- Y. Huang, S. Li, J. Chen, X. Zhang and Y. Chen, Appl. Surf. Sci., 2014, 293, 160–168 CrossRef CAS PubMed.
- Z. Wu, P. A. Webley and D. Zhao, J. Mater. Chem., 2012, 22, 11379–11389 RSC.
- B. Yuan, X. Wu, Y. Chen, J. Huang, H. Luo and S. Deng, Environ. Sci. Technol., 2013, 47, 5474–5480 CrossRef CAS PubMed.
- S.-Y. Lee, D.-I. Jang, S.-T. Bae and S.-J. Park, Int. J. Hydrogen Energy, 2014, 39, 12347–12352 CrossRef CAS PubMed.
- A. Chen, Y. Yu, Y. Zhang, W. Zang, Y. Yu, Y. Zhang, S. Shen and J. Zhang, Carbon, 2014, 80, 19–27 CrossRef CAS PubMed.
- Z. Wu, N. Hao, G. Xiao, L. Liu, P. Webley and D. Zhao, Phys. Chem. Chem. Phys., 2011, 13, 2495–2503 RSC.
- C.-C. Huang, Y.-H. Li, Y.-W. Wang and C.-H. Chen, Int. J. Hydrogen Energy, 2013, 38, 3994–4002 CrossRef CAS PubMed.
- L. Zou, L. Li, H. Song and G. Morris, Water Res., 2008, 42, 2340–2348 CrossRef CAS PubMed.
- C. Tsouris, R. Mayes, J. Kiggans, K. Sharma, S. Yiacoumi, D. DePaoli and S. Dai, Environ. Sci. Technol., 2011, 45, 10243–10249 CrossRef CAS PubMed.
- J. Jin, S. Tanaka, Y. Egashira and N. Nishiyama, Carbon, 2010, 48, 1985–1989 CrossRef CAS PubMed.
- K. Wang, Y. Wang, Y. Wang, E. Hosono and H. Zhou, J. Phys. Chem. C, 2008, 113, 1093–1097 Search PubMed.
- Z. Lei, N. Christov, L. L. Zhang and X. S. Zhao, J. Mater. Chem., 2011, 21, 2274–2281 RSC.
- Y. Song, L. Li, Y. Wang, C. Wang, Z. Guo and Y. Xia, ChemPhysChem, 2014, 15, 2084–2093 CrossRef CAS PubMed.
- C. Weinberger, S. Haffer, T. Wagner and M. Tiemann, Eur. J. Inorg. Chem., 2014, 2787–2792 CrossRef CAS PubMed.
- Y. Wan, X. Cui and Z. Wen, J. Hazard. Mater., 2011, 198, 216–223 CrossRef CAS PubMed.
- P. K. Tripathi, M. Liu, Y. Zhao, X. Ma, L. Gan, O. Noonan and C. Yu, J. Mater. Chem. A, 2014, 2, 8534–8544 CAS.
- C. He and X. Hu, Ind. Eng. Chem. Res., 2011, 50, 14070–14083 CrossRef CAS.
- C. He and X. Hu, Adsorption, 2012, 18, 337–348 CrossRef CAS.
- X. Peng, X. Hu, D. Fu and F. L. Lam, Appl. Surf. Sci., 2014, 294, 71–80 CrossRef CAS PubMed.
- L. Tang, Y. Cai, G. Yang, Y. Liu, G. Zeng, Y. Zhou, S. Li, J. Wang, S. Zhang and Y. Fang, Appl. Surf. Sci., 2014, 314, 746–753 CrossRef CAS PubMed.
- G. Tao, L. Zhang, Z. Hua, Y. Chen, L. Guo, J. Zhang, Z. Shu, J. Gao, H. Chen and W. Wu, Carbon, 2014, 66, 547–559 CrossRef CAS PubMed.
- A.-H. Lu, B. Spliethoff and F. Schüth, Chem. Mater., 2008, 20, 5314–5319 CrossRef CAS.
- L. Liu, F.-Y. Wang, G.-S. Shao and Z.-Y. Yuan, Carbon, 2010, 48, 2089–2099 CrossRef CAS PubMed.
- X. Wang, C. Liang and S. Dai, Langmuir, 2008, 24, 7500–7505 CrossRef CAS PubMed.
- G.-P. Hao, W.-C. Li, D. Qian, G.-H. Wang, W.-P. Zhang, T. Zhang, A.-Q. Wang, F. Schüth, H.-J. Bongard and A.-H. Lu, J. Am. Chem. Soc., 2011, 133, 11378–11388 CrossRef CAS PubMed.
- B. E. Wilson, S. G. Rudisill and A. Stein, Microporous Mesoporous Mater., 2014, 197, 174–179 CrossRef CAS PubMed.
- A. W. Christiansen, J. Appl. Polym. Sci., 2000, 75, 1760–1768 CrossRef CAS.
- R. B. Durairaj, Resorcinol: Chemistry, Technology and Applications, 2005, pp. 263–339 Search PubMed.
- J. Xu, A. Wang and T. Zhang, Carbon, 2012, 50, 1807–1816 CrossRef CAS PubMed.
- D. Liu, J.-H. Lei, L.-P. Guo, D. Qu, Y. Li and B.-L. Su, Carbon, 2012, 50, 476–487 CrossRef CAS PubMed.
- A. Prabhu, A. Al Shoaibi and C. Srinivasakannan, Mater. Lett., 2014, 136, 81–84 CrossRef CAS PubMed.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS PubMed.
- H. Chen, F. Sun, J. Wang, W. Li, W. Qiao, L. Ling and D. Long, J. Phys. Chem. C, 2013, 117, 8318–8328 CAS.
- Y. Zhang, Z. Qiang and B. D. Vogt, RSC Adv., 2014, 4, 44858–44867 RSC.
- J. Li, J. Gu, H. Li, Y. Liang, Y. Hao, X. Sun and L. Wang, Microporous Mesoporous Mater., 2010, 128, 144–149 CrossRef CAS PubMed.
- Y. Zhai, Y. Dou, X. Liu, S. S. Park, C.-S. Ha and D. Zhao, Carbon, 2011, 49, 545–555 CrossRef CAS PubMed.
- D. Zhao, J. Sun, Q. Li and G. D. Stucky, Chem. Mater., 2000, 12, 275–279 CrossRef CAS.
- Q. Huo, J. Feng, F. Schüth and G. D. Stucky, Chem. Mater., 1997, 9, 14–17 CrossRef CAS.
- S. Che, Y. Sakamoto, O. Terasaki and T. Tatsumi, Chem. Mater., 2001, 13, 2237–2239 CrossRef CAS.
- J. Kim, Chem. Commun., 1998, 259–260 RSC.
- P. Yang, D. Zhao, B. F. Chmelka and G. D. Stucky, Chem. Mater., 1998, 10, 2033–2036 CrossRef CAS.
- Y. Lu, R. Ganguli, C. A. Drewien, M. T. Anderson, C. J. Brinker, W. Gong, Y. Guo, H. Soyez, B. Dunn and M. H. Huang, Nature, 1997, 389, 364–368 CrossRef CAS.
- D. Zhao, P. Yang, N. Melosh, J. Feng, B. F. Chmelka and G. D. Stucky, Adv. Mater., 1998, 10, 1380–1385 CrossRef CAS.
- N. Melosh, P. Davidson and B. Chmelka, J. Am. Chem. Soc., 2000, 122, 823–829 CrossRef CAS.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Microporous Mesoporous Mater., 2009, 118, 218–223 CrossRef CAS PubMed.
- S. Tanaka, Y. Katayama, M. P. Tate, H. W. Hillhouse and Y. Miyake, J. Mater. Chem., 2007, 17, 3639–3645 RSC.
- F. H. Simanjuntak, J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Carbon, 2009, 47, 2531–2533 CrossRef CAS PubMed.
- S. Tanaka, A. Doi, N. Nakatani, Y. Katayama and Y. Miyake, Carbon, 2009, 47, 2688–2698 CrossRef CAS PubMed.
- Z. Qiang, Y. Zhang, J. A. Groff, K. A. Cavicchi and B. D. Vogt, Soft Matter, 2014, 10, 6068–6076 RSC.
- Z. Qiang, L. Zhang, G. E. Stein, K. A. Cavicchi and B. D. Vogt, Macromolecules, 2014, 47, 1109–1116 CrossRef CAS.
- Z. Qiang, Y. Zhang, Y. Wang, S. M. Bhaway, K. A. Cavicchi and B. D. Vogt, Carbon, 2015, 82, 51–59 CrossRef CAS PubMed.
- J. Li, J. Qi, C. Liu, L. Zhou, H. Song, C. Yu, J. Shen, X. Sun and L. Wang, J. Mater. Chem. A, 2014, 2, 4144–4149 CAS.
- X. Zhao, W. Li and S. Liu, Mater. Lett., 2014, 126, 174–177 CrossRef CAS PubMed.
- L. Li, T. Wang, Q. Liu, Y. Cao and J. Qiu, Carbon, 2012, 50, 5186–5195 CrossRef CAS PubMed.
- S.-H. Chai, P. F. Fulvio, P. C. Hillesheim, Z.-A. Qiao, S. M. Mahurin and S. Dai, J. Membr. Sci., 2014, 468, 73–80 CrossRef CAS PubMed.
- S. M. Mahurin, J. S. Lee, X. Wang and S. Dai, J. Membr. Sci., 2011, 368, 41–47 CrossRef CAS PubMed.
- S. P. Surwade, S.-H. Chai, J.-P. Choi, X. Wang, J. S. Lee, I. V. Vlassiouk, S. M. Mahurin and S. Dai, Langmuir, 2014, 30, 3606–3611 CrossRef CAS PubMed.
- S.-H. Hsu, Y.-F. Lin, T.-W. Chung, T.-Y. Wei, S.-Y. Lu, K.-L. Tung and K.-T. Liu, Sep. Purif. Technol., 2013, 109, 129–134 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., 2005, 117, 7215–7221 CrossRef PubMed.
- Y. Deng, T. Yu, Y. Wan, Y. Shi, Y. Meng, D. Gu, L. Zhang, Y. Huang, C. Liu and X. Wu, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS PubMed.
- S. Tanaka, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Commun., 2005, 2125–2127 RSC.
- C. How, M. A. Khan, S. Hosseini, T. Chuah and T. S. Choong, J. Ind. Eng. Chem., 2014, 20, 4286–4292 CrossRef CAS PubMed.
- G.-P. Hao, W.-C. Li, D. Qian, G.-H. Wang, W.-P. Zhang, T. Zhang, A.-Q. Wang, F. Schüth, H.-J. Bongard and A.-H. Lu, J. Am. Chem. Soc., 2011, 133, 11378–11388 CrossRef CAS PubMed.
- M. Florent, C. Xue, D. Zhao and D. Goldfarb, Chem. Mater., 2012, 24, 383–392 CrossRef CAS.
- S. E. Elaigwu, G. Kyriakou, T. J. Prior and G. M. Greenway, Mater. Lett., 2014, 123, 198–201 CrossRef CAS PubMed.
- D.-C. Guo, W.-C. Li, W. Dong, G.-P. Hao, Y.-Y. Xu and A.-H. Lu, Carbon, 2013, 62, 322–329 CrossRef CAS PubMed.
- S. Xu, Y. Luo, W. Zhong, Z. Xiao, Y. Luo, H. Ou and X.-Z. Zhao, Funct. Mater. Lett., 2014, 7, 1450055 CrossRef CAS.
- S. E. Elaigwu and G. M. Greenway, Mater. Lett., 2014, 115, 117–120 CrossRef CAS PubMed.
- S. Liu, Z. Huang and R. Wang, Mater. Res. Bull., 2013, 48, 2437–2441 CrossRef CAS PubMed.
- V. K. Saini, M. L. Pinto and J. Pires, Mater. Chem. Phys., 2013, 138, 877–885 CrossRef CAS PubMed.
- X. Tao, X. Chen, Y. Xia, H. Huang, Y. Gan, R. Wu and F. Chen, J. Mater. Chem. A, 2013, 1, 3295–3301 CAS.
- Y.-K. Lv, Y.-L. Feng, L.-H. Gan, M.-X. Liu, L. Xu, C. Liu, H.-W. Zheng and J. Li, J. Solid State Chem., 2012, 185, 198–205 CrossRef CAS PubMed.
- A. R. Menjoge, Q. Huang, B. Nohair, M. Eic, W. Shen, R. Che, S. Kaliaguine and S. Vasenkov, J. Phys. Chem. C, 2010, 114, 16298–16308 CAS.
- Z. Sun, Y. Liu, B. Li, J. Wei, M. Wang, Q. Yue, Y. Deng, S. Kaliaguine and D. Zhao, ACS Nano, 2013, 7, 8706–8714 CrossRef CAS PubMed.
- W. Xiong, M. Liu, L. Gan, Y. Lv, Y. Li, L. Yang, Z. Xu, Z. Hao, H. Liu and L. Chen, J. Power Sources, 2011, 196, 10461–10464 CrossRef CAS PubMed.
- Y. Fang, D. Gu, Y. Zou, Z. Wu, F. Li, R. Che, Y. Deng, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7987–7991 CrossRef CAS PubMed.
- Y. Han and J. Y. Ying, Angew. Chem., 2005, 117, 292–296 CrossRef PubMed.
- J. Liu, T. Yang, D.-W. Wang, G. Q. M. Lu, D. Zhao and S. Z. Qiao, Nat. Commun., 2013, 4, 2798, DOI:10.1038/ncomms3798.
- J. Wei, Y. Liang, X. Zhang, G. P. Simon, D. Zhao, J. Zhang, S. Jiang and H. Wang, Nanoscale, 2015, 7, 6247–6254 RSC.
- M. Li and J. Xue, J. Colloid Interface Sci., 2012, 377, 169–175 CrossRef CAS PubMed.
- S. B. Yoon, K. Sohn, J. Y. Kim, C.-H. Shin, J.-S. Yu and T. Hyeon, Adv. Mater., 2002, 14, 19 CrossRef CAS.
- Y. Li, Y. Yang, J. Shi and M. Ruan, Microporous Mesoporous Mater., 2008, 112, 597–602 CrossRef CAS PubMed.
- L. Guo, L. Zhang and J. Shi, Mater. Lett., 2011, 65, 1–3 CrossRef CAS PubMed.
- A. Chen, Y. Yu, H. Lv, Y. Wang, S. Shen, Y. Hu, B. Li, Y. Zhang and J. Zhang, J. Mater. Chem. A, 2013, 1, 1045–1047 CAS.
- F. Böttger-Hiller, P. Kempe, G. Cox, A. Panchenko, N. Janssen, A. Petzold, T. Thurn-Albrecht, L. Borchardt, M. Rose and S. Kaskel, Angew. Chem., Int. Ed., 2013, 52, 6088–6091 CrossRef PubMed.
- Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 886–891 CrossRef CAS PubMed.
- C.-H. Hsu, J.-Y. Jan, H.-P. Lin and P.-L. Kuo, New J. Chem., 2014, 38, 5521–5526 RSC.
- X. Chen, K. Kierzek, Z. Jiang, H. Chen, T. Tang, M. Wojtoniszak, R. J. Kalenczuk, P. K. Chu and E. Borowiak-Palen, J. Phys. Chem. C, 2011, 115, 17717–17724 CAS.
- X. Chen, K. Kierzek, K. Cendrowski, I. Pelech, X. Zhao, J. Feng, R. J. Kalenczuk, T. Tang and E. Mijowska, Colloids Surf., A, 2012, 396, 246–250 CrossRef CAS PubMed.
- H. Wang, L. Shi, T. Yan, J. Zhang, Q. Zhong and D. Zhang, J. Mater. Chem. A, 2014, 2, 4739–4750 CAS.
- B. Chang, Y. Li, Y. Guo, H. Yin, S. Zhang and B. Yang, J. Porous Mater., 2015, 22, 629–634 CrossRef CAS.
- K. Wang, W. Zhang, R. Phelan, M. A. Morris and J. D. Holmes, J. Am. Chem. Soc., 2007, 129, 13388–13389 CrossRef CAS PubMed.
- H.-J. Liu, X.-M. Wang, W.-J. Cui, Y.-Q. Dou, D.-Y. Zhao and Y.-Y. Xia, J. Mater. Chem., 2010, 20, 4223–4230 RSC.
- Z. Guo, D. Zhou, H. Liu, X. Dong, S. Yuan, A. Yu, Y. Wang and Y. Xia, J. Power Sources, 2015, 276, 181–188 CrossRef CAS PubMed.
- W. Zhang, Y. Tan, Y. Gao, J. Wu and B. Tang, J. Solid State Electrochem., 2015, 19, 593–598 CrossRef CAS.
- Y. Tan, W. Zhang, Y. Gao, J. Wu and B. Tang, J. Mater. Sci., 2015, 50, 4622–4628 CrossRef CAS.
- W. Li, F. Zhang, Y. Dou, Z. Wu, H. Liu, X. Qian, D. Gu, Y. Xia, B. Tu and D. Zhao, Adv. Energy Mater., 2011, 1, 382–386 CrossRef CAS PubMed.
- S.-H. Park and W.-J. Lee, J. Power Sources, 2015, 281, 301–309 CrossRef CAS PubMed.
- B.-H. Kim, K. S. Yang and J. P. Ferraris, Electrochim. Acta, 2012, 75, 325–331 CrossRef CAS PubMed.
- H. Liu, C.-Y. Cao, F.-F. Wei, Y. Jiang, Y.-B. Sun, P.-P. Huang and W.-G. Song, J. Phys. Chem. C, 2013, 117, 21426–21432 CAS.
- Z. Liu, D. Fu, F. Liu, G. Han, C. Liu, Y. Chang, Y. Xiao, M. Li and S. Li, Carbon, 2014, 70, 295–307 CrossRef CAS PubMed.
- R. Wang, T. Zhou, H. Wang, H. Feng and S. Ji, J. Power Sources, 2014, 269, 54–60 CrossRef CAS PubMed.
- X. Chen, K. Cendrowski, J. Srenscek-Nazzal, M. Rümmeli, R. J. Kalenczuk, H. Chen, P. K. Chu and E. Borowiak-Palen, Colloids Surf., A, 2011, 377, 150–155 CrossRef CAS PubMed.
- T.-H. Nguyen, Y.-Y. Yu, X. Wang, J.-Y. Wang and H. Song, Chem. Commun., 2013, 49, 10754–10756 RSC.
- H. Jiang, C. Li, T. Sun and J. Ma, Nanoscale, 2012, 4, 807–812 RSC.
- H. Jiang, T. Zhao, C. Li and J. Ma, Chem. Commun., 2011, 47, 8590–8592 RSC.
- K. Chen, X. Huang, C. Wan and H. Liu, Chem. Commun., 2015, 51, 7891–7894 RSC.
- F. Rodriguez-Reinoso and M. Molina-Sabio, Carbon, 1992, 30, 1111–1118 CrossRef CAS.
- Q. Qian, M. Machida and H. Tatsumoto, Bioresour. Technol., 2007, 98, 353–360 CrossRef CAS PubMed.
- Z. Hu and M. Srinivasan, Microporous Mesoporous Mater., 2001, 43, 267–275 CrossRef CAS.
- M. Sevilla, A. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 CAS.
- Z. Liu, F.-S. Zhang and J. Wu, Fuel, 2010, 89, 510–514 CrossRef CAS PubMed.
- A. S. Amarasekara and C. C. Ebede, Bioresour. Technol., 2009, 100, 5301–5304 CrossRef CAS PubMed.
- C. Wu, M. A. Nahil, N. Miskolczi, J. Huang and P. T. Williams, Environ. Sci. Technol., 2013, 48, 819–826 CrossRef PubMed.
- J. C. Acomb, C. Wu and P. T. Williams, Appl. Catal., B, 2014, 147, 571–584 CrossRef CAS PubMed.
- C. Zhuo and Y. A. Levendis, J. Appl. Polym. Sci., 2014 DOI:10.1002/app.39931.
- V. G. Pol and M. M. Thackeray, Energy Environ. Sci., 2011, 4, 1904–1912 CAS.
- Y. Huang, S. Li, H. Lin and J. Chen, Appl. Surf. Sci., 2014, 317, 422–431 CrossRef CAS PubMed.
- D. Saha, Y. Li, Z. Bi, J. Chen, J. K. Keum, D. K. Hensley, H. A. Grappe, H. M. Meyer III, S. Dai, M. P. Paranthaman and A. K. NasKar, Langmuir, 2014, 30, 900–910 CrossRef CAS PubMed.
- W.-J. Liu, H. Jiang, K. Tian, Y.-W. Ding and H.-Q. Yu, Environ. Sci. Technol., 2013, 47, 9397–9403 CrossRef CAS PubMed.
- J. Yao, L. Li, H. Song, C. Liu and X. Chen, Carbon, 2009, 47, 436–444 CrossRef CAS PubMed.
- Y. Zhai, Y. Dou, X. Liu, B. Tu and D. Zhao, J. Mater. Chem., 2009, 19, 3292–3300 RSC.
- I.-S. Park, M. Choi, T.-W. Kim and R. Ryoo, J. Mater. Chem., 2006, 16, 3409–3416 RSC.
- Y.-S. Lin and C. L. Haynes, Chem. Mater., 2009, 21, 3979–3986 CrossRef CAS.
- L. Zhi, Y. S. Hu, B. E. Hamaoui, X. Wang, I. Lieberwirth, U. Kolb, J. Maier and K. Müllen, Adv. Mater., 2008, 20, 1727–1731 CrossRef CAS PubMed.
- M. Dai, L. Song, J. T. LaBelle and B. D. Vogt, Chem. Mater., 2011, 23, 2869–2878 CrossRef CAS.
- H. Gu, D. Ding, P. Sameer, J. Guo, N. Yerra, Y. Huang, Z. Luo, T. C. Ho, N. Haldolaarachchige and D. P. Young, ECS Solid State Lett., 2013, 2, M65–M68 CrossRef CAS PubMed.
- A.-H. Lu, J.-J. Nitz, M. Comotti, C. Weidenthaler, K. Schlichte, C. W. Lehmann, O. Terasaki and F. Schuth, J. Am. Chem. Soc., 2010, 132, 14152–14162 CrossRef CAS PubMed.
- Z. Wu, W. Li, P. A. Webley and D. Zhao, Adv. Mater., 2012, 24, 485–491 CrossRef CAS PubMed.
- J. Zhu, M. Chen, N. Yerra, N. Haldolaarachchige, S. Pallavkar, Z. Luo, T. C. Ho, J. Hopper, D. P. Young and S. Wei, Nanoscale, 2013, 5, 1825–1830 RSC.
- J. Zhu, H. Gu, J. Guo, M. Chen, H. Wei, Z. Luo, H. A. Colorado, N. Yerra, D. Ding and T. C. Ho, J. Mater. Chem. A, 2014, 2, 2256–2265 CAS.
- W. H. Zhang, X. B. Lu, J. H. Xiu, Z. L. Hua, L. X. Zhang, M. Robertson, J. L. Shi, D. S. Yan and J. D. Holmes, Adv. Funct. Mater., 2004, 14, 544–552 CrossRef CAS PubMed.
- R. R. A. Rios, D. E. Alves, I. Dalmázio, S. F. V. Bento, C. L. Donnici and R. M. Lago, Mater. Res., 2003, 6, 129–135 CrossRef CAS.
- A. Contescu, C. Contescu, K. Putyera and J. Schwarz, Carbon, 1997, 35, 83–94 CrossRef CAS.
- H. Tamai, K. Shiraki, T. Shiono and H. Yasuda, J. Colloid Interface Sci., 2006, 295, 299–302 CrossRef CAS PubMed.
- B. Jarrais, A. R. Silva and C. Freire, Eur. J. Inorg. Chem., 2005, 4582–4589 CrossRef CAS PubMed.
- K. Xia, Q. Gao, C. Wu, S. Song and M. Ruan, Carbon, 2007, 45, 1989–1996 CrossRef CAS PubMed.
- X. Chen, M. Farber, Y. Gao, I. Kulaots, E. M. Suuberg and R. H. Hurt, Carbon, 2003, 41, 1489–1500 CrossRef CAS.
- M. Choi and R. Ryoo, J. Mater. Chem., 2007, 17, 4204–4209 RSC.
- Z. Li, G. D. Del Cul, W. Yan, C. Liang and S. Dai, J. Am. Chem. Soc., 2004, 126, 12782–12783 CrossRef CAS PubMed.
- L. Wang, Y. Zhao, K. Lin, X. Zhao, Z. Shan, Y. Di, Z. Sun, X. Cao, Y. Zou and D. Jiang, Carbon, 2006, 44, 1336–1339 CrossRef CAS PubMed.
- X. Wang, R. Liu, M. M. Waje, Z. Chen, Y. Yan, K. N. Bozhilov and P. Feng, Chem. Mater., 2007, 19, 2395–2397 CrossRef CAS.
- R. Xing, Y. Liu, Y. Wang, L. Chen, H. Wu, Y. Jiang, M. He and P. Wu, Microporous Mesoporous Mater., 2007, 105, 41–48 CrossRef CAS PubMed.
- Z. Li, W. Yan and S. Dai, Langmuir, 2005, 21, 11999–12006 CrossRef CAS PubMed.
- Z. Li and S. Dai, Chem. Mater., 2005, 17, 1717–1721 CrossRef CAS.
- G. Wu, Mater. Chem. Phys., 2004, 85, 81–87 CrossRef CAS PubMed.
- Z. Yue, W. Jiang, L. Wang, S. Gardner and C. Pittman, Carbon, 1999, 37, 1785–1796 CrossRef CAS.
- R. Leboda, J. Skubiszewska-Zieba and V. Bogillo, Langmuir, 1997, 13, 1211–1217 CrossRef CAS.
- Y. Yan, J. Wei, F. Zhang, Y. Meng, B. Tu and D. Zhao, Microporous Mesoporous Mater., 2008, 113, 305–314 CrossRef CAS PubMed.
- A. Vinu, K. Z. Hossian, P. Srinivasu, M. Miyahara, S. Anandan, N. Gokulakrishnan, T. Mori, K. Ariga and V. V. Balasubramanian, J. Mater. Chem., 2007, 17, 1819–1825 RSC.
- S. Jun, M. Choi, S. Ryu, H.-Y. Lee and R. Ryoo, Stud. Surf. Sci. Catal., 2003, 146, 37–40 CrossRef CAS.
- P. A. Bazuła, A.-H. Lu, J.-J. Nitz and F. Schüth, Microporous Mesoporous Mater., 2008, 108, 266–275 CrossRef PubMed.
- A. Gil, G. de la Puente and P. Grange, Microporous Mater., 1997, 12, 51–61 CrossRef CAS.
- Z. Wu, P. A. Webley and D. Zhao, Langmuir, 2010, 26, 10277–10286 CrossRef CAS PubMed.
- D. M. Burke, M. A. Morris and J. D. Holmes, Sep. Purif. Technol., 2013, 104, 150–159 CrossRef CAS PubMed.
- R. Xing, Y. Liu, Y. Wang, L. Chen, H. Wu, Y. Jiang, M. He and P. Wu, Microporous Mesoporous Mater., 2007, 105, 41–48 CrossRef CAS PubMed.
- J. Pang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2010, 46, 6935–6937 RSC.
- R. Liu, X. Wang, X. Zhao and P. Feng, Carbon, 2008, 46, 1664–1669 CrossRef CAS PubMed.
- X. Wang, R. Liu, M. M. Waje, Z. Chen, Y. Yan, K. N. Bozhilov and P. Feng, Chem. Mater., 2007, 19, 2395–2397 CrossRef CAS.
- S. Suganuma, K. Nakajima, M. Kitano, H. Kato, A. Tamura, H. Kondo, S. Yanagawa, S. Hayashi and M. Hara, Microporous Mesoporous Mater., 2011, 143, 443–450 CrossRef CAS PubMed.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato and S. Hayashi, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS PubMed.
- S. Kito, A. Satsuma, T. Ishikura, M. Niwa, Y. Murakami and T. Hattori, Catal. Today, 2004, 97, 41–47 CrossRef CAS PubMed.
- K. Hou, A. Zhang, L. Gu, M. Liu and X. Guo, J. Colloid Interface Sci., 2012, 377, 18–26 CrossRef CAS PubMed.
- L. She, J. Li, Y. Wan, X. Yao, B. Tu and D. Zhao, J. Mater. Chem., 2011, 21, 795–800 RSC.
- Q. Wang, H. Li, L. Chen and X. Huang, Carbon, 2001, 39, 2211–2214 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- M.-M. Titirici, A. Thomas and M. Antonietti, J. Mater. Chem., 2007, 17, 3412–3418 RSC.
- A. Stein, Z. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS PubMed.
- J.-W. Lang, X.-B. Yan, W.-W. Liu, R.-T. Wang and Q.-J. Xue, J. Power Sources, 2012, 204, 220–229 CrossRef CAS PubMed.
- S. Matzner and H. Boehm, Carbon, 1998, 36, 1697–1703 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu, N. Kodiweera, P. E. Stallworth, S. Greenbaum and T. J. Bandosz, Carbon, 2009, 47, 1576–1584 CrossRef CAS PubMed.
- J. M. Rosas, R. Ruiz-Rosas, J. Rodríguez-Mirasol and T. Cordero, Carbon, 2012, 50, 1523–1537 CrossRef CAS PubMed.
- A. Puziy, O. Poddubnaya, A. Martınez-Alonso, F. Suárez-Garcıa and J. Tascon, Carbon, 2003, 41, 1181–1191 CrossRef CAS.
- A. Sánchez-Sánchez, F. Suárez-García, A. Martínez-Alonso and J. Tascón, Appl. Surf. Sci., 2014, 299, 19–28 CrossRef PubMed.
- N. Job, R. Pirard, J. Marien and J.-P. Pirard, Carbon, 2004, 42, 3217–3227 CrossRef CAS PubMed.
- A. Stein, Z. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS PubMed.
- P. Valle-Vigón, M. Sevilla and A. B. Fuertes, Mater. Chem. Phys., 2013, 139, 281–289 CrossRef PubMed.
- A. Sánchez-Sánchez, F. Suárez-García, A. Martínez-Alonso and J. M. Tascón, Carbon, 2014, 70, 119–129 CrossRef PubMed.
- S. Ding, N. Liu, X. Li, L. Peng, X. Guo and W. Ding, Langmuir, 2010, 26, 4572–4575 CrossRef CAS PubMed.
- S. Ding, S. Zheng, M. Xie, L. Peng, X. Guo and W. Ding, Microporous Mesoporous Mater., 2011, 142, 609–613 CrossRef CAS PubMed.
- G. P. Mane, S. N. Talapaneni, C. Anand, S. Varghese, H. Iwai, Q. Ji, K. Ariga, T. Mori and A. Vinu, Adv. Funct. Mater., 2012, 22, 3596–3604 CrossRef CAS PubMed.
- S. Ding, S. Zheng, M. Xie, L. Peng, X. Guo and W. Ding, Microporous Mesoporous Mater., 2011, 142, 609–613 CrossRef CAS PubMed.
- X. Zhao, Q. Zhang, B. Zhang, C.-M. Chen, A. Wang, T. Zhang and D. S. Su, J. Mater. Chem., 2012, 22, 4963–4969 RSC.
- V. Schwartz, H. Xie, H. M. Meyer, S. H. Overbury and C. Liang, Carbon, 2011, 49, 659–668 CrossRef CAS PubMed.
- H. Wang, X. Bo, Y. Zhang and L. Guo, Electrochim. Acta, 2013, 108, 404–411 CrossRef CAS PubMed.
- W. Shen and W. Fan, J. Mater. Chem. A, 2013, 1, 999–1013 CAS.
- R. Pietrzak, Fuel, 2009, 88, 1871–1877 CrossRef CAS PubMed.
- A. Vinu, K. Ariga, T. Mori, T. Nakanishi, S. Hishita, D. Golberg and Y. Bando, Adv. Mater., 2005, 17, 1648–1652 CrossRef CAS PubMed.
- A. Vinu, Adv. Funct. Mater., 2008, 18, 816–827 CrossRef CAS PubMed.
- D. Zhang, L. Zheng, Y. Ma, L. Lei, Q. Li, Y. Li, H. Luo, H. Feng and Y. Hao, ACS Appl. Mater. Interfaces, 2014, 6, 2657–2665 CAS.
- M. S. Sam, H. O. Lintang, M. M. Sanagi, S. L. Lee and L. Yuliati, Spectrochim. Acta, Part A, 2014, 124, 357–364 CrossRef CAS PubMed.
- Y. Xia, Z. Yang and R. Mokaya, J. Phys. Chem. B, 2004, 108, 19293–19298 CrossRef CAS.
- P. F. Fulvio, M. Jaroniec, C. Liang and S. Dai, J. Phys. Chem. B, 2008, 112, 13126–13133 CAS.
- Y. Shin, G. E. Fryxell, M. H. Engelhard and G. J. Exarhos, Inorg. Chem. Commun., 2007, 10, 1541–1544 CrossRef CAS PubMed.
- Y. Shin, C. Wang, M. Englehard and G. E. Fryxell, Microporous Mesoporous Mater., 2009, 123, 345–348 CrossRef CAS PubMed.
- J. Yu, M. Guo, F. Muhammad, A. Wang, F. Zhang, Q. Li and G. Zhu, Carbon, 2014, 69, 502–514 CrossRef CAS PubMed.
- J. Yu, M. Guo, F. Muhammad, A. Wang, G. Yu, H. Ma and G. Zhu, Microporous Mesoporous Mater., 2014, 190, 117–127 CrossRef CAS PubMed.
- A. Lu, A. Kiefer, W. Schmidt and F. Schüth, Chem. Mater., 2004, 16, 100–103 CrossRef CAS.
- Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 1553–1558 CrossRef CAS PubMed.
- C.-M. Yang, C. Weidenthaler, B. Spliethoff, M. Mayanna and F. Schüth, Chem. Mater., 2005, 17, 355–358 CrossRef CAS.
- A. Vinu, S. Anandan, C. Anand, P. Srinivasu, K. Ariga and T. Mori, Microporous Mesoporous Mater., 2008, 109, 398–404 CrossRef CAS PubMed.
- W. Li, D. Chen, Z. Li, Y. Shi, Y. Wan, G. Wang, Z. Jiang and D. Zhao, Carbon, 2007, 45, 1757–1763 CrossRef CAS PubMed.
- J. Wang and Q. Liu, J. Phys. Chem. C, 2007, 111, 7266–7272 CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS PubMed.
- A. Bagreev, J. A. Menendez, I. Dukhno, Y. Tarasenko and T. J. Bandosz, Carbon, 2005, 43, 208–210 CrossRef CAS PubMed.
- X. Wang, D.-E. Jiang and S. Dai, Chem. Mater., 2008, 20, 4800–4802 CrossRef CAS.
- Z. Wu, P. A. Webley and D. Zhao, J. Mater. Chem., 2012, 22, 11379–11389 RSC.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS PubMed.
- B. Xu, S. Hou, F. Zhang, G. Cao, M. Chu and Y. Yang, J. Electroanal. Chem., 2014, 712, 146–150 CrossRef CAS PubMed.
- Z. Wu, P. A. Webley and D. Zhao, J. Mater. Chem., 2012, 22, 11379–11389 RSC.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS PubMed.
- X. Wang, C.-G. Liu, D. Neff, P. F. Fulvio, R. T. Mayes, A. Zhamu, Q. Fang, G. Chen, H. M. Meyer and B. Z. Jang, J. Mater. Chem. A, 2013, 1, 7920–7926 CAS.
- T.-D. Nguyen, K. E. Shopsowitz and M. J. MacLachlan, J. Mater. Chem. A, 2014, 2, 5915–5921 CAS.
- L. Zhao, N. Baccile, S. Gross, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M.-M. Titirici, Carbon, 2010, 48, 3778–3787 CrossRef CAS PubMed.
- Q. Jiang, Z. Y. Wu, Y. M. Wang, Y. Cao, C. F. Zhou and J. H. Zhu, J. Mater. Chem., 2006, 16, 1536–1542 RSC.
- Y. Wang, C. Zhang, S. Kang, B. Li, Y. Wang, L. Wang and X. Li, J. Mater. Chem., 2011, 21, 14420–14423 RSC.
- A. Chen, Y. Yu, Y. Zhang, T. Xing, Y. Wang, Y. Zhang and J. Zhang, J. Hazard. Mater., 2014, 279, 280–288 CrossRef CAS PubMed.
- A. Chen, C. Liu, Y. Yu, Y. Hu, H. Lv, Y. Zhang, S. Shen and J. Zhang, J. Hazard. Mater., 2014, 276, 192–199 CrossRef CAS PubMed.
- E. Kroke and M. Schwarz, Coord. Chem. Rev., 2004, 248, 493–532 CrossRef CAS PubMed.
- J. Wang, D. R. Miller and E. G. Gillan, Carbon, 2003, 41, 2031–2037 CrossRef CAS.
- Y. Shao, X. Wang, M. Engelhard, C. Wang, S. Dai, J. Liu, Z. Yang and Y. Lin, J. Power Sources, 2010, 195, 4375–4379 CrossRef CAS PubMed.
- J. Song, T. Xu, M. L. Gordin, P. Zhu, D. Lv, Y. B. Jiang, Y. Chen, Y. Duan and D. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS PubMed.
- Z. Zhao, Y. Dai, J. Lin and G. Wang, Chem. Mater., 2014, 26, 3151–3161 CrossRef CAS.
- Y. Wan, X. Qian, N. Jia, Z. Wang, H. Li and D. Zhao, Chem. Mater., 2007, 20, 1012–1018 CrossRef.
- J. Wang, H. Liu, X. Gu, H. Wang and D. S. Su, Chem. Commun., 2014, 50, 9182–9184 RSC.
- S. Abbet, A. Sanchez, U. Heiz, W.-D. Schneider, A. Ferrari, G. Pacchioni and N. Rösch, J. Am. Chem. Soc., 2000, 122, 3453–3457 CrossRef CAS.
- Y. Shin, G. E. Fryxell, C. A. Johnson Ii and M. M. Haley, Chem. Mater., 2007, 20, 981–986 CrossRef.
- M. Jaroniec, J. Choma, J. Gorka and A. Zawislak, Chem. Mater., 2007, 20, 1069–1075 CrossRef.
- A. Stein, Z. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS PubMed.
- Y. Deng, T. Yu, Y. Wan, Y. Shi, Y. Meng, D. Gu, L. Zhang, Y. Huang, C. Liu and X. Wu, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS PubMed.
- H.-P. Lin, C.-Y. Chang-Chien, C.-Y. Tang and C.-Y. Lin, Microporous Mesoporous Mater., 2006, 93, 344–348 CrossRef CAS PubMed.
- R. Liu, Y. Ren, Y. Shi, F. Zhang, L. Zhang, B. Tu and D. Zhao, Chem. Mater., 2007, 20, 1140–1146 Search PubMed.
- P. Gao, A. Wang, X. Wang and T. Zhang, Chem. Mater., 2008, 20, 1881–1888 CrossRef CAS.
- J. Zhou, J. He, T. Wang, D. Sun, G. Zhao, X. Chen, D. Wang and Z. Di, J. Mater. Chem., 2008, 18, 5776–5781 RSC.
- M. Jaroniec, J. Phys. Chem. C, 2008, 112, 11657–11660 Search PubMed.
- M. Jaroniec, J. Choma, J. Gorka and A. Zawislak, Chem. Mater., 2007, 20, 1069–1075 CrossRef.
- M. Jaroniec, J. Gorka, J. Choma and A. Zawislak, Carbon, 2009, 47, 3034–3040 CrossRef CAS PubMed.
- Y. Yang, Y. Lu, M. Lu, J. Huang, R. Haddad, G. Xomeritakis, N. Liu, A. P. Malanoski, D. Sturmayr and H. Fan, J. Am. Chem. Soc., 2003, 125, 1269–1277 CrossRef CAS PubMed.
- S. B. Yoon, J. Y. Kim and J.-S. Yu, Chem. Commun., 2002, 1536–1537 RSC.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- Y. Wan, Y. Shi and D. Zhao, Chem. Commun., 2007, 897–926 RSC.
- R. Liu, Y. Ren, Y. Shi, F. Zhang, L. Zhang, B. Tu and D. Zhao, Chem. Mater., 2007, 20, 1140–1146 Search PubMed.
- X. Qian, Y. Wan, Y. Wen, N. Jia, H. Li and D. Zhao, J. Colloid Interface Sci., 2008, 328, 367–373 CrossRef CAS PubMed.
- X. Yang, G. Zhang, M. Zhong, D. Wu and R. Fu, Langmuir, 2014, 30, 9183–9189 CrossRef CAS PubMed.
- Y. Chi, W. Geng, L. Zhao, X. Yan, Q. Yuan, N. Li and X. Li, J. Colloid Interface Sci., 2012, 369, 366–372 CrossRef CAS PubMed.
- Y. Tokudome, K. Nakane and M. Takahashi, Carbon, 2014, 77, 1104–1110 CrossRef CAS PubMed.
- P. Li, Y. Song, Z. Tang, G. Yang and J. Yang, J. Colloid Interface Sci., 2014, 413, 154–158 CrossRef CAS PubMed.
- N. Liu, H. Song and X. Chen, J. Mater. Chem., 2011, 21, 5345–5351 RSC.
- Y. Deng, C. Liu, T. Yu, F. Liu, F. Zhang, Y. Wan, L. Zhang, C. Wang, B. Tu and P. A. Webley, Chem. Mater., 2007, 19, 3271–3277 CrossRef CAS.
- B.-L. Su, A. Vantomme, L. Surahy, R. Pirard and J.-P. Pirard, Chem. Mater., 2007, 19, 3325–3333 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., 2008, 120, 379–382 CrossRef PubMed.
- J. Pang, Q. Hu, Z. Wu, J. E. Hampsey, J. He and Y. Lu, Microporous Mesoporous Mater., 2004, 74, 73–78 CrossRef CAS PubMed.
- M. Enterría, F. Suárez-García, A. Martínez-Alonso and J. M. Tascón, Microporous Mesoporous Mater., 2014, 190, 156–164 CrossRef PubMed.
- K. P. Gierszal and M. Jaroniec, J. Am. Chem. Soc., 2006, 128, 10026–10027 CrossRef CAS PubMed.
- Y. Oda, K. Fukuyama, K. Nishikawa, S. Namba, H. Yoshitake and T. Tatsumi, Chem. Mater., 2004, 16, 3860–3866 CrossRef CAS.
- Y. Deng, C. Liu, D. Gu, T. Yu, B. Tu and D. Zhao, J. Mater. Chem., 2008, 18, 91–97 RSC.
- J. Zhang, Y. Deng, J. Wei, Z. Sun, D. Gu, H. Bongard, C. Liu, H. Wu, B. Tu and F. Schüth, Chem. Mater., 2009, 21, 3996–4005 CrossRef CAS.
- Y. Deng, Y. Cai, Z. Sun, D. Gu, J. Wei, W. Li, X. Guo, J. Yang and D. Zhao, Adv. Funct. Mater., 2010, 20, 3658–3665 CrossRef CAS PubMed.
- L. Liu, F.-Y. Wang, G.-S. Shao, T.-Y. Ma and Z.-Y. Yuan, Carbon, 2010, 48, 2660–2664 CrossRef CAS PubMed.
- Y. Deng, J. Liu, C. Liu, D. Gu, Z. Sun, J. Wei, J. Zhang, L. Zhang, B. Tu and D. Zhao, Chem. Mater., 2008, 20, 7281–7286 CrossRef CAS.
- Y. Li, B. Yuan, J. Fu, S. Deng and X. Lu, J. Colloid Interface Sci., 2013, 408, 181–190 CrossRef CAS PubMed.
- Y. Li, J. Fu, S. Deng and X. Lu, J. Colloid Interface Sci., 2014, 424, 104–112 CrossRef CAS PubMed.
- W. Zhou, C. Wang, Q. Zhang, H. D. Abruña, Y. He, J. Wang, S. X. Mao and X. Xiao, Adv. Energy Mater., 2015 DOI:10.1002/aenm.201401752.
- Y. Zhou, L. Tang, G. Zeng, J. Chen, Y. Cai, Y. Zhang, G. Yang, Y. Liu, C. Zhang and W. Tang, Biosens. Bioelectron., 2014, 61, 519–525 CrossRef CAS PubMed.
- Y. Yang, Y. Ren, C. Sun and S. Hao, Green Chem., 2014, 16, 2273–2280 RSC.
- D. D. Asouhidou, K. S. Triantafyllidis, N. K. Lazaridis, K. A. Matis, S.-S. Kim and T. J. Pinnavaia, Microporous Mesoporous Mater., 2009, 117, 257–267 CrossRef CAS PubMed.
- X. Zhuang, Y. Wan, C. Feng, Y. Shen and D. Zhao, Chem. Mater., 2009, 21, 706–716 CrossRef CAS.
- H. Hadoun, Z. Sadaoui, N. Souami, D. Sahel and I. Toumert, Appl. Surf. Sci., 2013, 280, 1–7 CrossRef CAS PubMed.
- Y. Zhai, D. Pang, H. Chen, B. Xiang, J. Chen, C. Li, G. Zeng and L. Qiu, Appl. Surf. Sci., 2013, 280, 590–597 CrossRef CAS PubMed.
- Y. Kado, K. Imoto, Y. Soneda and N. Yoshizawa, J. Power Sources, 2014, 271, 377–381 CrossRef CAS PubMed.
- A. Vinu, K. Z. Hossian, P. Srinivasu, M. Miyahara, S. Anandan, N. Gokulakrishnan, T. Mori, K. Ariga and V. V. Balasubramanian, J. Mater. Chem., 2007, 17, 1819–1825 RSC.
- H. Qin, P. Gao, F. Wang, L. Zhao, J. Zhu, A. Wang, T. Zhang, R. A. Wu and H. Zou, Angew. Chem., Int. Ed., 2011, 50, 12218–12221 CrossRef CAS PubMed.
- T. Yan, H. Chen, F. Jiang and X. Wang, J. Chem. Eng. Data, 2014, 59, 508–515 CrossRef CAS.
- M. Hartmann, A. Vinu and G. Chandrasekar, Chem. Mater., 2005, 17, 829–833 CrossRef CAS.
- A. Vinu, K. Hossain, G. S. Kumar and K. Ariga, Carbon, 2006, 44, 530–536 CrossRef CAS PubMed.
- J. Goscianska, A. Olejnik and R. Pietrzak, J. Taiwan Inst. Chem. Eng., 2014, 45, 347–353 CrossRef CAS PubMed.
- L. Guo, L. Zhang, J. Zhang, J. Zhou, Q. He, S. Zeng, X. Cui and J. Shi, Chem. Commun., 2009, 6071–6073 RSC.
- G. Tao, L. Zhang, Z. Hua, Y. Chen, L. Guo, J. Zhang, Z. Shu, J. Gao, H. Chen and W. Wu, Carbon, 2014, 66, 547–559 CrossRef CAS PubMed.
- L. Ji, F. Liu, Z. Xu, S. Zheng and D. Zhu, Environ. Sci. Technol., 2010, 44, 3116–3122 CrossRef CAS PubMed.
- E. Garcia-Bordeje, M. Lazaro, R. Moliner, P. Alvarez, V. Gómez-Serrano and J. Fierro, Carbon, 2006, 44, 407–417 CrossRef CAS PubMed.
- A. F. Pérez-Cadenas, F. Kapteijn, J. A. Moulijn, F. J. Maldonado-Hodar, F. Carrasco-Marín and C. Moreno-Castilla, Carbon, 2006, 44, 2463–2468 CrossRef PubMed.
- Y. Shin, G. E. Fryxell, W. Um, K. Parker, S. V. Mattigod and R. Skaggs, Adv. Funct. Mater., 2007, 17, 2897–2901 CrossRef CAS PubMed.
- T. Phenrat, Y. Liu, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2009, 43, 1507–1514 CrossRef CAS.
- Y. Pang, G. Zeng, L. Tang, Y. Zhang, Y. Liu, X. Lei, Z. Li, J. Zhang, Z. Liu and Y. Xiong, Chem. Eng. J., 2011, 175, 222–227 CrossRef CAS PubMed.
- D.-M. Yun, H.-H. Cho, J.-W. Jang and J.-W. Park, Water Res., 2013, 47, 1858–1866 CrossRef CAS PubMed.
- L. Tang, G.-D. Yang, G.-M. Zeng, Y. Cai, S.-S. Li, Y.-Y. Zhou, Y. Pang, Y.-Y. Liu, Y. Zhang and B. Luna, Chem. Eng. J., 2014, 239, 114–122 CrossRef CAS PubMed.
- L. Zhou, X. Liu, J. Li, N. Wang, Z. Wang and Y. Zhou, Chem. Phys. Lett., 2005, 413, 6–9 CrossRef CAS PubMed.
- X. Liu, J. Li, L. Zhou, D. Huang and Y. Zhou, Chem. Phys. Lett., 2005, 415, 198–201 CrossRef CAS PubMed.
- Y. Shen and J. Bai, Chem. Commun., 2010, 46, 1308–1310 RSC.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2012, 24, 471–479 CrossRef CAS.
- M. Plaza, C. Pevida, A. Arenillas, F. Rubiera and J. Pis, Fuel, 2007, 86, 2204–2212 CrossRef CAS PubMed.
- F. Su, C. Lu, A.-J. Chung and C.-H. Liao, Appl. Energy, 2014, 113, 706–712 CrossRef CAS PubMed.
- T. Li and N. L. Rosi, Chem. Commun., 2013, 49, 11385–11387 RSC.
- Q. Li, J. Yang, D. Feng, Z. Wu, Q. Wu, S. S. Park, C.-S. Ha and D. Zhao, Nano Res., 2010, 3, 632–642 CrossRef CAS.
- S. Feng, W. Li, Q. Shi, Y. Li, J. Chen, Y. Ling, A. M. Asiri and D. Zhao, Chem. Commun., 2014, 50, 329–331 RSC.
- S. C. Lee, B. Y. Choi, T. J. Lee, C. K. Ryu, Y. S. Ahn and J. C. Kim, Catal. Today, 2006, 111, 385–390 CrossRef CAS PubMed.
- S. Walspurger, L. Boels, P. D. Cobden, G. D. Elzinga, W. G. Haije and R. W. van den Brink, ChemSusChem, 2008, 1, 643–650 CrossRef CAS PubMed.
- X. P. Wang, J. J. Yu, J. Cheng, Z. P. Hao and Z. P. Xu, Environ. Sci. Technol., 2007, 42, 614–618 CrossRef.
- M. R. Reddy, Z. Xu, G. Lu and J. D. Da Costa, Ind. Eng. Chem. Res., 2008, 47, 2630–2635 CrossRef CAS.
- W. Liu, N. W. Low, B. Feng, G. Wang and J. C. Diniz da Costa, Environ. Sci. Technol., 2009, 44, 841–847 CrossRef PubMed.
- H. Gupta and L.-S. Fan, Ind. Eng. Chem. Res., 2002, 41, 4035–4042 CrossRef CAS.
- G. S. Grasa and J. C. Abanades, Ind. Eng. Chem. Res., 2006, 45, 8846–8851 CrossRef CAS.
- P. Kowalczyk, R. Hołyst, M. Terrones and H. Terrones, Phys. Chem. Chem. Phys., 2007, 9, 1786–1792 RSC.
- R. Ströbel, J. Garche, P. Moseley, L. Jörissen and G. Wolf, J. Power Sources, 2006, 159, 781–801 CrossRef PubMed.
- C.-C. Yang, Y. J. Li and W.-H. Chen, Int. J. Hydrogen Energy, 2010, 35, 2336–2343 CrossRef CAS PubMed.
- S. Giraudet and Z. Zhu, Carbon, 2011, 49, 398–405 CrossRef CAS PubMed.
- A. Pandolfo and A. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS PubMed.
- T. Younos and K. E. Tulou, Journal of Contemporary Water Research & Education, 2005, 132, 3–10 Search PubMed.
- L. Zou, L. Li, H. Song and G. Morris, Water Res., 2008, 42, 2340–2348 CrossRef CAS PubMed.
- H.-Y. Liu, K.-P. Wang and H. Teng, Carbon, 2005, 43, 559–566 CrossRef CAS PubMed.
- S. M. Zhu, H. S. Zhou, M. Hibino and I. Honma, J. Mater. Chem., 2003, 13, 1115–1118 RSC.
- H. Li, L. Zou, L. Pan and Z. Sun, Environ. Sci. Technol., 2010, 44, 8692–8697 CrossRef CAS PubMed.
- D. Zhang, X. Wen, L. Shi, T. Yan and J. Zhang, Nanoscale, 2012, 4, 5440–5446 RSC.
- K.-L. Yang, T.-Y. Ying, S. Yiacoumi, C. Tsouris and E. S. Vittoratos, Langmuir, 2001, 17, 1961–1969 CrossRef CAS.
- C. J. Gabelich, T. D. Tran and I. M. Suffet, Environ. Sci. Technol., 2002, 36, 3010–3019 CrossRef CAS.
- P. Xu, J. E. Drewes, D. Heil and G. Wang, Water Res., 2008, 42, 2605–2617 CrossRef CAS PubMed.
- J. O. áKiggans Jr, J. Mater. Chem., 2010, 20, 8674–8678 RSC.
- K. Sharma, Y. Kim, J. Gabitto, R. Mayes, S. Yiacoumi, H. Bilheux, L. Walker, S. Dai and C. Tsouris, Langmuir, 2015, 31(3), 1038–1047 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- W. Li, D. Chen, Z. Li, Y. Shi, Y. Wan, J. Huang, J. Yang, D. Zhao and Z. Jiang, Electrochem. Commun., 2007, 9, 569–573 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- A. Malinauskas, J. Malinauskiene and A. Ramanavičius, Nanotechnology, 2005, 16, R51 CrossRef CAS PubMed.
- J. Xu, K. Wang, S.-Z. Zu, B.-H. Han and Z. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed.
- Y. Yan, Q. Cheng, G. Wang and C. Li, J. Power Sources, 2011, 196, 7835–7840 CrossRef CAS PubMed.
- V. Ruiz, C. Blanco, E. Raymundo-Piñero, V. Khomenko, F. Béguin and R. Santamaría, Electrochim. Acta, 2007, 52, 4969–4973 CrossRef CAS PubMed.
- K. Jurewicz, K. Babeł, A. Źiółkowski and H. Wachowska, Electrochim. Acta, 2003, 48, 1491–1498 CrossRef CAS.
- D. Hulicova, J. Yamashita, Y. Soneda, H. Hatori and M. Kodama, Chem. Mater., 2005, 17, 1241–1247 CrossRef CAS.
- D. Hulicova, M. Kodama and H. Hatori, Chem. Mater., 2006, 18, 2318–2326 CrossRef CAS.
- M. Li and J. Xue, J. Phys. Chem. C, 2014, 118, 2507–2517 CAS.
- D.-W. Wang, F. Li, Z.-G. Chen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2008, 20, 7195–7200 CrossRef CAS.
- X. Zhao, A. Wang, J. Yan, G. Sun, L. Sun and T. Zhang, Chem. Mater., 2010, 22, 5463–5473 CrossRef CAS.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- F. Ma, H. Zhao, L. Sun, Q. Li, L. Huo, T. Xia, S. Gao, G. Pang, Z. Shi and S. Feng, J. Mater. Chem., 2012, 22, 13464–13468 RSC.
- X. Zhao, Q. Zhang, C.-M. Chen, B. Zhang, S. Reiche, A. Wang, T. Zhang, R. Schlögl and D. S. Su, Nano Energy, 2012, 1, 624–630 CrossRef CAS PubMed.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS PubMed.
- G. Hasegawa, M. Aoki, K. Kanamori, K. Nakanishi, T. Hanada and K. Tadanaga, J. Mater. Chem., 2011, 21, 2060–2063 RSC.
- J. Jiang, Q. Gao, K. Xia and J. Hu, Microporous Mesoporous Mater., 2009, 118, 28–34 CrossRef CAS PubMed.
- X. He, K. Xie, R. Li and M. Wu, Mater. Lett., 2014, 115, 96–99 CrossRef CAS PubMed.
- W. Xing, C. Huang, S. Zhuo, X. Yuan, G. Wang, D. Hulicova-Jurcakova, Z. Yan and G. Lu, Carbon, 2009, 47, 1715–1722 CrossRef CAS PubMed.
- B. Xu, F. Wu, R. Chen, G. Cao, S. Chen, Z. Zhou and Y. Yang, Electrochem. Commun., 2008, 10, 795–797 CrossRef CAS PubMed.
- H.-J. Liu, W.-J. Cui, L.-H. Jin, C.-X. Wang and Y.-Y. Xia, J. Mater. Chem., 2009, 19, 3661–3667 RSC.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., 2008, 120, 379–382 CrossRef PubMed.
- D. Banham, F. Feng, J. Burt, E. Alsrayheen and V. Birss, Carbon, 2010, 48, 1056–1063 CrossRef CAS PubMed.
- J.-Z. Wang, L. Lu, M. Choucair, J. A. Stride, X. Xu and H.-K. Liu, J. Power Sources, 2011, 196, 7030–7034 CrossRef CAS PubMed.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., 2011, 123, 6026–6030 CrossRef PubMed.
- Y. Cao, X. Li, I. A. Aksay, J. Lemmon, Z. Nie, Z. Yang and J. Liu, Phys. Chem. Chem. Phys., 2011, 13, 7660–7665 RSC.
- T. Lin, Y. Tang, Y. Wang, H. Bi, Z. Liu, F. Huang, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 1283–1290 CAS.
- G. Zhou, D.-W. Wang, F. Li, P.-X. Hou, L. Yin, C. Liu, G. Q. M. Lu, I. R. Gentle and H.-M. Cheng, Energy Environ. Sci., 2012, 5, 8901–8906 CAS.
- J. Wang, Z. Yao, C. W. Monroe, J. Yang and Y. Nuli, Adv. Funct. Mater., 2013, 23, 1194–1201 CrossRef CAS PubMed.
- M. He, L.-X. Yuan, W.-X. Zhang, X.-L. Hu and Y.-H. Huang, J. Phys. Chem. C, 2011, 115, 15703–15709 CAS.
- Y. Yang, G. Yu, J. J. Cha, H. Wu, M. Vosgueritchian, Y. Yao, Z. Bao and Y. Cui, ACS Nano, 2011, 5, 9187–9193 CrossRef CAS PubMed.
- K. T. Lee, R. Black, T. Yim, X. Ji and L. F. Nazar, Adv. Energy Mater., 2012, 2, 1490–1496 CrossRef CAS PubMed.
- Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- Y. Fu, Y. S. Su and A. Manthiram, Angew. Chem., 2013, 125, 7068–7073 CrossRef PubMed.
- J. Gao, M. A. Lowe, Y. Kiya and H. C. D. Abruña, J. Phys. Chem. C, 2011, 115, 25132–25137 CAS.
- Z. Lin, Z. Liu, W. Fu, N. J. Dudney and C. Liang, Angew. Chem., 2013, 125, 7608–7611 CrossRef PubMed.
- Z. Li, L. Yuan, Z. Yi, Y. Sun, Y. Liu, Y. Jiang, Y. Shen, Y. Xin, Z. Zhang and Y. Huang, Adv. Energy Mater., 2014, 4 DOI:10.1002/aenm.201301473.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- C. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724–4730 CrossRef CAS.
- X. Li, Y. Cao, W. Qi, L. V. Saraf, J. Xiao, Z. Nie, J. Mietek, J.-G. Zhang, B. Schwenzer and J. Liu, J. Mater. Chem., 2011, 21, 16603–16610 RSC.
- J. Song, T. Xu, M. L. Gordin, P. Zhu, D. Lv, Y. B. Jiang, Y. Chen, Y. Duan and D. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS PubMed.
- P. Kichambare, J. Kumar, S. Rodrigues and B. Kumar, J. Power Sources, 2011, 196, 3310–3316 CrossRef CAS PubMed.
- Y. Shao, X. Wang, M. Engelhard, C. Wang, S. Dai, J. Liu, Z. Yang and Y. Lin, J. Power Sources, 2010, 195, 4375–4379 CrossRef CAS PubMed.
- J. Zang, C. X. Guo, F. Hu, L. Yu and C. M. Li, Anal. Chim. Acta, 2011, 683, 187–191 CrossRef CAS PubMed.
- N. Jia, Z. Wang, G. Yang, H. Shen and L. Zhu, Electrochem. Commun., 2007, 9, 233–238 CrossRef CAS PubMed.
- J. C. Ndamanisha, L. Guo and G. Wang, Microporous Mesoporous Mater., 2008, 113, 114–121 CrossRef CAS PubMed.
- J. C. Ndamanisha and L. Guo, Bioelectrochemistry, 2009, 77, 60–63 CrossRef CAS PubMed.
- B. Wu, C. Miao, L. Yu, Z. Wang, C. Huang and N. Jia, Sens. Actuators, B, 2014, 195, 22–27 CrossRef CAS PubMed.
- F. Tan, Q. Zhao, F. Teng, D. Sun, J. Gao, X. Quan and J. Chen, Mater. Lett., 2014, 129, 95–97 CrossRef CAS PubMed.
- R. Liu, X. Wang, X. Zhao and P. Feng, Carbon, 2008, 46, 1664–1669 CrossRef CAS PubMed.
- M. Hara, ChemSusChem, 2009, 2, 129–135 CrossRef CAS PubMed.
- M. Okamura, A. Takagaki, M. Toda, J. N. Kondo, K. Domen, T. Tatsumi, M. Hara and S. Hayashi, Chem. Mater., 2006, 18, 3039–3045 CrossRef CAS.
- K. Nakajima, M. Okamura, J. N. Kondo, K. Domen, T. Tatsumi, S. Hayashi and M. Hara, Chem. Mater., 2008, 21, 186–193 CrossRef.
- N. Gokulakrishnan, G. Peru, S. Rio, J. Blach, B. Léger, D. Grosso, E. Monflier and A. Ponchel, J. Mater. Chem. A, 2014, 2, 6641–6648 CAS.
- D. S. Su, J. J. Delgado, X. Liu, D. Wang, R. Schlögl, L. Wang, Z. Zhang, Z. Shan and F. S. Xiao, Chem.–Asian J., 2009, 4, 1108–1113 CrossRef CAS PubMed.
- L. Liu, Q.-F. Deng, B. Agula, X. Zhao, T.-Z. Ren and Z.-Y. Yuan, Chem. Commun., 2011, 47, 8334–8336 RSC.
- L. Liu, Q.-F. Deng, B. Agula, T.-Z. Ren, Y.-P. Liu, B. Zhaorigetu and Z.-Y. Yuan, Catal. Today, 2012, 186, 35–41 CrossRef CAS PubMed.
- L. Liu, Q.-F. Deng, Y.-P. Liu, T.-Z. Ren and Z.-Y. Yuan, Catal. Commun., 2011, 16, 81–85 CrossRef CAS PubMed.
- Y. Wang, J. Zhang, X. Wang, M. Antonietti and H. Li, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CrossRef CAS PubMed.
- C. Han, X. Bo, Y. Zhang, M. Li and L. Guo, J. Power Sources, 2014, 272, 267–276 CrossRef CAS PubMed.
- D.-S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J.-S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- J. Wang, H. Liu, X. Gu, H. Wang and D. S. Su, Chem. Commun., 2014, 50, 9182–9184 RSC.
- J. Lu, X. Bo, H. Wang and L. Guo, Electrochim. Acta, 2013, 108, 10–16 CrossRef CAS PubMed.
- Y. Cui, J. Huang, X. Fu and X. Wang, Catal. Sci. Technol., 2012, 2, 1396–1402 CAS.
- K. Kwon, Y. J. Sa, J. Y. Cheon and S. H. Joo, Langmuir, 2011, 28, 991–996 CrossRef PubMed.
- C. Anand, S. V. Priya, G. Lawrence, G. P. Mane, D. S. Dhawale, K. S. Prasad, V. V. Balasubramanian, M. A. Wahab and A. Vinu, Catal. Today, 2013, 204, 164–169 CrossRef CAS PubMed.
- W. Wei, C. Yu, Q. Zhao, X. Qian, G. Li and Y. Wan, Appl. Catal., B, 2014, 146, 151–161 CrossRef CAS PubMed.
- C. Coromelci-Pastravanu, M. Ignat, E. Popovici and V. Harabagiu, J. Hazard. Mater., 2014, 278, 382–390 CrossRef CAS PubMed.
- M.-J. Kim, H.-J. Chae, K.-S. Ha, K.-E. Jeong, C.-U. Kim, S.-Y. Jeong and T.-W. Kim, J. Porous Mater., 2014, 21, 365–377 CrossRef CAS.
- S. Suganuma, K. Nakajima, M. Kitano, H. Kato, A. Tamura, H. Kondo, S. Yanagawa, S. Hayashi and M. Hara, Microporous Mesoporous Mater., 2011, 143, 443–450 CrossRef CAS PubMed.
- P. V. Shanahan, L. Xu, C. Liang, M. Waje, S. Dai and Y. Yan, J. Power Sources, 2008, 185, 423–427 CrossRef CAS PubMed.
- I. Simakova, O. Simakova, P. Mäki-Arvela and D. Y. Murzin, Catal. Today, 2010, 150, 28–31 CrossRef CAS PubMed.
- Y. Yan, S. Liu, F. Hao, P. Liu and H. A. Luo, Catal. Commun., 2014, 50, 9–12 CrossRef CAS PubMed.
- H. C. Wang, B. L. Li, J. T. Li, P. Lin, X. B. Bian, J. Li, B. Zhang and Z. X. Wan, Appl. Surf. Sci., 2011, 257, 4325–4330 CrossRef CAS PubMed.
- C. X.-Y. Zhou Shu-Hui, J. Inorg. Mater., 2014, 29, 584–588 Search PubMed.
- C. Kresge, M. Leonowicz, W. Roth, J. Vartuli and J. Beck, Nature, 1992, 359, 710–712 CrossRef CAS PubMed.
- S. A. Jenekhe and X. L. Chen, Science, 1999, 283, 372–375 CrossRef CAS.
- D. M. Antonelli and J. Y. Ying, Angew. Chem., Int. Ed. Engl., 1996, 35, 426–430 CrossRef CAS PubMed.
- A.-H. Lu, B. Spliethoff and F. Schüth, Chem. Mater., 2008, 20, 5314–5319 CrossRef CAS.
- C. M. Ghimbeu, L. Vidal, L. Delmotte, J.-M. le Meins and C. Vix-Guterl, Green Chem., 2014, 16, 3079–3088 RSC.
- J. Zhang, L.-B. Kong, J.-J. Cai, H. Li, Y.-C. Luo and L. Kang, Microporous Mesoporous Mater., 2010, 132, 154–162 CrossRef CAS PubMed.
- X. Zhang, Y. Li and C. Cao, J. Mater. Chem., 2012, 22, 13918–13921 RSC.
- Z. Hong, L. Zhou, J. Li and J. Tang, Electrochim. Acta, 2013, 109, 671–677 CrossRef CAS PubMed.
- N. D. Petkovich and A. Stein, Chem. Soc. Rev., 2013, 42, 3721–3739 RSC.
- K. Sreenivasan, J. Mater. Sci., 2007, 42, 7575–7578 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |