Helical poly(isocyanides): past, present and future
Erik
Schwartz†
,
Matthieu
Koepf‡
,
Heather J.
Kitto
,
Roeland J. M.
Nolte
and
Alan E.
Rowan
*
Radboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, Nijmegen, 6525, AJ, The Netherlands. E-mail: a.rowan@science.ru.nl
First published on 21st September 2010
Abstract
Stable helical polymers with a preferred handedness are compounds that offer intriguing characteristics. This review describes the progress in the synthesis of helical polyisocyanides and the investigations to determine their structural properties, such as helical pitch and handedness, by spectroscopic measurements and high resolution AFM. This review is not intended to be comprehensive; its purpose is to highlight recent studies that allow a better understanding of the main aspects of helical polyisocyanides.
 Erik Schwartz |
Erik Schwartz was born on 14 August 1980, in The Netherlands. He studied chemistry at the Radboud University Nijmegen, where he obtained his master degree in 2005, graduating cum laude. He obtained his PhD at the same university working on the synthesis and characterisation of helical polyisocyanopeptides, under the supervision of Professors Roeland Nolte, Alan Rowan and Jeroen Cornelissen. Currently he is a postdoctoral fellow in the group of Professor Valery Fokin at the Scripps Research Institute in La Jolla, California. He received both a RUBICON fellowship (NWO) and a fellowship from Schering-Plough. He is the first recipient of this Schering-Plough award, recently established to support a PhD student from The Netherlands as post-doctoral researcher at a university within the United States. |
 Matthieu Koepf |
Matthieu Koepf completed his PhD in organic chemistry in 2007 under the supervision of Dr. Jennifer A. Wytko and Dr. Jean Weiss, at the Université Louis Pasteur (Strasbourg, France). He started a first postdoctoral research period at the Radboud University (Nijmegen, The Netherlands) under the supervision of Prof. Roeland Nolte and Prof. Alan Rowan, working on polyisocyanopeptide chemistry. He has been affiliated to the Arizona State University (Tempe, the United States) as a postdoctoral fellow in the group of Prof. Thomas Moore since February 2010, focusing on the development of bio-inspired catalysts for water oxidation. |
 Heather J. Kitto |
Heather Kitto completed her PhD in inorganic phosphorus chemistry in 2005 under the supervision of Professor S. Bruce Wild at the Australian National University. She then undertook a postdoctoral fellowship at the Radboud University Nijmegen (The Netherlands) under the guidance of Professors Alan Rowan, Roeland Nolte and Jeroen Cornelissen where she worked on the synthesis, structural characterisation and fluorescent properties of water-soluble, helical polyisocyanides. In 2010 she returned to Australia, where she is currently working in industry. |
 Roeland J. M. Nolte |
Roeland J. M. Nolte is Professor of Organic Chemistry at the Radboud University Nijmegen, The Netherlands, and Director of the Institute for Molecules and Materials. He is a member of the Royal Netherlands Academy of Science and holds a special Royal Academy of Science Chair in Chemistry. His research interests span topics at the interfaces of Supramolecular Chemistry, Macromolecular Chemistry, and Biomimetic Chemistry, in which he focuses on the design of catalysts and (macro) molecular materials. His contributions to science have been recognized with numerous award lectureships and several national and international prices including the Izatt-Christensen Award for Excellence in Macrocyclic Chemistry, the first Royal Netherlands Academy of Science Chair in Chemistry, and a knighthood in 2003. He has served on the editorial boards of many scientific journals, including Science (Washington) and Chemical Communications (as Chairman). |
 Alan E. Rowan |
Professor Dr. Alan Rowan (pictured not eating melon) studied at the University of Liverpool, England, where he obtained a BSc 1st Honours in Chemistry and then a PhD in Physical Organic Chemistry. In 1992 he moved to New Zealand where he completed a Post doctoral study in the field of Supramolecular chemistry with Prof C. Hunter. In 1994 he moved to Nijmegen the Netherlands as a Marie Curie Fellow, and then as Assistant and Associate Professor with Prof R. J. Nolte. In 2005, he set up a new department of Molecular Materials in the Institute for Molecules and Materials, Nijmegen. His interests are in the relationship between molecular architecture and function, in self-assembling and macromolecular (bio)-organic and magnetic materials. |
1. Introduction
In the search for new materials in the field of electronics, biosensing and catalysis, materials that not only possess the structural integrity and flexibility of naturally occurring compounds but also possess their functionality are considered to be of great potential. In nature, the formation of well-defined structures is usually accompanied by a loss of entropy. This loss must be compensated by either an increase in favourable enthalpic interactions or a gain in entropy of the environment, which has been achieved by the evolution of specific structural motifs common to the entire biological world. The key structural components of these bio-macromolecules are mainly based on two robust architectures: the α-helix and the β-sheet. In these biological architectures, the entropic loss encountered by these peptide segments upon folding is repaid by favourable steric, hydrophobic, electrostatic and hydrogen-bonding interactions within the secondary structure. Nature has therefore developed a large pool of biomacromolecules that carry out complicated tasks such as information storage, support of tissue, transport of energy, and the performance of localised chemical transformations. In the delicate interplay, these interactions give rise to the tertiary structure of nature's main building blocks, the proteins, and hence to their functions.The α-helix and the β-sheet often have a profound influence on the biological activity of a given molecule. The discovery of helical proteins1 and the helical nature of DNA by Pauling, and Watson, Crick and Franklin in 19532 has made an increasing number of scientists over the past decades turn their eyes to nature in their efforts to design and synthesise helical structures from various building blocks. Pioneering work of Natta3 resulted in the discovery that highly isotactic polypropylene adopts a helical conformation in the crystalline state. Following this, many chemists took the challenge to develop stable helical polymers with a preferred handedness in solution to study their unique properties and their potential in the fields of material and supramolecular sciences, although, to date, nucleic acids and proteins still outclass man-made materials in terms of functionality. In polymer chemistry, helical architectures have been studied since the publication of the first paper by Pino and Lorenzi.4 The first examples were, however, mainly isotactic polymers, which exhibited only short-range helices and were, in solution, totally dynamic in nature.5,6Polymers that maintain a stable helical structure in solution, like biomacromolecules, are of great interest since they can display optical activity solely based on their main-chain conformation. In principle, the helical polymers can be divided into two major classes. The first are polymers, such as polyisocyanates7,8 or polyacetelynes9,10 that have a low helix inversion barrier. Their handedness can, for example, be controlled by the presence of a chiral stimulant interacting with the polymer backbone. The second class are polymers with a high helix inversion barrier, such as sterically restricted poly(methacrylate ester)s,11 polychlorals,12 polyguanidines13,14 and polyisocyanides.15,16 A preferred helical sense of these polymers can be achieved by the polymerisation of chiral monomers or by using a chiral catalyst. Helical polymers are considered to be stable when their helical inversion barrier exceeds approximately 85 kJ mol−1. This review will focus on polyisocyanides, which were the first polymers to be reported possessing a stable helical conformation. After the development of the first catalytic system for isocyanides by Millich,16 Nolte and Drenth et al. demonstrated in 1974 that poly(tert-butylisocyanide) can be successfully resolved into the right- and left-handed helices; these helices do not undergo racemisation, even at elevated temperatures (Fig. 1).17
 | ||
| Fig. 1 Formation of the stable helical poly(tert-butylisocyanide) by the nickel(II) catalysed polymerisation of tert-butylisocyanide. The left-handed (M) and right-handed (P) helices can be resolved by chromatography using a chiral support. | ||
After this first example, a vast amount of polyisocyanides with a preferred handedness have been developed. Polyisocyanides display their helical structure due to the restricted rotation around the single bonds connecting the main carbon atoms, referred to as atropisomerism. The purpose of this review, more than 35 years after their discovery and 16 years after the last review (in 1994)15 solely attributed to polyisocyanides, is to highlight the most interesting features and (fundamental) aspects of helical polyisocyanides. The emphasis will be on the achievements in recent years with the focus on the characteristic properties of the polymers. A complete progress in the field of helical polymers and foldamers can be found in comprehensive reviews by Nakano and Okamoto,18 Cornelissen et al.,19 Ito and Suginome,20 Moore et al.21 and Yashima et al.22,23
2. Synthesis and catalysis
Polyisocyanides, also known as polyisonitriles or polyiminomethylenes, are prepared by the polymerisation of isocyanides. The first reports on isocyanides appeared in the 1860s from Gautier,24 Hofmann25 and Lieke,26 who were immediately struck by the characteristic and repulsive odour of (liquid) isocyanides.27 The original procedures for the preparation of isocyanides, from either silver cyanide and alkyl iodide (Gautier) or the reaction of CCl2 with an amine (Hofmann), were not generally applicable for the synthesis of large quantities. Therefore, only a few isocyanides were reported before the end of the 1950s, at which point Hagedorn28 presented the preparation of isocyanides by the dehydration of a formamide, shortly followed by reports from Ugi and co-workers.29,30The latter approach from Ugi opened the way to an easy and straightforward synthesis of isocyanides and is still the leading method of today. The most common dehydration agents used are di- or triphosgene, although various other agents have also been utilised, including the Burgess reagent (inner salt of N-(triethylammoniumsulfonyl)carbamate),31,32phosphoryl trichloride,33thionyl chloride34,35 or cyanuric chloride.36,37
Shortly after the discovery of isocyanides, it was realised that they readily polymerise; a rather remarkable feature given that sufficient steric bulk is introduced upon formation of the polymer chain. The driving force of polymerisation is the conversion of a formally divalent carbon atom in the monomer to a tetravalent carbon in the polymer, which yields a heat of polymerisation of around 81 kJ mol−1.15 A special characteristic of polyisocyanides is the fact that every carbon atom in the polymer backbone bears a substituent and as a consequence, the side chains experience sufficient steric hindrance causing the polymer to adopt a non-planar conformation.
The first catalytic system proposed for the polymerisation of isocyanides was developed by Millich, who used acid coated glass together with a radical initiator or air to obtain polyisocyanides.16 Although this technique has been applied to polyisocyanides derived from alanine units (see below),38 the common and most used preparation method of polyisocyanides nowadays involves transition metal complexes, namely, Ni(II), Pd(II)–Pt(II) or Rh(III) complexes (Chart 1). The Pd(II)–Pt(II) heteronuclear complex is mainly used for the polymerisation of aryl isocyanides, whereas in cases where there are bulky substituents at the ortho position of the isocyanide monomer, the rhodium catalyst 7 was found to be the most effective.39,40
 | ||
| Chart 1 Various metal complexes used for the polymerisation of isocyanides. | ||
Nolte and co-workers showed that for ethyl isocyanide, Ni(acac)2 (1) is by far the most efficient catalyst along with other Ni(II) salts, such as 2 and 3.41 Based on kinetic measurements and experiments with optically active isocyanides, the so-called “merry-go-round” mechanism was proposed for the Ni(II) catalysed polymerisation of isocyanides. The details of this mechanism have been thoroughly reviewed,19 but a brief summary is described here. The polymerisation is initiated by a nucleophile (e.g., an amine or an alcohol) and, during the reaction, the isocyanide monomers coordinate to the nickel centre and are incorporated into the growing chain by a series of consecutive α-insertions. This results in a helical polymeric backbone (Fig. 2) in which the fifth side group is positioned above the first one, the ninth on top of the fifth and so on. When achiral isocyanides are used, the intermediate, which is formed after attack of the nucleophile (R′) on one of the coordinated isocyanide molecules, has no preference to attack either the left or the right neighbouring isocyanide. Consequently, for an achiral monomer and initiator, an equal amount of left- (M) and right-handed (P) helical polyisocyanides will be generated. Stereoselectivity (i.e., a preferred helical handedness) can be introduced when a chiral initiator42 or a chiral isocyanide is used.43,44
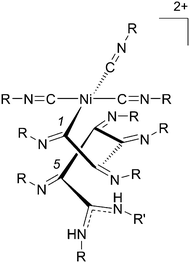 | ||
| Fig. 2 Structure of the growing helical polyisocyanide chain. R′ (most often an alcohol or amine) is the nucleophile that is required to start the polymerisation reaction. | ||
Although the ‘merry-go-round’ mechanism allows the explanation of many polymerisation characteristics and polymer properties, and recently X-ray spectroscopic and diffraction studies provided the first structural characterisation of a crucial intermediate in the ‘merry-go-round’ mechanism,45 the actual mechanism of polymerisation is most likely more complex, as can be concluded from detailed mechanistic studies performed by Deming and Novak. They revealed, with the help of electron spin resonance (ESR), cyclic voltammetry (CV) and magnetic susceptibility measurements, that a Ni(I) species is also present during the polymerisation reaction. In addition, it was found that the presence of O2 has a dramatic influence on the outcome of the reaction; in the absence of O2 the polymerisation proceeds slowly, whereas at higher oxygen concentrations (above 1 atmosphere) the isocyanide is converted into an isocyanate. It was proposed that when an excess of 10 equivalents of isocyanide is added to NiCl2, the isocyanide reduces the Ni(II) complex to an inactive Ni(I) species, which is then reactivated by oxidation with O2.46 To verify this hypothesis and to prevent this deactivation pathway, Deming and Novak developed the electron deficient η3-allylnickel trifluoroacetate 4 (and its chiral equivalent 5) as a catalyst (Chart 1). In this complex, the electron density of the nickel centre is minimised by the presence of a trifluoroacetate anion to promote nucleophilic attack on the isocyanide; the allyl group acts as an internal initiator. The catalyst proved to be a highly active system and, in a non-coordinating solvent, displayed living chain-growth behaviour.46,47 More detailed kinetic and mechanistic investigations employing this type of Ni-catalyst assumed that Ni(I), and not Ni(II), is most likely the active species,48 although X-ray absorption spectroscopy (XAS) points in a different direction.45 A mechanism for polymerisation of isocyanides with 4 was formulated by Deming and Novak in which Ni(I) is formed upon reduction of the Ni(II) complex (4) by the isocyanide. Under atmospheric conditions, O2 then acts as a spin trap for the formed isocyanide radical allowing the Ni(I) to remain active. If the reaction is carried out under an N2 atmosphere, after every insertion the Ni(II) species will be reformed and hence the reaction proceeds at a slower rate. In line with this observation, it was found that under N2 conditions the polymerisation was found to be first order with respect to the monomer, whereas under an O2 atmosphere the rate was zero order with respect to the monomer.48,49
As an alternative to using nickel as a catalyst for the polymerisation of isocyanides, a heteronuclear Pd(II)–Pt(II) complex (6, Chart 1), initially developed by Takahashi and co-workers,50,51 can be used. By using this catalytic system, aryl isocyanides can be polymerised in good yields in refluxing THF. Alkyl isocyanides, however, are not polymerised using these conditions. Most likely, the polymerisation mechanism involves the coordination of the isocyanide to the Pd metal and the subsequent migratory insertion of the isocyanide into the carbon–Pd bond. The chain is terminated by the nucleophilic attack of a chlorine ion on the Pd centre leading to the insertion of the last isocyanide. It was found that the isocyanide exclusively inserts into the carbon–Pd bond and not the carbon–Pt bond. The Pt metal, however, plays an important role since only a single insertion, and thus no polymerisation, was observed for a similar mononuclear Pd complex, even in the presence of a 100-fold excess of the isocyanide. Other catalytic systems including radical and anionic polymerisation reactions or related transition metal complexes (e.g. Co or Cu) have been investigated as discussed by Suginome and Ito20 and they showed, in some cases, fruitful polymerisation of isocyanides.
3. Structural characterisation
The helical conformation of polyisocyanides has been the subject of intensive debates in the last decades. The initial studies by Millich prompted the proposal that polymers of α-phenylethyl isocyanide possess a highly organised polymer backbone structure containing four repeat units per turn (a so-called four-over-one helix, denoted as 41) and a helical pitch of 4.1–4.2 Å.52,53 These studies were readily confirmed by Nolte and Drenth et al., who, in addition, assigned the handedness of poly(tert-butylisocyanide) on the basis of Circular Dichroism (CD) spectroscopy measurements.54 Additional theoretical studies on the conformation of oligomers of tert-butylisocyanide indicated that a helical conformation was favoured with an increasing number of monomer units. The average dihedral N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CN angle in the hexamer was found to be 78.6°, which amounts to 3.75 monomer units per helical turn.55 When the oligomer length was increased to 16, the calculation resulted in a dihedral angle of 84.3°, corresponding to 3.60 units per helical turn. Substitution of the tert-butyl group by a methyl, ethyl, or isopropyl group offered a smaller dihedral angle and more units per helical turn. In the case of poly(methylisocyanide), calculations revealed that the methyl group was too small to lead to a fixed dihedral angle and hence no atropisomerism was anticipated.
C–CN angle in the hexamer was found to be 78.6°, which amounts to 3.75 monomer units per helical turn.55 When the oligomer length was increased to 16, the calculation resulted in a dihedral angle of 84.3°, corresponding to 3.60 units per helical turn. Substitution of the tert-butyl group by a methyl, ethyl, or isopropyl group offered a smaller dihedral angle and more units per helical turn. In the case of poly(methylisocyanide), calculations revealed that the methyl group was too small to lead to a fixed dihedral angle and hence no atropisomerism was anticipated.
In depth calculations on polyisocyanides were carried out by Kollmar and Hoffmann57 and Clericuzio et al.58 On the basis of molecular orbital calculations on three polyisocyanides models, (HNC)n, (CH3NC)n and (C(CH3)3NC)n (Fig. 3a), Kollmar and Hoffmann proposed that for a polymer containing a small side chain (i.e.(HCN)n), the electronic repulsion between the N lone pairs forces the polyisocyanide backbone to adopt a non-planar conformation.57 When the side groups are sufficiently bulky (i.e.(C(CH3)3NC)n), a non-planar conformation also occurs, however, in this case the non-planarity is dictated by steric repulsion of the side groups. In the intermediate case (i.e.(CH3NC)n), a balance between the two repulsive interactions will occur. According to their calculations, the helical angle that is adopted in a polyisocyanide backbone varies from a fairly broad range of values for (HCN)n, to a narrow range around the 4-fold helix as the steric bulk of the side group increases to tert-butyl. For intermediate steric bulk, two helical minima with different degrees of helicity were proposed.
 | ||
| Fig. 3 (a) Illustration of the different (repulsive) interactions in a polyisocyanide chain. (b) Two proposed structures of polyisocyanides: the 41 helix and the ‘syndio’ conformation. (c) Chemical structure of the polyisocyanide derived from phenylalanine as synthesised by Yamada and co-workers.56 (d) Various stereoisomeric possibilities for a triad in a polyisocyanide. | ||
Besides the helical conformation, a so-called ‘syndio’ conformation for polyisocyanides was discussed on the basis of ab initio calculations by Clericuzio and co-workers.58 They found that for a series of polyisocyanides conformations that were close to the 41 helical architecture were stable, but these conformations were not at the absolute minimum in energy. The conformation of minimum energy, the ‘syndio’ conformation, would correspond to a regularly alternating disposition of the substituents on the imine groups of the polymer backbone; the dimeric sections are alternatively trans-planar (E,E) and close to 90° (Z,Z), as is shown in Fig. 3b. The conformation is slightly symmetrical but non-helical with an alternating configuration of the side groups and an alternating 180° ± 90° conformation of the backbone dihedral NC–CN angles. The driving force for this conformation would lie partly in the large preference for the trans-planar units; the rotation around the NC–CN central bond in the ethane diimine with an E,E configuration shows an (s-cis)–(s-trans) energy difference of around 34 kJ mol−1.
Both theoretical studies were experimentally supported by work from Yamada, who revealed that increasing the steric bulk from ethyl to tert-butyl substituents led to an increase in helix stability in a polyisocyanide derived from phenylalanine (Fig. 3c).56 On the basis of 13C NMR investigations it was revealed that the syndio conformation was most likely present in oligomeric compounds derived from phenylisocyanide,59 whereas in larger polymers of this type, random coil conformations were found.60
Alternative conformations of polyisocyanides were suggested by Green and co-workers, which, on the basis of comprehensive measurements, such as viscosity, light scattering and NMR spectroscopy, showed that certain polyisocyanides can adopt an irregular conformation in which the stereo-irregularity is associated with a syn–anti isomerism about the imine bond (Fig. 3d).61 In agreement with this suggestion, Takahashi showed that the polymerisation of isocyanide 8 with catalyst 2 resulted in a polyisocyanide 9 with a low specific rotation of [α]D = +354 (Fig. 4).62 Upon annealing the polymer in THF under reflux conditions, the specific optical rotation increased dramatically to [α]D = +1038. When isocyanide 8 was subjected to the Pd–Pt catalyst 6 in refluxing THF, a specific rotation for polymer 10 of [α]D = +1070 was found, which did not increase nor decrease after annealing in THF under reflux conditions. It was suggested by the authors that the initial stereo-irregularity in the polymers formed by the nickel-catalysed polymerisation is associated with the existence of both syn- and anti-isomers of the imino groups in the backbone. Upon annealing, the irregular conformation would reorganise into the thermodynamically stable, stereoregular form by syn–antiisomerisation of the imino group. In parallel, the polymerisation at high temperature with catalyst 6 would immediately lead to the energetically favourable stereoregular conformation.
 | ||
| Fig. 4 The polymerisation of 8 with either the Ni salt 2 or the heteronuclear Pt–Pd catalyst 6; rt = room temperature. | ||
Extensive studies on poly(4-carboxyphenyl isocyanides) reported by Yashima and co-workers elegantly demonstrated that some polyisocyanides can even present a dynamic behaviour (Fig. 5). In their studies the authors showed that the optically inactive poly(4-carboxyphenyl isocyanide) can adopt a preferred helical handedness upon the development of ionic interactions with chiral amines, such as (R)-2-amino-2-phenylethanol, (R)-1-phenylethanamine or (1R,2S)-1-amino-2,3-dihydro-1H-inden-2-ol, in water.63 The induced helix conformation remained unaltered after removal of the chiral amines; even after additional modification of the pendant groups no loss of macromolecular helicity was observed.64,65 The solvent, however, plays an important role in the stabilisation of the polymer conformation; the helical structures of 11 induced by chiral amines in water or aqueous organic solutions (>50 vol% water) can be effectively memorised after complete removal of the chiral amines (11b, Fig. 5), whereas in DMSO66 or DMSO–water mixtures67 (<30 vol% water), the helicity is not maintained after removal of the amines (11a, Fig. 5). The temperature was also found to play a critical role in the stabilisation of the induced helical conformations of poly(4-carboxyphenyl isocyanides). In water, a helix could be induced that, after removal of the optically active amines, maintained its conformation at ambient temperatures, however, at elevated temperatures the helix readily unfolded. It was postulated that a combination of hydrophobic and chiral ionic interactions were responsible for the helix formation and the memory effect in water since, in DMSO, induced helices were unable to maintain their helical structure.63 Based on their observations, the authors suggested that the aforementioned iminosyn–anti isomerism takes place during the helicity induction process in each solvent.
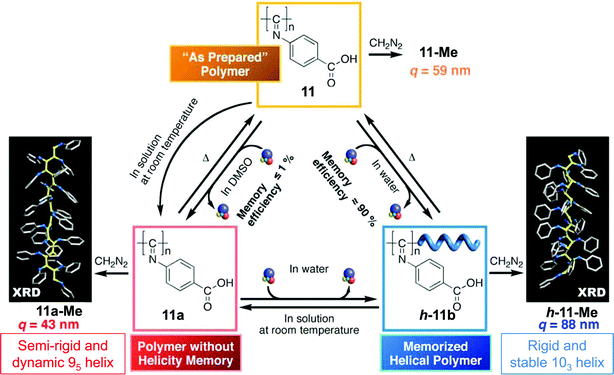 | ||
| Fig. 5 Schematic illustration of the helicity induction of polymer 11 in DMSO and water and their mutual interconversions and main characteristics. (Adapted with permission from ref. 23 and 68, Copyright 2009, American Chemical Society.) | ||
Recently a detailed and comprehensive investigation into the mechanism of the helical induction by absorption, vibrational and electronic CD, IR and NMR spectroscopies, combined with XRD studies on the cholesteric LC (model) polymer 11-Me (the methyl ester of 11) was reported (Fig. 5).68 These studies suggest that the most plausible helical structure of 11a-Me is a 95 helix with a persistence length of q = 43 nm and with two monomers constituting the repeat unit, whereas that of the helical memorised polymer h-11-Me is a 103 helix consisting of one monomer per repeat unit, with a persistence length q = 88 nm (Fig. 5.)
For stable (static) polyisocyanides the determination of absolute helical handedness is crucial and several methods have been developed to get this information. X-Ray diffraction (XRD) analyses on liquid crystalline (LC) phases of the polymers have been extensively used by Yashima and co-workers in the aforementioned studies.
The helix sense can also be determined by using CD spectroscopy, as was reported for poly(tert-butylisocyanide) in an early study (Chart 2). For this simple isocyanide the presence of only one single type of UV active group (e.g. the imino chromophore) enabled Nolte and co-workers to determine the screw sense of the polymer backbone on the basis of calculations.54 By making use of a porphyrin as a spectator group,69–71 Takahashi and co-workers could advantageously use the exciton-coupled CD method to determine the screw sense of poly(aryl isocyanides) for which the superposition of the absorption spectra of aryl and imino groups would complicate the direct interpretation of the CD signal below λ = 400 nm.72 The incorporation of achiral porphyrin monomers as a central block between two blocks of the enantiopure methyl derivatised isocyanides in the triblock copolymer architecture 12 (Chart 2) resulted in the transfer of chirality from the menthyl-derivatized blocks to the central porphyrin block, which adopted a stable helical conformation with a preferred handedness as shown by CD spectroscopy. Since the polyisocyanide architecture enforces an extremely short distance between the stacked porphyrin chromophores, an exciton coupled bisignate Cotton effect was observed in the Soret band absorption range in the CD spectrum. From the sign of this cotton effect the screw sense of the polymer could be determined using the exciton chirality method.69–71 By using similar exciton coupled CD effects, the screw sense of polyisocyanide 13, functionalised with diazo moieties, could also be determined (Chart 2).73
 | ||
| Chart 2 Chemical structures of poly(tert-butylisocyanide) and polyisocyanides 12 and 13, for which the helical senses were determined by the introduction of a spectator chromophore. | ||
The observation of helical polymers at the molecular level using scanning probe microscopy74–76 allows for the direct determination of the screw sense of the backbone. The resolution of sub-molecular structures remains, however, a challenge due to the soft nature of organic polymers. Taking advantage of the close 2D packing that could be achieved on atomically flat surfaces for polyacetylenes77–79 and polyisocyanides,80,81 Yashima and co-workers recently reported high resolution imaging of these helical polymers. In their work, the authors induced the formation of large 2D polymer crystals on highly oriented pyrolytic graphite (HOPG), by the exposition of the samples to solvent vapours, which permitted to increase the polymer surface mobility and thus favoured the rearrangement of the chains into well packed domains. This elegant method allowed the helical polymers to be visualised with a resolution close to 1 nm with AFM and the chirality of the macromolecules to be investigated at the single chain level. Upon polymerisation of the enantiomerically pure isocyanide monomer 14, using different solvents, both diastereomeric right- and left-handed helical polyisocyanides could be obtained (Fig. 6).80 When 14 was polymerised in, for example, CCl4 or toluene at room temperature, a positive Cotton effect for polymer 15a was observed and the self-assembled 2D domains showed mostly the presence of a right-handed helix (Fig. 6). In contrary, polymer 15b, obtained from the polymerisation of monomer 14 in THF at room temperature or toluene at 100 °C, exhibited a negative Cotton effect and the high resolution AFM images showed left-handed helices. This remarkable behaviour is explained by the critical influence of the temperature and of the solvents on the hydrogen bounds that can develop between the monomers. In apolar solvents or at low temperature, the polymerisation proceeds under kinetic control and hydrogen bonding between the pendant amide residues and the growing chain of the polymer is thought to play a role. In more polar solvents or at high temperatures, hydrogen bonding will be suppressed and the thermodynamic product is obtained. These ideas were confirmed by the fact that the polymerisation of a related isocyanide, in which the amide linkage is replaced by an ester and hence no hydrogen bonding can take place, leads to the formation of materials presenting similar chiroptical properties independent of the polymerisation conditions.81
 | ||
| Fig. 6 High-resolution AFM phase images of 2D self-assembled helices of 15 obtained from CCl4 and toluene at 100 °C, followed by annealing in toluene at 100 °C for six days on HOPG. The bars in the AFM phase images denote a one-handed helical array. The polymers form a stiff 154 helix with a helical pitch of ca. 1.3 nm. (Adapted with permission from ref. 80 and 82, Copyright 2006 and 2009, respectively, American Chemical Society.) | ||
Polymerisation of 14 with the Pt–Pd catalyst 6, which is known to induce a living polymerisation, produced both left- and right-handed helices, which could be separated into acetone-soluble (right-handed, 15c) and acetone-insoluble (left-handed, 15d) fractions. In combination with XRD analysis, high resolution AFM showed that both 15c and 15d possess a 154 helix with a helical pitch of around 1.3 nm. Interestingly, the helical sense excess could be directly calculated to be between 97 and 99% from the AFM image.82 Given that, even after work-up, the Pd end group remains connected to the polymer, the polymerisation can be reinitiated. The isolated polymers 15c and 15d were therefore subjected to another polymerisation reaction in which the blocks were further copolymerised with the corresponding chiral monomers.83 The block copolymerisation proceeded in an almost perfect, selective manner in which the macroinitiator blocks determine the overall helical sense.
4. Selected examples of polyisocyanides
4.1 Polyisocyanopeptides
A range of polyisocyanides derived from chiral peptide residues have been synthesised over the years since the first report on their synthesis in the 1980s,84,85 by the polymerisation of the isocyanopeptides with the nickel catalysts 2 or 3 (Chart 3).86 It was found that the helical conformation of the polyisocyanide backbone can be effectively stabilised if a well-defined hydrogen bonded network is present between the peptide side chains at positions n and n + 4. In such helices the peptidic pendants are thus stacked above each other at a distance of approximately 4.6 Å (Fig. 7). The kinetic nature of the polymerisation reaction was revealed by the stepwise addition of the isocyanopeptide monomer 16a to the catalyst 3. After the addition of 8 equivalents of monomer to a solution of the catalyst in CH2Cl2 a steep increase in the CD signal was observed and, at the same time, a shift of the amide protons could be observed in the 1H NMR spectrum, characteristic for the formation of hydrogen bonding interactions between the amide.38 Polyisocyanide 22, derived from trialanine, contains two amide groups per side chain that adopt a β-sheet-like packing along the isocyanide backbone, mimicking the interactions present in naturally occurring β-helices.87,88 Detailed IR and 1H NMR spectroscopic investigations showed that nearly all amide groups present in polymers 18–25 participate in hydrogen bonding in a similar way as observed in the X-ray crystal structure of the monomer 16a (Fig. 7). Ordered arrays of hydrogen bonds along the polymeric backbone, however, were not observed for polyisocyanide 21, which is derived from alanine–glycine units.86,89 It is remarkable that in contrast to 21, polyisocyanide 20, derived from glycine–alanine (i.e., the glycine is closest to the polymer backbone), did give a well-defined helical structure suggesting that the steric bulk in the second amino acid is of great importance to direct and stabilise the hydrogen bonding network. Analogous to the denaturation of proteins, the hydrogen bonds in these polymers can be disrupted leading to the unfolding of the helix. This unfolding is, however, only promoted by strong acids, such as trifluoroacetic acid (TFA), and not by hydrogen bonding solvents, such as methanol and DMSO, thus demonstrating the robust character of the hydrogen bonded arrays in the polymer backbone.86,90Powder X-ray diffraction (PXRD) experiments showed that, in the solid state, the rigid polyisocyanopeptides are organised in a pseudo-hexagonal arrangement. The acidified samples, which were studied for comparison, in contrast, only gave broad signals pointing to a decreased level of organisation in the polymer structure. The peptide-derived polyisocyanides are stable in solution at room temperature and, as a result of their rigidity, individual macromolecules can easily be visualised by AFM.86,89,91 By measuring the contour lengths and by careful analysis of the chain curvatures it was possible to determine the molecular weight, the polydispersity, and the persistence length of the polymers. The persistence length of 19a was found to be 76 nm,91 highlighting that these polymers are more rigid than double-stranded DNA (persistence length of 53 nm92). An accurate value of 1.6 nm for the height of the fibres was obtained by AFM measurements under chloroform vapour,93 which corresponds well with the values obtained for the polymer chains derived from molecular modelling and Powder X-ray Diffraction (PXRD) measurements.86 | ||
| Chart 3 Various polyisocyanides derived from di- and tri- peptidic isocyanides. | ||
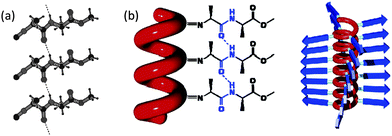 | ||
| Fig. 7 (a) X-Ray structure of isocyanopeptide 16a showing three molecules and the hydrogen-bonding network. (b) Schematic drawing of the hydrogen bonding array between the amide groups in polymer 18a and side view of the polymer showing the stacked arrangement of the β-strands. | ||
The assignment of the helical sense of peptide-derived polyisocyanides by CD spectroscopy is hampered by the overlap of signals arising from the polymer backbone and the side chains. For an L-alanine-based polyisocyanide containing a spectator group (i.e., a diazo chromophore) in the side chains, a right-handed helical geometry (P-helix) was found.73 Since the helical sense in polyisocyanides is kinetically controlled, this handedness was tentatively assigned to all L-alanine derived polyisocyanides. A strong positive Cotton effect around λ = 315 nm indicates the presence of a right-handed helix and for these polymers the presence of a well-defined hydrogen bonding array was visible in the IR. When hydrogen bonds are absent or less defined (e.g., polymer 21), the Cotton effect appears at lower wavelength, has the opposite sign and has a lower intensity. From IR and NMR spectroscopic studies it was concluded that polyisocyanides 18, 19 and 22 retain their hydrogen bonded helical conformation for significant periods of time94 even when they are dissolved in water after hydrolysis of the methyl ester functions. The thermal denaturation of these water-soluble polymers was also studied in water. It was demonstrated by using variable temperature (VT) CD spectroscopy that the denaturation process proceeds in a cooperative fashion.86
Recently, using vibrational pump-probe spectroscopy, the hydrogen-bonding array in polymer 19a was studied and these investigations showed that self-trapped vibrational states exist within these polymers. Self-trapping, which may play a role in the energy-transport mechanism in proteins and enzymes, makes it possible to transport the vibrational energy along a hydrogen-bonded chain in the form of a dispersionless wave packet. As a consequence, the energy generated at one site of the protein could therefore be transported to another site without any dispersion of the energy packet.95,96 In polyisocyanopeptides vibrational self-trapping was evidenced for polymers possessing a well-defined hydrogen bonded network along the side chain, however, when the hydrogen bonding network was disrupted by the incorporation of the non-hydrogen bonding monomer in the polymer chain, the resulting copolymer showed no evidence for self-trapping.97,98
As previously shown by Millich for several aliphatic isocyanides,16 the isocyanopeptide polymerisation can be initiated by Brønsted acids. In this case remarkably long polymers chains with lengths up to 14 µm can be grown.92,99 For the polymerisation of 17a, at a trifluoroacetic acid (TFA) concentration of 1 mM, kinetic studies revealed a large entropy of activation, with a value of −170 J mol−1 K−1; this indicates a very high degree of organisation in the transition state.38 At higher TFA concentrations, hydrolysis of the monomer to the formamide was observed instead of the formation of a polymer. A polymerisation mechanism was proposed for the acid catalysis, which, in the first instance, involves the formation of a helical oligomer. This oligomer then acts as a template for the incorporation of the next monomer through a supramolecular complex (Fig. 8, route A). The supramolecular complex is destabilised at higher acid concentrations, thereby favouring the hydrolysis of the monomer and hence formation of the corresponding formamide (route B).38 Since no polymer was found upon the addition of TFA to 16a, steric repulsion is believed to prevent the formation of a helical template in this case. Of particular interest is the fact that the reaction of the alanine derived isocyanide monomers is highly stereospecific. Since acid initiated polymerisation has a living character, the polymer chain retains its activity and thus can be further polymerised after a first block has been grown. When 17a was added to the macroinitiator block 19a, the block copolymerisation took place, as was expected. The addition of the enantiomeric pure monomer 17b to 19a completely blocked the propagation of the polymerisation of 19a, even with only 1% of 17b added. The diastereomer 16a, but not 16b, could be incorporated into the growing polymer, although 16a itself, without 17a present, could not be polymerised with TFA. These subtle differences demonstrate the critical effect of the configuration of the first chiral centre of the monomer on the polymerisation reaction and the high stereospecificity of the transition state. Interestingly, when the nickel complex 3 was used as the catalyst, all monomers and combinations thereof could be readily polymerised.
 | ||
| Fig. 8 (Route A) Mechanism of the acid initiated polymerisation of 17a, showing the helical template formation and the subsequent polymerisation. (Route B) The side reaction, occurring at higher acid concentrations, to the corresponding formamide. (Adapted with permission from ref. 38, Copyright 2005, Wiley-VCH.) | ||
Polyisocyanides 23 and 24 derived from β-amino acids were also found to form well-defined rod-like polymers with a preferred helical handedness (Chart 3).100 In contrast to the polymers derived from α-amino acids, the helical structures of the poly(isocyano-β-peptides) are dynamic in nature. The initial kinetically formed polymer is unstable and readily converted into a more thermodynamically stable helix over time, evidenced from CD studies. For polymer 23, the CD pattern irreversibly inverted and the specific rotation value evolved from −87.5 to 27.8°. Additional IR and variable temperature CD studies showed that the transformation was accompanied by a slight change in helical pitch and that the altered helical polymer possessed a better defined hydrogen pattern. Upon addition of TFA to both the original and altered polymers of 23, the chiroptical activity was lost, indicating a non-helical conformation. Upon neutralisation of the solutions, only the energetically favourable altered conformation was recovered, showing that that these polyisocyanides posses a dynamic character, as is also seen by the helix induction of polyisocyanide 11 with chiral amines (see above).
Using nickel functionalised macroinitiators, polyisocyanides blocks could also be grown on non-isocyanide based polymer blocks, leading to various architectures. A polystyrenenickel carbene complex could be synthesised, allowing the formation of a polystyrene-block-polyisocyanopeptide in which the methyl ester of the polyisocyanide block could be further saponified leading to the assembly of helical superstructures with interesting morphologies in aqueous environments.101 In a similar way, block copolymers with carbosilane wedges were prepared.102 Instead of rod–coil block copolymers, such as the polystyrene-block-polyisocyanopeptide, rod–rod copolymers were also prepared by the nickel catalyzed polymerisation of N-carboxy anhydride derivatised amino acids (NCAs) followed by the polymerisation of isocyanopeptides, resulting in a polypeptide–polyisocyanide rod–rod block copolymer.103
The stable and well-defined helical conformation of polyisocyanopeptides can be advantageously used to organize chromophoric and electroactive groups in extended arrays. Various functionalised isocyanopeptides have therefore been prepared and polymerised to give homo- and heteropolymers with interesting properties.104–106 Additional examples (e.g., those containing porphyrins, thiophenes and perylenes) have been described in the literature but these will not be discussed here.107,108 Although the versatility of this synthetic strategy has been demonstrated through the syntheses of polyisocyanides exposing chromophores, a more modular approach, which involves the post-modification of a polyisocyanide scaffold offers more possibilities. This approach relies on the use of the well-established copper catalysed azide–alkyne cycloaddition reaction (CuAAC).109,110 Using this strategy, the laborious synthetic procedure to obtain the chromophoric isocyanide monomers can be circumvented by making use of an easy-to-obtain polyisocyanide building block; one that can be scaled up to larger quantities. To this end, polymer scaffold 25 with two alanine groups and a terminal acetylene functionality in its side chain was developed. It was found that the stereochemistry had a major influence on the solubility: polymer 25a was insoluble in most common organic solvents, whereas its diastereoisomer 25b was completely soluble in, for example, dichloromethane, which allowed for its post-modification with functionalised azides.111
The grafting of ethylene glycol azides to polymer scaffold 25b was found to result in the formation of water-soluble polymers.112 The co-grafting of the scaffold with the same azide in conjunction with a perylene azide gave, for the first time, chromophoric water-soluble polyisocyanopeptides, which exhibited the same CD spectrum in water as in dichloromethane. By using two chromophoric azides, a coumarin dye and a perylene dye, random copolymers were formed in which the absorption and emission from both chromophores were present. Furthermore, interactions between the chromophores were observed, as evidenced by the partial quenching and blue-shifted emission of the coumarin molecules that are in close proximity to a perylene molecule in the perylene–coumarin functionalised polymer. Recently, the method was inverted by making use of an azide-appended polymer scaffold that was post-functionalised with a rhodium acetylene dye.113 This shows that two different chromophores can now be readily incorporated into the polymers and opens the way to polymeric materials with a wide range of optical properties. Preparation of a thiolated polymer, by the introduction of a cysteine residue, has also been reported and broadens the general approach of post-modification of polyisocyanopeptides. In this last example, both highly efficient grafting strategies based on Michael's addition as well as dynamic covalent approaches have been successfully developed.114
4.2. Poly(arylisocyanides)
Amabilino, Veciana and co-workers have investigated in detail the polymerisation of the chiral isocyanides 26 and 27. Interestingly the authors showed that that diastereomers of polymers 26 and 27 could be formed by kinetically inhibiting the growth of the normally occurring helix by the addition of the slowly polymerizing isocyanide 28, of the same chirality, as the co-monomer (Chart 4).115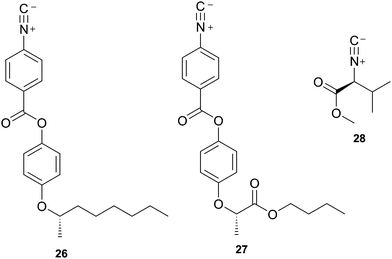 | ||
| Chart 4 Chemical structures of 26 (S), 27 (S) and 28 (S). | ||
The authors also revealed that, apart from steric bulk, other interactions between the monomers can also influence the polymerisation in a defined manner.116,117 The phenyl benzoate-containing isocyanides 26 and 27 are promesogenic compounds and hence 26 is able to induce cholesteric phases in nematic liquid crystals, while 27 is able to induce chiral smectic C phases in smectic C liquid crystals.116,117 Upon polymerisation of the isocyanide monomers, in most cases the handedness of the polymers was the same as that of the monomer induced LC phases induced by the corresponding monomer. The long range chiral induction by the stereogenic centre in the tail was explained by the occurrence of a stereoselective interaction of the incoming monomer with the growing polymer in a similar fashion as observed in the LC phase. Most likely, the rigid nature of the phenyl benzoate group allows the transfer of chirality from the side chain to the isocyanide functionality (i.e., the growing polymer chain). Using CD spectroscopy, it was shown that the CD signal related to the imine backbone rapidly diminished and inverted in sign when the chiral centre in the alkyl tail was placed further away from the promesogenic group. The role of the non-covalent interactions on the stereoselectivity was further explored by performing polymerisations at various concentrations and in different solvents.117 Variable temperature CD measurements revealed that the polymer of 26 has a stable conformation up to at least 55 °C. Introduction of a nitro-group onto the phenyl ring in close proximity to the stereogenic centre led to a less stable conformation, as was demonstrated by the dramatic and irrecoverable loss of optical activity at 55 °C. This observation confirms that the formed polyisocyanide is the kinetically determined product.118
The influence of the length and the rigidity of the rod-like spacers connecting the polyimine backbone to the stereocentres, in polymers 29a–g, were investigated in detail (Chart 5). It was concluded that some semi-rigid, twisted conformation must be adopted by the spacer in order to be able to effectively transfer the chiral information to a helical polymer backbone. The most efficient transfer of chirality was observed for the phenyl benzoate spacer 29a, whereas a flexible linker between the two phenyl groups showed no chiral induction at all. Remarkably, an induced predominant screw sense in the polymer main chain was still observed for the ‘extended benzoate spacer’ 29g, in which the stereocenter is as far as 21 Å away from the polymer backbone.119,120
 | ||
| Chart 5 Schematic representations of the series of polymers developed by Amabilino and co-workers to study the influence of spacer length and rigidity on the transfer of chirality from the side chain to the main-chain polymer. | ||
The same group also reported the synthesis of electroactive polyisocyanides bearing tetrathiafulvalene (TTF) derivatives.121,122Polymer 30 (Chart 6) was found to possess three extreme univalent states (UVSs) and two very wide mixed-valence states (MVSs), which are fully interconvertible as a result of fully reversible redox processes (Fig. 9). CD spectroscopy revealed that the polymer had very different chiroptical properties compared with the monomer; these properties are induced by the stereocentre located more than 18 Å from the polyimine backbone. The different redox states (neutral, cation or dicationic) of the polymer were investigated by both cyclic voltammetry (CV) and UV-Vis spectroscopy. The CV studies revealed the presence of two waves at 0.61 and 0.93 V, corresponding to the cationic and dication states, respectively. In the CD spectrum, characteristic Cotton effects were observed for the three different states. In general, these studies showed that redox systems can be incorporated in a well-defined manner and hence allow these polymers to be used as multistate redox-switchable organic materials in molecular devices.122 Upon oxidation of polymer 31, a related TTF-functionalised polymer, charge transfer between the TTF moieties was observed upon oxidation of the polymer.121
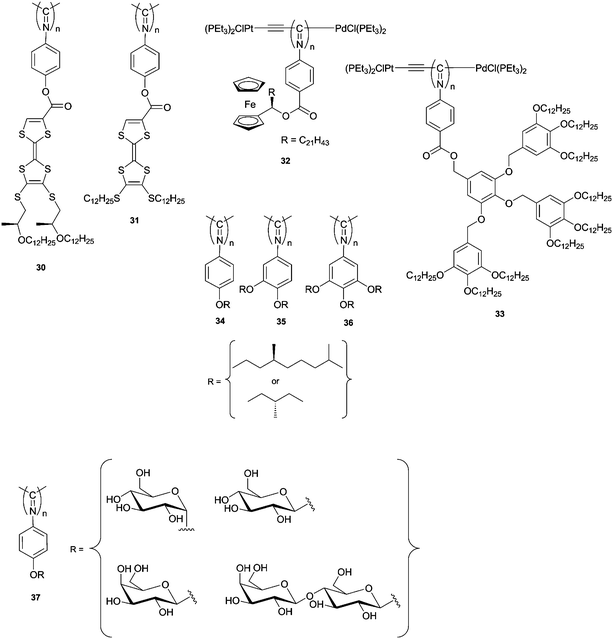 | ||
| Chart 6 Molecular structures of polyisocyanides 30–37. | ||
 | ||
| Fig. 9 Schematic representation of the three univalent and the two mixed-valence redox states of polymer 30. The colours in the scheme correspond to the actual colour of the polymer solutions. (Adapted with permission from ref. 112, Copyright 2005, Wiley-VCH.) | ||
Redox active polyisocyanides have also been reported by Takahashi and co-workers,123 who synthesised a chiral ferrocenyl polyisocyanide 32 (Chart 6) using catalyst 6. The recorded redox cycles of polymer 32 were completely reversible with a half-wave potential of approximately 0.6 V. The CD spectrum of the polymer exhibited a strong positive Cotton effect at λ = 360 nm, assigned to the n–π* transition of the imine group, and a strong negative effect at λ = 250 nm, assigned to the π–π* transition of the benzene rings. Upon electrochemical oxidation of the polymer at 1 or 1.5 V, the CD spectrum showed a decrease in the intensity of the positive Cotton effect to 40% of the initial value and the disappearance of the negative Cotton effect. In the UV-Vis spectrum, new absorption bands at λ = 240–310 and 620 nm appeared upon oxidation, which were attributed to the ferrocenium chromophore. Similar behaviour was observed by the chemical oxidation of the polymer with [NO][PF6] followed by reduction with [(C5Me5)2Fe]. The reversible loss of CD signal upon oxidation suggests a reversible unfolding of the helical backbone due to the electrostatic repulsion between the ferrocenium ions. Reduction of the ferrocenium ions leads to the refolding of the polymer into its initial helical conformation.
Polyisocyanides with achiral dendronised phenyl side groups (e.g., polymer 33) have been reported by Iyoda and co-workers; their synthesis shows that it is also possible to polymerize isocyanides with extremely bulky groups by use of the Pd–Pt complex 6.124,125 A comparable strategy has been used recently by Yashima and co-workers who introduced chiral side chains onto a poly(phenylisocyanide) and studied the influence of the stereogenic centres on the polymer properties.126 No Cotton effect in the CD spectrum was observed in the polymer with only one alkoxy chain (34), regardless of the position of the stereogenic centre. Although increasing the number of alkoxy chains to two (35) resulted in a small Cotton effect, three alkoxy chains (36) were required for an intense Cotton effect to be observed.
The bio-inspired sugar polyisocyanides 37 were synthesised to study the effect of saccharide arrays along the polyisocyanide backbone on molecular recognition phenomena. The polymerisation of acetylated glucose, galactose and lactose phenylisocyanides and subsequent deacetylation resulted in polymers with a preferred handedness and an array of defined saccharide units.127,128 Binding studies towards lectin, together with fluorescence spectroscopy, showed that 37 contributes to small specific interactions with lectins, different from the interactions found in multivalent glycoclusters attached to flexible acrylamide glycopolymers. This difference most likely arises from the fact that the saccharide cannot reorient in a favourable way to interact with the lectins due to the rigidity of the backbone in the polyisocyanide construct.
5. Conclusion and outlook
Over the years a remarkable range of helical polyisocyanides have been synthesised and studied. New monomers and specific catalysts have been developed offering access to polymers with a wide variety of properties. It is clear from this review that polyisocyanide chemistry is still flourishing some 35 years after the discovery of these compounds. This is primarily due to the unique nature of the polymers, which are only now becoming fully appreciated. Although metal complexes are the most used catalysts for the polymerisation of isocyanides, the first reported catalyst (i.e., acid) is still of importance since it can offer polymers with exceptionally long lengths and high stereospecificity, thus allowing new insight into the polymerisation mechanism. The helical architecture of the polymers has been thoroughly investigated but details remain unclear. Emergent techniques such as high-resolution AFM and Vibrational Circular Dichroism may in the future provide deeper insights into the principles of helix formation. Polyisocyanides in which the functionalities are precisely positioned along the well-defined polymer backbone, can act as, for instance, nanowires along which excitons can easily migrate or as a chiral catalyst. It can therefore be foreseen that polyisocyanides will continue to raise the interest of (bio)chemists and physicists and leading to various applications, e.g. in devices.Acknowledgements
The Technology Foundation STW, NanoNed, The Council for the Chemical Sciences of the Netherlands Organisation for Scientific Research, and the Royal Academy for Arts and Sciences are acknowledged for financial support.References
- L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CAS.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CAS.
- G. Natta, P. Pino, P. Corradini, F. Danusso, E. Mantica, G. Mazzanti and G. Moraglio, J. Am. Chem. Soc., 1955, 77, 1708–1710 CrossRef CAS.
- P. Pino and G. P. Lorenzi, J. Am. Chem. Soc., 1960, 82, 4745–4747 CrossRef CAS.
- M. M. Green and S. K. Jha, Chirality, 1997, 9, 424–427 CrossRef CAS.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1989, 28, 21–37 CrossRef.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CrossRef CAS.
- M. M. Green, J. W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3139–3154 CAS.
- E. Yashima and K. Maeda, in Foldamers: Structure, Properties, and Applications, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, 2007, pp. 331–366 Search PubMed.
- J. W. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745–754 CrossRef CAS.
- Y. Okamoto and E. Yashima, Prog. Polym. Sci., 1990, 15, 263–298 CrossRef CAS.
- K. Ute, K. Hirose, H. Kashimoto, K. Hatada and O. Vogl, J. Am. Chem. Soc., 1991, 113, 6305–6306 CrossRef CAS.
- H. Z. Tang, B. M. Novak, J. T. He and P. L. Polavarapu, Angew. Chem., Int. Ed., 2005, 44, 7298–7301 CrossRef CAS.
- H. Z. Tang, Y. J. Lu, G. L. Tian, M. D. Capracotta and B. M. Novak, J. Am. Chem. Soc., 2004, 126, 3722–3723 CrossRef CAS.
- R. J. M. Nolte, Chem. Soc. Rev., 1994, 23, 11–19 RSC.
- F. Millich, Chem. Rev., 1972, 72, 101–113 CrossRef CAS.
- R. J. M. Nolte, A. J. M. Van Beijnen and W. Drenth, J. Am. Chem. Soc., 1974, 96, 5932–5933 CrossRef CAS.
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS.
- J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS.
- M. Suginome and Y. Ito, Adv. Polym. Sci., 2004, 171, 77–136 CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS.
- E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166–1180 CrossRef CAS.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS.
- A. Gautier, Ann. Chem. Pharm., 1867, 142, 289–294 Search PubMed.
- A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1870, 3, 63 CrossRef.
- W. Lieke, Ann. Chem. Pharm., 1859, 112, 316–321 Search PubMed.
- M. C. Pirrung and S. Ghorai, J. Am. Chem. Soc., 2006, 128, 11772–11773 CrossRef CAS.
- I. Hagedorn, U. Eholzer and A. Lüttringhaus, Chem. Ber., 1960, 93, 1584–1590 CrossRef CAS.
- I. Ugi and R. Meyr, Angew. Chem., Int. Ed. Engl., 1958, 70, 702–703 Search PubMed.
- I. Ugi and U. Fetzer, Chem. Ber., 1961, 94, 2239–2243 CAS.
- S. M. Creedon, H. K. Crowley and D. G. McCarthy, J. Chem. Soc., Perkin Trans. 1, 1998, 1015–1018 RSC.
- S. Khapli, S. Dey and D. Mal, J. Indian Inst. Sci., 2001, 81, 461–476 Search PubMed.
- H. Böhme and G. Fuchs, Chem. Ber., 1970, 103, 2775–2779 CrossRef.
- H. M. Walborsky and G. E. Niznik, J. Org. Chem., 1972, 37, 187 CrossRef CAS.
- J. M. Vandereijk, R. J. M. Nolte and W. Drenth, Recl. Trav. Chim. Pays-Bas, 1978, 97, 46–49 CAS.
- R. Wittmann, Angew. Chem., Int. Ed., 1961, 73, 219 Search PubMed.
- A. Porcheddu, G. Giacomelli and M. Salaris, J. Org. Chem., 2005, 70, 2361–2363 CrossRef CAS.
- G. A. Metselaar, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2005, 44, 1990–1993 CrossRef CAS.
- K. Onitsuka, T. Mori, M. Yamamoto, F. Takei and S. Takahashi, Macromolecules, 2006, 39, 7224–7231 CrossRef CAS.
- K. Onitsuka, M. Yamamoto, T. Mori, F. Takei and S. Takahashi, Organometallics, 2006, 25, 1270–1278 CrossRef CAS.
- R. J. M. Nolte, R. W. Stephany and W. Drenth, Recl. Trav. Chim. Pays-Bas, 1973, 92, 83–91 CAS.
- P. C. J. Kamer, R. J. M. Nolte and W. Drenth, J. Am. Chem. Soc., 1988, 110, 6818–6825 CrossRef CAS.
- W. Drenth and R. J. M. Nolte, Acc. Chem. Res., 1979, 12, 30–35 CrossRef CAS.
- P. C. J. Kamer, M. C. Cleij, R. J. M. Nolte, T. Harada, A. M. F. Hezemans and W. Drenth, J. Am. Chem. Soc., 1988, 110, 1581–1587 CrossRef CAS.
- G. A. Metselaar, E. Schwartz, R. de Gelder, M. C. Feiters, S. Nikitenko, G. Smolentsev, G. E. Yalovega, A. V. Soldatov, J. Cornelissen, A. E. Rowan and R. J. M. Nolte, ChemPhysChem, 2007, 8, 1850–1856 CrossRef CAS.
- T. J. Deming and B. M. Novak, Macromolecules, 1991, 24, 326–328 CrossRef CAS.
- T. J. Deming and B. M. Novak, Macromolecules, 1991, 24, 6043–6045 CrossRef CAS.
- T. J. Deming and B. M. Novak, J. Am. Chem. Soc., 1993, 115, 9101–9111 CrossRef CAS.
- T. J. Deming and B. M. Novak, Macromolecules, 1993, 26, 7092–7094 CrossRef CAS.
- K. Onitsuka, K. Yanai, F. Takei, T. Joh and S. Takahashi, Organometallics, 1994, 13, 3862–3867 CrossRef CAS.
- K. Onitsuka, T. Joh and S. Takahashi, Angew. Chem., Int. Ed. Engl., 1992, 104, 851–852 CrossRef.
- F. Millich and G. K. Baker, Macromolecules, 1969, 2, 122–128 CrossRef CAS.
- F. Millich and R. G. Sinclair, II, J. Polym. Sci., Polym. Symp., 1968, 22, 33–43 Search PubMed.
- A. J. M. Van Beijnen, R. J. M. Nolte, W. Drenth and A. M. F. Hezemans, Tetrahedron, 1976, 32, 2017–2019 CrossRef CAS.
- C. J. M. Huige, A. M. F. Hezemans, R. J. M. Nolte and W. Drenth, Recl. Trav. Chim. Pays-Bas, 1993, 112, 33–37 CAS.
- Y. Yamada, T. Kawai, J. Abe and T. Iyoda, J. Polym. Sci., Part A: Polym. Chem., 2001, 40, 399–408.
- C. Kollmar and R. Hoffmann, J. Am. Chem. Soc., 1990, 112, 8230–8238 CrossRef CAS.
- M. Clericuzio, G. Alagona, C. Ghio and P. Salvadori, J. Am. Chem. Soc., 1997, 119, 1059–1071 CrossRef CAS.
- L. Spencer, W. B. Euler, D. D. Traficante, M. Kim and W. Rosen, Magn. Reson. Chem., 1998, 36, 398–402 CrossRef CAS.
- J. T. Huang, J. X. Sun, W. B. Euler and W. Rosen, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 439–446 CrossRef CAS.
- M. M. Green, R. A. Gross, F. C. Schilling, K. Zero and C. Crosby, III, Macromolecules, 1988, 21, 1839–1846 CrossRef CAS.
- F. Takei, K. Onitsuka and S. Takahashi, Macromolecules, 2005, 38, 1513–1516 CrossRef CAS.
- M. Ishikawa, K. Maeda, Y. Mitsutsuji and E. Yashima, J. Am. Chem. Soc., 2004, 126, 732–733 CrossRef CAS.
- Y. Hase, Y. Mitsutsuji, M. Ishikawa, K. Maeda, K. Okoshi and E. Yashima, Chem.–Asian J., 2007, 2, 755–763 CrossRef CAS.
- T. Miyabe, Y. Hase, H. Iida, K. Maeda and E. Yashima, Chirality, 2009, 21, 44–50 CrossRef CAS.
- M. Ishikawa, K. Maeda and E. Yashima, J. Am. Chem. Soc., 2002, 124, 7448–7458 CrossRef CAS.
- Y. Hase, M. Ishikawa, R. Muraki, K. Maeda and E. Yashima, Macromolecules, 2006, 39, 6003–6008 CrossRef CAS.
- Y. Hase, K. Nagai, H. Iida, K. Maeda, N. Ochi, K. Sawabe, K. Sakajiri, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2009, 131, 10719–10732 CrossRef CAS.
- S. Matile, N. Berova, K. Nakanishi, S. Novkova, I. Philipova and B. Blagoev, J. Am. Chem. Soc., 1995, 117, 7021–7022 CrossRef CAS.
- S. Matile, N. Berova, K. Nakanishi, J. Fleischhauer and R. W. Woody, J. Am. Chem. Soc., 1996, 118, 5198–5206 CrossRef CAS.
- N. Berova, K. Nakanishi and R. W. Woody, Circular Dichroism: Principles and Applications, Wiley-VCH, New York, 2nd edn, 2000 Search PubMed.
- F. Takei, H. Hayashi, K. Onitsuka, N. Kobayashi and S. Takahashi, Angew. Chem., Int. Ed., 2001, 40, 4092–4094 CrossRef CAS.
- J. J. L. M. Cornelissen, N. A. J. M. Sommerdijk and R. J. M. Nolte, Macromol. Chem. Phys., 2002, 203, 1625–1630 CrossRef CAS.
- A. D. Schlüter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864–883 CrossRef CAS.
- S. S. Sheiko and M. Moller, Chem. Rev., 2001, 101, 4099–4124 CrossRef CAS.
- J. Kumaki, S. Sakurai and E. Yashima, Chem. Soc. Rev., 2009, 38, 737–746 RSC.
- S.-I. Sakurai, S. Ohsawa, K. Nagai, K. Okoshi, J. Kumaki and E. Yashima, Angew. Chem., Int. Ed., 2007, 46, 7605–7608 CrossRef CAS.
- S.-I. Sakurai, K. Okoshi, J. Kumaki and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 1245–1248 CrossRef CAS.
- S.-I. Sakurai, K. Okoshi, J. Kumaki and E. Yashima, J. Am. Chem. Soc., 2006, 128, 5650–5651 CrossRef CAS.
- T. Kajitani, K. Okoshi, S. I. Sakurai, J. Kumaki and E. Yashima, J. Am. Chem. Soc., 2006, 128, 708–709 CrossRef.
- T. Kajitani, K. Okoshi and E. Yashima, Macromolecules, 2008, 41, 1601–1611 CrossRef CAS.
- H. Onouchi, K. Okoshi, T. Kajitani, S. I. Sakurai, K. Nagai, J. Kumaki, K. Onitsuka and E. Yashima, J. Am. Chem. Soc., 2008, 130, 229–236 CrossRef CAS.
- Z. Q. Wu, K. Nagai, M. Banno, K. Okoshi, K. Onitsuka and E. Yashima, J. Am. Chem. Soc., 2009, 131, 6708–6718 CrossRef CAS.
- H. G. J. Visser, R. J. M. Nolte and W. Drenth, Macromolecules, 1985, 18, 1818–1825 CrossRef CAS.
- J. M. Vandereijk, R. J. M. Nolte, W. Drenth and A. M. F. Hezemans, Macromolecules, 1980, 13, 1391–1397 CrossRef CAS.
- J. J. L. M. Cornelissen, J. J. J. M. Donners, R. de Gelder, W. S. Graswinckel, G. A. Metselaar, A. E. Rowan, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 2001, 293, 676–680 CAS.
- C. Branden and J. Tooze, Introduction to Protein Structure, Garland Publishing, New York, 2nd edn, 1999, vol. 84, p.288 Search PubMed.
- G. A. Metselaar, P. J. H. M. Adams, R. J. M. Nolte, J. L. L. M. Cornelissen and A. E. Rowan, Chem.–Eur. J., 2007, 13, 950–960 CrossRef CAS.
- J. J. L. M. Cornelissen, W. S. Graswinckel, P. J. H. M. Adams, G. H. Nachtegaal, A. P. M. Kentgens, N. A. J. M. Sommerdijk and R. J. M. Nolte, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 4255–4264 CrossRef CAS.
- J. J. L. M. Cornelissen, W. S. Graswinckel, A. E. Rowan, N. A. J. M. Sommerdijk and R. J. M. Nolte, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1725–1736 CrossRef CAS.
- P. Samori, C. Ecker, I. Goessl, P. A. J. de Witte, J. J. L. M. Cornelissen, G. A. Metselaar, M. B. J. Otten, A. E. Rowan, R. J. M. Nolte and J. P. Rabe, Macromolecules, 2002, 35, 5290–5294 CrossRef CAS.
- C. Rivetti, M. Guthold and C. Bustamante, J. Mol. Biol., 1996, 264, 919–932 CrossRef CAS.
- W. Zhuang, C. Ecker, G. A. Metselaar, A. E. Rowan, R. J. M. Nolte, P. Samori and J. P. Rabe, Macromolecules, 2005, 38, 473–480 CrossRef CAS.
- J. J. J. M. Donners, R. J. M. Nolte and N. A. J. M. Sommerdijk, J. Am. Chem. Soc., 2002, 124, 9700–9701 CrossRef CAS.
- A. S. Davydov, J. Theor. Biol., 1973, 38, 559–569 CrossRef CAS.
- A. Scott, Phys. Rep., 1992, 217, 1–67 CrossRef.
- P. Bodis, E. Schwartz, M. Koepf, J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and S. Woutersen, J. Chem. Phys., 2009, 131, 124503 CrossRef.
- E. Schwartz, P. Bodis, M. Koepf, J. J. L. M. Cornelissen, A. E. Rowan, S. Woutersen and R. J. M. Nolte, Chem. Commun., 2009, 4675–4677 RSC.
- M. B. J. Otten, C. Ecker, G. A. Metselaar, A. E. Rowan, R. J. M. Nolte, P. Samorì and J. P. Rabe, ChemPhysChem, 2004, 5, 128–130 CrossRef CAS.
- S. J. Wezenberg, G. A. Metselaar, A. E. Rowan, J. J. L. M. Cornelissen, D. Seebach and R. J. M. Nolte, Chem.–Eur. J., 2006, 12, 2778–2786 CrossRef CAS.
- J. J. L. M. Cornelissen, M. Fischer, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 1998, 280, 1427–1430 CrossRef CAS.
- J. J. L. M. Cornelissen, R. van Heerbeek, P. C. J. Kamer, J. N. H. Reek, N. A. J. M. Sommerdijk and R. J. M. Nolte, Adv. Mater., 2002, 14, 489–492 CrossRef CAS.
- A. Kros, W. Jesse, G. A. Metselaar and J. Cornelissen, Angew. Chem., Int. Ed., 2005, 44, 4349–4352 CrossRef CAS.
- C. E. Finlayson, R. H. Friend, M. B. J. Otten, E. Schwartz, J. Cornelissen, R. J. M. Nolte, A. E. Rowan, P. Samorì, V. Palermo, A. Liscio, K. Peneva, K. Mullen, S. Trapani and D. Beljonne, Adv. Funct. Mater., 2008, 18, 3947–3955 CrossRef CAS.
- V. Palermo, M. B. J. Otten, A. Liscio, E. Schwartz, P. A. J. de Witte, M. A. Castriciano, M. M. Wienk, F. Nolde, G. De Luca, J. Cornelissen, R. A. J. Janssen, K. Mullen, A. E. Rowan, R. J. M. Nolte and P. Samorì, J. Am. Chem. Soc., 2008, 130, 14605–14614 CrossRef CAS.
- R. Dabirian, V. Palermo, A. Liscio, E. Schwartz, M. B. J. Otten, C. E. Finlayson, E. Treossi, R. H. Friend, G. Calestani, K. Mullen, R. J. M. Nolte, A. E. Rowan and P. Samorì, J. Am. Chem. Soc., 2009, 131, 7055–7063 CrossRef CAS.
- V. Palermo, E. Schwartz, C. E. Finlayson, A. Liscio, M. B. J. Otten, S. Trapani, K. Müllen, D. Beljonne, R. H. Friend, R. J. M. Nolte, A. E. Rowan and P. Samorì, Adv. Mater., 2010, 22, E81–E88 CrossRef CAS.
- E. Schwartz, S. L. Gac, J. J. L. M. Cornelissen, R. J. M. Nolte and A. E. Rowan, Chem. Soc. Rev., 2010, 39, 1576–1599 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- E. Schwartz, H. J. Kitto, R. de Gelder, R. J. M. Nolte, A. E. Rowan and J. J. L. M. Cornelissen, J. Mater. Chem., 2007, 17, 1876–1884 RSC.
- H. J. Kitto, E. Schwartz, M. Nijemeisland, M. Koepf, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, J. Mater. Chem., 2008, 18, 5615–5624 RSC.
- E. Schwartz, M. Koepf, H. J. Kitto, M. Espelt, V. J. Nebot-Carda, R. De Gelder, R. J. M. Nolte, J. J. L. M. Cornelissen and A. E. Rowan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4150–4164 CrossRef CAS.
- S. Le Gac, E. Schwartz, M. Koepf, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem.–Eur. J., 2010, 16, 6176–6186 CrossRef CAS.
- D. B. Amabilino, E. Ramos, J. L. Serrano and J. Veciana, Adv. Mater., 1998, 10, 1001–1005 CrossRef CAS.
- E. Ramos, J. Bosch, J. L. Serrano, T. Sierra and J. Veciana, J. Am. Chem. Soc., 1996, 118, 4703–4704 CrossRef CAS.
- D. B. Amabilino, E. Ramos, J.-L. Serrano, T. Sierra and J. Veciana, J. Am. Chem. Soc., 1998, 120, 9126–9134 CrossRef CAS.
- D. B. Amabilino, J. L. Serrano and J. Veciana, Chem. Commun., 2005, 322–324 RSC.
- D. B. Amabilino, E. Ramos, J. L. Serrano, T. Sierra and J. Veciana, Polymer, 2005, 46, 1507–1521 CrossRef CAS.
- J. P. Chen, J. P. Gao and Z. Y. Wang, Polym. Int., 1997, 44, 83–87 CrossRef CAS.
- E. Gomar-Nadal, L. Mugica, J. Vidal-Gancedo, J. Casado, J. T. L. Navarrete, J. Veciana, C. Rovira and D. B. Amabilino, Macromolecules, 2007, 40, 7521–7531 CrossRef CAS.
- E. Gomar-Nadal, J. Veciana, C. Rovira and D. B. Amabilino, Adv. Mater., 2005, 17, 2095–2098 CrossRef CAS.
- N. Hida, F. Takei, K. Onitsuka, K. Shiga, S. Asaoka, T. Iyoda and S. Takahashi, Angew. Chem., Int. Ed., 2003, 42, 4349–4352 CrossRef CAS.
- Y. Q. Tian, Y. Li and T. Iyoda, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1871–1880 CrossRef CAS.
- Y. Q. Tian, K. Kamata, H. Yoshida and T. Iyoda, Chem.–Eur. J., 2006, 12, 584–591 CrossRef.
- T. Kajitani, H. Lin, K. Nagai, K. Okoshi, H. Onouchi and E. Yashima, Macromolecules, 2009, 42, 560–567 CrossRef CAS.
- T. Hasegawa, S. Kondoh, K. Matsuura and K. Kobayashi, Macromolecules, 1999, 32, 6595–6603 CrossRef CAS.
- T. Hasegawa, K. Matsuura, K. Ariga and K. Kobayashi, Macromolecules, 2000, 33, 2772–2775 CrossRef CAS.
Footnotes |
| † Present address: Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California, USA. |
| ‡ Present address: Department of Chemistry & Biochemistry, Arizona State University, PO Box 871604, Tempe, Arizona 85287, USA. |
| This journal is © The Royal Society of Chemistry 2011 |