
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking high-efficiency energy storage and conversion with biocompatible electrodes: the key role of interfacial interaction assembly and structural design†
Jeongyeon
Ahn†
 a,
Hyeseoung
Lim†
b,
Jongkuk
Ko
*b and
Jinhan
Cho
*acd
a,
Hyeseoung
Lim†
b,
Jongkuk
Ko
*b and
Jinhan
Cho
*acd
aDepartment of Chemical and Biological Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
bSchool of Chemical, Biological, and Battery Engineering, Gachon University, 1342 Seongnam-daero, Seongnam si, Gyeonggi-do 13120, Republic of Korea. E-mail: kojk@gachon.ac.kr
cKU-KIST Graduate School of Converging Science and Technology, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
dSoft Hybrid Materials Research Center, Advanced Materials Research Division, Korea Institute of Science and Technology (KIST), 5 Hwarang-ro 14-gil, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: jinhan71@korea.ac.kr
First published on 7th August 2024
Abstract
Biocompatible electrodes, situated at the intersection of bioelectronics and soft electronics, hold the promise of groundbreaking advancements in human–machine interaction and bio-inspired applications. Their development relies on achieving stable, robust deposition of electrically and/or electrochemically active components on biocompatible substrates, ensuring operational stability under various mechanical stresses. However, despite notable progress, most biocompatible electrodes still struggle to simultaneously achieve high mechanical flexibility, electrical conductivity, electrochemical activity, and long-term stability at the same time. These challenges present critical barriers to the development of more advanced biocompatible devices, particularly in the field of energy storage and conversion. The key lies in optimizing the complementary interfacial interactions between active components (i.e., electrical and/or electrochemical components) and biocompatible substrates, and between adjacent active components, as well as in the structural design of the electrodes. In this perspective, we review recent approaches for preparing textile- and hydrogel-based biocompatible electrodes that can achieve high electrical conductivity without compromising favorable properties of biocompatible substrates (i.e., textile and hydrogel) for energy storage and conversion devices. In particular, we highlight the critical role of the interfacial interactions between electrode components and demonstrate how these interactions significantly enhance the energy performance and operational stability.
 Jeongyeon Ahn | Jeongyeon Ahn is a PhD candidate under Prof. Jinhan Cho at the Department of Chemical and Biological Engineering in Korea University. Currently, his current research is focused on the development of novel textile energy electrodes and their energy storage applications using a ligand replacement reaction-based layer-by-layer assembly. |
 Hyeseoung Lim | Hyeseoung Lim is an integrated BS/MS candidate under Prof. Jongkuk Ko at the School of Chemical, Biological and Battery Engineering in Gachon University. Currently, her research interest has focused on development of soft actuators for various robotic applications. |
 Jongkuk Ko | Jongkuk Ko is an assistant professor at the School of Chemical, Biological and Battery Engineering in Gachon University. He received his MS and PhD degree in Chemical and Biological Engineering from Seoul National University in 2012 and 2018, respectively. Then, he had post-doc courses at the Department of Chemical & Biological Engineering in Korea University from 2018 to 2021 and at the Max Planck Institute for Intelligent Systems from 2021 to 2023, respectively. His research focuses on interfacial engineering of organic and inorganic functional materials for energy conversion devices. |
 Jinhan Cho | Jinhan Cho is a professor at the Department of Chemical & Biological Engineering in Korea University since 2010. His research career started in POSTECH and Seoul National University, where completed MS and PhD degrees in 1997 and 2001, respectively. Then, he had post-doc courses at Max Planck Institute of Colloids and Interface and University of Melbourne. In 2003–2005, he was a senior researcher in LG Chemistry R&D center. In 2006–2010, he had worked as an assistant professor in Kookmin University. His research interests have been focused on studying the surface chemistry and electrochemical properties of various electrode materials. |
1. Introduction
Since the dawn of the ubiquitous networking era in the 1990s, electronic devices in today's society have evolved rapidly, overcoming the limitations of traditional bulky and immobile electronics. Driven by a growing demand for mobility and seamless integration, these devices are undergoing a transformative shift towards portable, wearable, and even implantable forms. Simultaneously, considerable research has been dedicated to the development of a suitable power supply, including energy storage and conversion, with high energy efficiency, mechanical flexibility, and long-term operational stability. These advances are essential for adequately powering the functions of these evolving electronic devices. At the core of these changes in both form and functionality, the development of soft electrodes has emerged as a pivotal factor.1–13 Therefore, substantial research endeavors have been devoted to realizing this vision, yielding the remarkable technical advances and the commercialization of innovative products. In particular, soft electrodes based on electrically and/or electrochemically active organics or carbon-based components (i.e., carbon nanotubes (CNTs) and graphene) have exhibited significant promise, offering cost-effective and highly flexible devices.14–17 However, despite immense efforts and technical progress, the performance and operational stability of active organics or carbon components pose significant challenges compared to their rigid metal counterparts in inorganic materials. Furthermore, ensuring the biological and operational stability in the biocompatibility of traditional soft electrodes on the living tissue and/or within the living body remains a complex issue, further complicating their development.Within the diverse realm of soft electrodes, biocompatible electrodes stand out as a unique category.18–22 Unlike their potentially harsh counterparts to living biological systems, especially those based on plastic substrates and conducting polymers, biocompatible electrodes are specifically designed to have a safe and efficient interface with the living body. Moreover, it is imperative for these biocompatible electrodes to possess the capability to maintain optimal flexibility and electrical/electrochemical activity under various external mechanical stresses, such as bending, twisting, compression, and stretching. Due to these favorable properties to human body, biocompatible electrodes play a pivotal role in biomedical technology, supporting applications such as medical implantable devices, wearable health monitoring devices, electrochemical drug delivery systems, and bioactuators. In these contexts, the development of biocompatible and flexible electrodes is essential to mitigate the harmful side effects that conventional electrodes may cause when in direct contact with skin or soft tissues.
To create these electrodes, a variety of biocompatible polymers, including cellulose-based paper, silicon-based poly(dimethyl siloxane) (PDMS), poly(vinyl alcohol) (PVA) and specialized hydrogels derived from poly(ethylene glycol) (PEG), poly(2-methoxyethyl acrylate) (PMEA),23 poly(tetrahydrofurfuryl acrylate) (PTHF),24 and poly(acrylic acid) (PAA), have been utilized as host materials. The subsequent step involves the uniform and robust deposition of biocompatible and electrically and/or electrochemically active components onto these host substrates. For example, Yi Cui's group achieved excellent energy storage performance by incorporating CNTs with electrochemically active properties into a biocompatible paper substrate by solution processing.13 In addition, the Cosnier group reported that the solution deposition of glucose oxidase (GOx) enzymes, capable of converting biochemical energy into electricity, on conductive CNT supports resulted in an electrode suitable as an anode for biofuel cells (BFCs).19,25 Moreover, these advances can have been extended to hydrogel actuators capable of converting electricity into mechanical energy through the deposition of solution-processable conductive components on both sides of hydrogel substrate.
Although some of these energy storage and conversion electrodes have been developed using solution processable active components, in many cases, the electrodes for biological devices are typically prepared by the vacuum deposition of a metal layer on biocompatible substrates.26–28 For instance, Stephanie Lacour's group reported the fabrication of implantable compliant electrodes from a vacuum-deposited gold micro-electrode array embedded in PDMS microchannels for monitoring the bladder nerve function.26 However, this vacuum deposition process faces significant challenges in uniformly depositing the metal layer on biocompatible substrates with varying forms, such as porous or curved structures. This challenge is even more pronounced when working with hydrophilic hydrogel substrates, which are highly promising candidates due to their ability to seamlessly interface with living organisms.29,30 The porous structure and wet surface properties of hydrogels make the robust deposition of conductive layers difficult.31–37 Although conductive components can be successfully deposited onto dried hydrogel films, these films undergo significant swelling in a water environment and shrink when dried. This cycle of swelling and shrinking causes the conductive components to detach easily from the hydrogel film.
To address these issues, significant research efforts have been dedicated to developing conductive hydrogels, with a key focus on integrating conductive components into hydrophilic polymer networks through crosslinking.38–48 One notable study by Zhao et al. demonstrated the creation of highly stretchable and conductive hydrogels by incorporating silver flakes and liquid metal into a hydrophilic polymer network (polyvinyl alcohol-borax gel).48 However, the electrical conductivity of these hydrogels (∼700 S cm−1) was much lower than that of bulk metal, which is essentially required for widespread applications in energy storage and conversion electrodes. Therefore, to more effectively utilize hydrogels for various biocompatible energy electrodes, urgent attention needs to be paid to fully unlock their potential.
In this perspective, we offer a comprehensive review of recent advances and innovative interfacial design strategies in the field of biocompatible energy electrodes, primarily focusing on energy conversion (with a focus on BFCs and soft actuators) electrodes, as depicted in Fig. 1. Our main objective is to scrutinize noteworthy challenges in the field of biocompatible energy electrodes and to explore potential strategies to overcome critical hurdles in achieving high-performance energy electrodes. In particular, our attention is directed toward the hydrogel current collector-based energy electrodes, a relatively unexplored avenue in current research. This article begins with an introduction to various approaches for fabricating biocompatible electrical conductors, highlighting the preparative methods. We then review recent breakthroughs in biocompatible energy storage and conversion electrodes using various interfacial interaction-driven assembly techniques. Emphasis is placed on how careful control of the interfacial interactions between biocompatible substrate and conductive materials can significantly improve the energy performance and operational stability of the electrodes. In addition, we show that these controls are instrumental in improving the overall energy performance of the devices. In summary, we provide a brief outlook on the prospective path of interfacial engineering, envisioning the realization of high-performance energy electrodes in the near future.
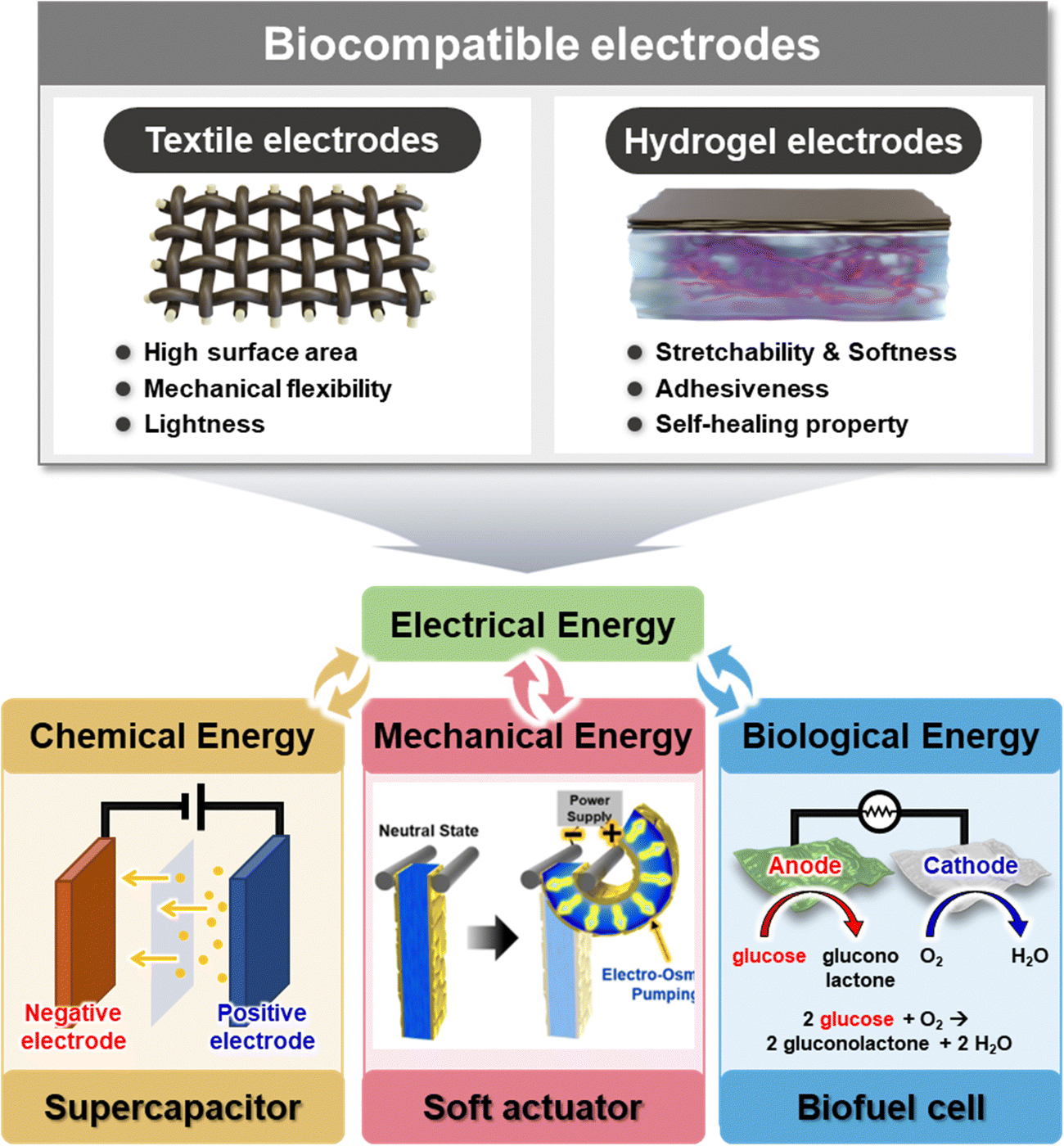 | ||
| Fig. 1 Schematic illustration of textile-based and hydrogel-based biocompatible electrodes for the application of various energy storage and conversion devices, such as supercapacitor, biofuel cell, and soft actuator. | ||
2. Biocompatible electrodes
2.1. Requirements of composing materials
Biocompatible electrodes, which are essential components for various bio-related applications requiring contact with or implantation in the human body, have been extensive studied by introducing diverse materials for the substrates and conductive components. However, achieving a high degree of performance and operational stability of the electrodes without adverse effects on the body remains a critical challenge. Therefore, careful selection and structural design of both substrate and conductive materials are crucial for the successful development of biocompatible electrodes.18,20–22,49To guarantee safe and effective operation when interfaced with biological tissues, biocompatible electrodes should meet several material requirements, including biocompatibility, electrical properties, mechanical properties, and chemical stability. In particular, biocompatibility is the primary requirement, referring to a material's ability to maintain its structure and function when interacting with biological cells or tissues, without causing foreign body responses or undesirable effects.50–53 This means that biocompatible materials must not release harmful substances or provoke immune responses that could lead to inflammation, rejection, or fibrosis, ultimately minimizing toxicity, immune response, and carcinogenicity to tissues. Specifically, minimal cell death is critical when designing electrodes for biomedical devices to ensure long-term, reliable operation under robust physiological conditions. Recent studies have suggested a series of methods to evaluate biocompatibility of materials, including in vitro tests, in vivo tests, degradation tests, aging tests, following guidelines from the International Organization of Standardization (e.g., ISO 10993) and the Food and Drug Administration. The biocompatibility of materials can be assessed using a combination of these methods. Various natural (e.g., cellulose, silk, gelatin, collagen, ions) and synthetic (e.g., PDMS, PI, PVA, PAA, Au, Ti) materials have been suggested to compose substrate and conductive parts of biocompatible electrodes.
In addition to biocompatibility, electrical properties such as electrical conductivity and electrochemical activity are crucial when designing biocompatible electrodes, especially for various energy storage and conversion applications. The electrical conductivity of electrodes directly affects the performance and effectiveness of energy conversion and storage devices. High electrical conductivity (e.g., conductivity of bulk metals >105 S cm−1) enables efficient electrical energy transfer and conversion, minimizes energy loss, and enhances operational stability. Electrochemical activity is another critical property that determines the effectiveness and functionality of biocompatible electrodes for energy conversion devices. Particularly, areal performance parameters, such as areal capacitance (capacitance per unit area, F cm−2), power, and energy densities, have been considered as important factors to reflect the practical utilization of electrodes within the body.
Mechanical properties of the materials also play an important role in determining the performance and functionality of the biocompatible electrodes.54–56 Particularly, flexibility and stretchability of the electrodes enable the electrodes to conform to the dynamic and irregular surfaces of biological tissues without sacrificing the performance. This conformability also minimizes mechanical mismatch and enhances patient comfort, especially in long-term wearable and implantable devices. In addition, softness and compliance of the materials reduce tissue damage and irritation, promoting better integration with the biological systems. Mechanical durability under physiological conditions is essential for the long-term operation stability of the electrodes under various mechanical stresses.
Chemical stability is another critical requirement to prevent the corrosion and degradation of the materials in the physiological environment.21,22,57,58 Corrosion-resistant materials, such as noble metals (e.g., Pt, Au, Ir) have been utilized to ensure the long-term functionality without compromising safety. Hydrolytic stability is also vital, as hydrolytic reactions in the presence of bodily fluids can lead to device failure and potential health risks. Additionally, surface properties, including smoothness and appropriate levels of hydrophilicity or hydrophobicity, influence the interaction between the electrode and the tissue, affecting both performance and biocompatibility.
Achieving all the critical material requirements for biocompatible electrodes remains a notable challenge. However, promising advancements have been made through the introduction of unique structures and materials, such as textile-based and hydrogel-based electrodes. Textile-based electrodes, composed of numerous fibrils and/or fibers forming a highly entangled three-dimensional (3D) porous structure, offer exceptional physical properties. These include a substantially high surface area, mechanical flexibility, and lightness, which cannot be achieved by conventional flat-structured electrodes. The high specific surface area-to-volume ratio is particularly crucial for the functioning of energy device in limited areas and/or volumes.
In terms of materials, hydrogel-based electrodes exhibit novel functionalities due to their distinctive properties. Specifically, the diverse chemical structures of the polymers in hydrogels offer a wide range of tunable properties. By tailoring parameters like monomer and crosslink types, composition, and concentration, hydrogels can achieve remarkable chemical diversity. This translates to unique properties such as stretchability, tissue-like softness, transparency, adhesiveness, self-healing, and stimuli-responsiveness, which cannot be easily realized by other types of soft materials. Therefore, future research should focus on transforming these materials with unique properties into biocompatible electrodes that satisfy abovementioned critical requirements.
2.2. Fabrication method of biocompatible electrodes
Various biocompatible polymers, including cellulose-based paper, silicon-based PDMS, PVA and specialized hydrogels derived from PEG, PMEA, PTHF, and PAA, have been developed for use as host materials for biocompatible electrodes. The next step involves the uniform and robust deposition of biocompatible, electrically and/or electrochemically active components onto these host substrates.Various deposition methods have also been proposed for the fabrication of biocompatible electrodes. Conductive materials, such as metals or graphene, can be deposited onto the substrates using either physical or chemical vapor deposition processes, which inevitably require high vacuum conditions. Alternatively, conductive materials can be processed into slurries and applied onto substrates using solution-based methods, including dip coating, spin coating, spray coating, or inkjet printing (Fig. 2). These methods successfully integrate a variety of biocompatible components, including multifunctional sensors, an active-passive circuit, a light emitting diode, wireless power transmitter coils, high-frequency inductors, capacitors, and antennas into various wearable or implantable biomedical devices.26,27,59–62 However, while these approaches work well on flat, dried surfaces of host materials, they face significant challenges when applied to textile-based and hydrogel-based electrodes. Specifically, these conventional methods struggle to uniformly and robustly deposit electrode layers on biocompatible substrates with diverse forms, such as porous or curved structures (for textile substrates) and wetted surfaces (for hydrogel substrates). Therefore, achieving robust deposition of electrically active components on biocompatible substrates, regardless of their size or shape, is essential for ensuring the high electrical and/or electrochemical performance of the resulting electrodes.
 | ||
| Fig. 2 (a) Gold microelectrode array prepared on PDMS substrate and its fabrication method. Adapted with permission.26 Copyright 2012, Royal Society of Chemistry. (b) Gold nanonetwork microeelctrode array on PI film for the electrocorticogram (ECoG) monitoring system. Adapted with permission.59 Copyright 2020, Wiley-VCH. (c) Schematic illustration of MXene electrode printed on paper substrate and (d) the sheet resistance changes of the printed electrodes as a function of printed paths. (e) Printed electrode array on a paper substrate. Adapted under the terms of the CC-BY Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).63 Copyright 2019, Springer Nature. (f) Schematic illustration and SEM image of black phosphorous/carbon nanotubes electrodes coated on ionic gel by hot press method. Adapted with permission.64 Copyright 2019, Wiley-VCH. | ||
A recently reported approach to converting insulating textiles into metallized textiles involves metal electroplating using an external current flow. In this process, the uniform application of the external current to the substrate during metal electroplating is crucial for forming a high-quality conducting or functional layer. However, for metal electroplating on non-conductive flexible substrates including natural textiles, a conductive seed layer with sufficient electrical conductivity must first be coated onto the insulating textiles. This can be achieved through various deposition methods, such as autocatalytic metal deposition, metal sputtering, and a layer-by-layer (LbL) assembly of conductive components.63,65–69 Once the conductive seed layer is coated onto the insulating textile substrates, precise control of the externally applied electric field during metal electroplating can facilitate the formation of a highly uniform metallized layer on the textile surface.
Numerous research efforts have also been directed towards creating the conductive hydrogel electrodes by depositing electrode layers onto hydrogel film surfaces through methods such as printing, transfer, cluster beam implantation, and chemical reduction.64,70–75 Although hydrogel is one among the most promising host materials for biocompatible electrodes, their wet surface properties pose significant challenges for the robust deposition of conductive components. Achieving high electrical conductivities without compromising the unique properties of hydrogels has been difficult. In particular, hydrogel-based electrodes often suffer from delamination of the electrode layer under mechanical deformation and compromised mechanical properties due to poor interfacial interactions between the conductive components and the hydrogels. To address these issues, significant research efforts have been dedicated to developing conductive hydrogels by incorporating conductive components (e.g., mobile ions, conductive polymers, or conductive particles) within hydrogel matrix.38–48 However, these conductive hydrogels (i.e., ionically conductive hydrogels, conductive polymer-based hydrogels, and nanocomposite hydrogels) often face a trade-off between electrical conductivity and mechanical properties. This trade-off arises from the undesirable effects of higher concentrations of conductive components in the hydrogel matrix. Consequently, the reported hydrogels are not suitable for fabricating host electrodes (or current collectors) with bulk metal-like electrical conductivity, which is essential for widespread applications in energy storage and conversion electrodes.
The successful development of next-generation biocompatible electrodes must effectively unravel the diverse and complex correlations between all electrode constituents and host materials. Particularly, establishing appropriate interfacial interactions between electrode materials and host materials during electrode deposition is highly desirable. The fine control of these interfacial interactions can substantially enhance the performance and stability of biocompatible electrodes. Among various methods, a LbL assembly method, which leverages complementary interactions between different materials, provides versatile approaches for depositing a variety of nanocomposites composed of organic and inorganic materials.76–86
Recently, a ligand replacement reaction-based LbL (LRR-LbL) assembly method was reported.9,87–91 This method allows the direct assembly of hydrophobic metal (or metal oxide) nanoparticles (NPs) with hydrophilic small-molecule linkers through covalent bonding interactions. The use of small-molecule linkers with smaller molecular weights enables both the high NP-packing density and the reduced contact resistance in the assembled layers (Fig. 3a). Ko et al. demonstrated textile electrodes with metal-like conductivities by uniformly and robustly coating the entire textile structures with electrode layers via a small molecule linker (tris-(2-aminoethyl)amine, TREN)-assisted LRR-LbL assembly of hydrophobic Au NPs (stabilized by tetraoctylammonium bromide, TOABr) (Fig. 3b).87 In addition, applying this approach to hydrogel substrates induces continuous LRR assembly of Au NPs in the lateral and vertical dimensions on the hydrogel surface. This process results in highly conductive, mechanically compliant, and robust metallic layers on the hydrogel surfaces (Fig. 4).90,91 An additional deswelling process can generate wrinkled electrode structures, which significantly enhance the electrical and mechanical properties of biocompatible electrodes.
 | ||
| Fig. 3 (a) Schematic illustration of LRR-LbL assembly for the fabrication of textile electrodes. Adapted with permission.9 Copyright 2023, Wiley-VCH. (b) Photographic images of LRR-LbL assembly based (TREN/TOABr-Au NP)10 coated textile electrode. (c) Sheet resistances of (TREN/TOABr-Au NP)n-coated textile electrodes as a function of bilayer number, n. (d) FE-SEM images and EDX mapping images of the (TREN/TOABr-Au NP)10-coated textile electrodes. (e) Electrical property changes of (TREN/TOABr-Au NP)10-coated textile electrodes as a function of bending cycles. Adapted under the terms of the CC-BY Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).87 Copyright 2017, Springer Nature. | ||
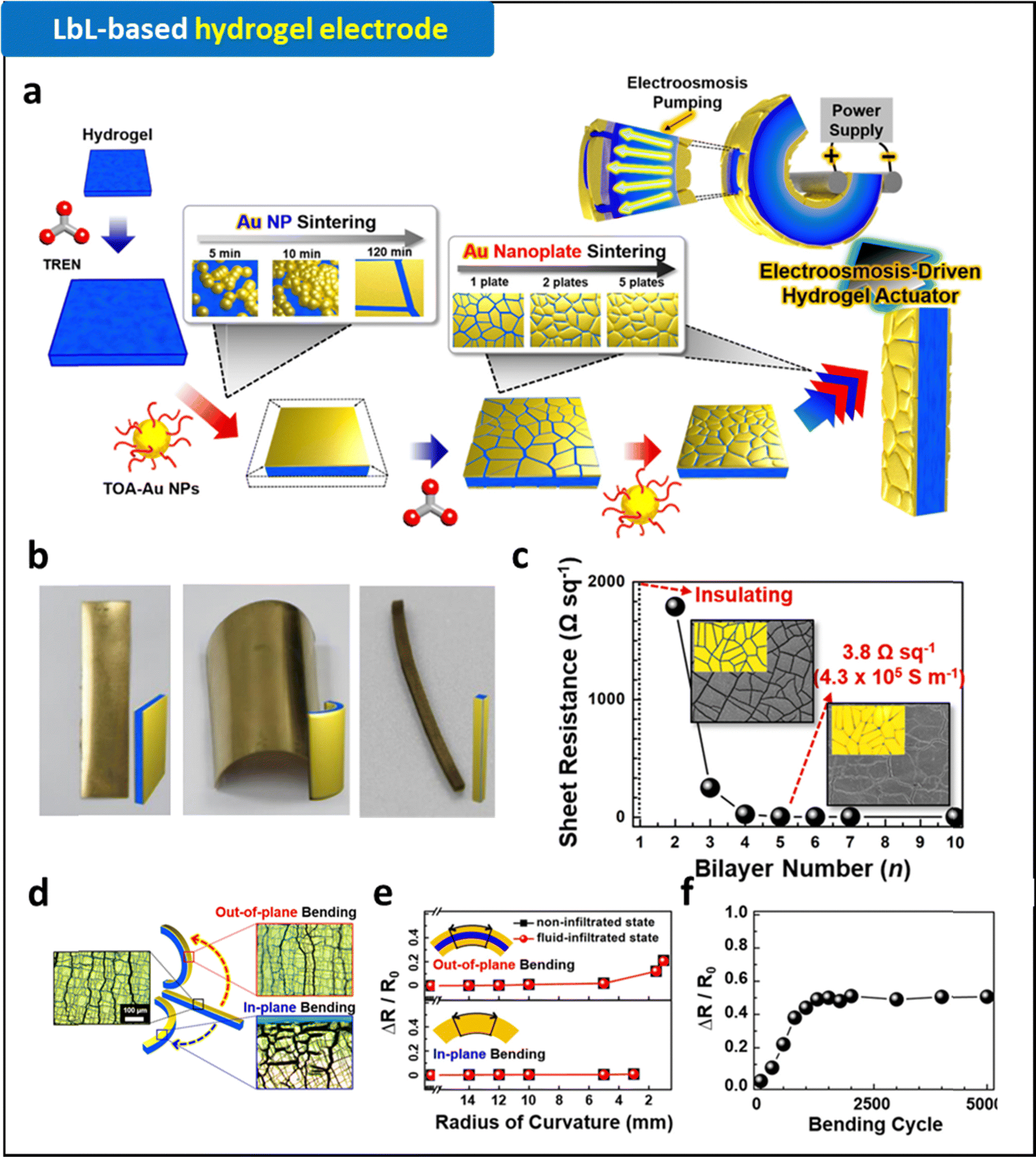 | ||
| Fig. 4 (a) Schematic illustration of LRR-LbL assembly for the fabrication of hydrogel electrodes. (b) Photographic images of (Au NP)n-coated PAA-co-PAN hydrogels with different shapes fabricated by LRR-LbL assembly. (c) Changes of sheet resistances of (Au NP)n-coated PAA-co-PAN hydrogels as a function of bilayer number, n. (d) Schematic and optical images of (Au NP)5-coated hydrogel electrodes with different bending conditions. Electrical stability of (Au NP)5-coated hydrogel electrodes as a function of (e) bending curvature and (f) bending cycle. Adapted with permission.90 Copyright 2020, American Chemical Society. | ||
2.3. Electrical and mechanical properties of biocompatible electrodes
Biocompatible electrodes exhibit severely different electrical and mechanical properties depending on the host materials, structures, and fabrication methods, which ultimately determine their device performance and functionalities as summarized in Table 1. It is crucial to impart electrical properties to biocompatible substrates without compromising their intrinsic mechanical advantages, such as porosity, flexibility, stretchability, or self-healing capabilities. For textile-based electrodes, conductive materials like carbon nanotubes, graphene, metal nanoparticles, and/or conducting polymers can be incorporated using various methods. For instance, slurries or inks containing conductive filers and polymeric binders can be applied to 3D-structured textile substrates through dip coating, spray coating or inkjet printing. While these solution-based processes are simple and suitable for large-area systems, achieving uniform deposition on the highly porous structure is challenging, as inner to outer surface coating is difficult to control. Additionally, polymer-based additives may act as internal contact resistances due to their insulating properties. Beyond physical adsorption methods, conductive or electrochemically active materials can be directly grown onto the surface via the chemical reduction of metal precursors. However, the rapid reduction reaction makes it difficult to control the thickness at the nanometer scale, and impurities such as reducing agents or catalysts hinder the achievement of high electrical conductivity. Furthermore, such methods which do not consider the interfacial interactions between textile surfaces and conductive materials can lead to desorption or failure of the deposited layer under mechanical deformation. For hydrogel-based electrodes, incorporation of mobile ions or embedding conductive materials within hydrogel matrix have shown a limited degree of electrical conductivity, which result in poor electric signal-to-noise ratio inside human body. Although attempts to form electrode layers on hydrogel surfaces based on conventional approaches such as printing, hot pressing, and chemical reduction can enhance electrical properties of hydrogel electrodes, stiff mechanical properties as well as delamination of thick electrode layers under mechanical deformations can have detrimental effects on biological tissues when interfaced with the human body.| Type | Fabrication method | Materials | Conductivity (S cm−1) | Other properties | Ref. |
|---|---|---|---|---|---|
| Textile-based electrodes | Drop casting | PEDOT:PSS | 0.14 | 92 | |
| Hydrothermal activation | rGO | 0.04 | 93 | ||
| Electroless plating, Electroplating | Cu | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
– 94.9% after 2000 bending cycles | 94 | |
| LRR-LbL assembly | Au | 210000 |
– 96% after 10000 bending cycles |
87 | |
| Hydrogel-based electrodes | Mobile ions | 2.74 M NaCl | <0.5 | – Modulus ∼ 100 kPa | 38 |
| – Stretchability > 1000% | |||||
| – Transparency ∼ 98.9% | |||||
| Mobile ions/embedding | NaCl, MXene | Ionic: 1.63 | – Strength ∼ 100 MPa | 95 | |
| Electrical: 200 | – Electrochemical-osmotic activity | ||||
| Embedding | Polyaniline nanofiber | 0.12 | – Modulus ∼ 310 kPa | 96 | |
| – Electrochemical activity | |||||
| Embedding | Ag flakes/liquid metal | 700 | – Modulus ∼ 20 kPa | 48 | |
| – Stretchability > 400% | |||||
| – Self-healing | |||||
| Hot pressing | Graphitic carbon nitride/nitrogen-doped graphene | 780 | – Modulus ∼ 1330 MPa | 72 | |
| – Stretchability ∼ 20% | |||||
| – Electrochemical activity | |||||
| Chemical reduction | Pt | 4000 | – Modulus ∼ 150 MPa | 71 | |
| – Electrochemical activity | |||||
| LRR-LbL assembly | Interconnected cracked Au assembly | 4310 | – Modulus ∼ 4 MPa | 90 | |
| – Stretchability ∼ 20% | |||||
| – Electroosmosis | |||||
| LRR-LbL assembly | Wrinkled Au nanomembrane | 41000 |
– Modulus ∼ 4.2 MPa | 91 | |
| – Stretchability ∼ 110% | |||||
| – Electroosmosis | |||||
Recently, it was reported that LRR-LbL assembly method enabled conformal and robust deposition of electrically and electrochemically active components on various types of textiles, resulting in substantial electrical conductivities under high mechanical deformations (2.1 × 105 S cm−1 maintaining 96% of the initial value after 10000 bending cycles),87 which is highly beneficial for energy storage and conversion devices. Additionally, hydrogel-based electrodes prepared by continuous LRR-LbL assembly method achieved exceptional electrical conductivity under diverse mechanical stresses (∼3600 S cm−1 at fully bent state),90 demonstrating several magnitudes higher electrical conductivities compared to previously reported hydrogel electrodes. Moreover, it should also be noted that the formation of wrinkled electrodes structures in these hydrogel-based electrodes can further enhance their mechanical and electrical properties (1.2 × 104 and 3.5 × 104 S cm−1 at 110% stretched state and fully bent state).91
3. Applications to energy electrodes
3.1. Biocompatible supercapacitors
Miniaturized bioelectronic devices, such as biosensors and continuous glucose monitoring systems, inevitably rely heavily on consistent and biologically stable power sources for uninterrupted functionality. While lithium-ion batteries are renowned for their high energy density, they suffer from relatively low power density and contain toxic materials such as heavy metal-based cathodes, organic liquid electrolytes, and metal (Cu and Al) current collectors.97–99 Additionally, their bulky size and rigid structure make them unsuitable for skin-adherent biosensing devices and for implantation in soft organs or tissues.Specifically, the requirements for biocompatible energy storage devices include several key criteria: (1) all components, ranging from active materials to electrolytes and current collectors, must be made of biocompatible materials. (2) These devices must provide energy and power outputs that can meet the needs of various bioelectronic applications while ensuring sustained and reliable performance under physiological conditions. Considering the challenges associated with replacing worn-out devices that can place significant stress on the body, the ability to maintain a stable electrical supply over extended periods of time is critical. (3) To accommodate the dynamic movements of biological tissues and organs, it is highly desirable for these energy storage devices to be mechanically flexible and stretchable to some degree. As a result, numerous research efforts have been dedicated to developing biocompatible energy storage devices that can meet these stringent requirements.
In this context, supercapacitors have garnered attention as biocompatible energy storage devices owing to their high power density and long-term stability. These supercapacitors are generally classified into two types based on their charge-storage mechanisms: (1) electric double-layer capacitors (EDLCs) with carbon electrodes and (2) rapid redox reactions at the surface of high-capacitance active components.100,101 Their ability to be rapidly charged and discharged, driven by reactions at the electrode/electrolyte interfaces, renders them advantageous for integration with other energy harvesting devices. This integration facilitates efficient storage and utilization of excess energy generated by human tissues. Moreover, supercapacitors typically employ aqueous solutions containing charged ions as electrolytes, and in some cases, human fluid, making them compatible with the human body.15,102 Despite their fast redox mechanisms, they still suffer from relatively low energy densities, which need to be improved for more extensive applications.
Recently, there has been a growing emphasis on areal performance parameters, such as areal capacitance (capacitance per unit area, F cm−2), power and energy densities, as key metrics for evaluating practical and flexible supercapacitors. Therefore, a central challenge in this field is the development of supercapacitors with higher power and energy densities per unit area than existing models. To address this challenge, researchers have explored various 3D porous fibers such as cotton, silk, or yarn as substrates for current collectors in biocompatible energy electrodes.69,92–94,103–112 These fibers offer flexibility, lightness, and environmental friendliness, along with porous structures and large surface areas that provide abundant sites for active material adsorption, enabling high mass loading and areal energy/power density. However, their intrinsic insulating properties require the introduction of electrical conductivity through coating with carbon materials, conductive polymers, or metals. In this case, it is crucial to ensure that the coated conductive layers do not block up the porous structure of the fibers or delaminate during mechanical deformation. For example, Park et al. fabricated a current collector by dip coating a stretchable fabric substrate into poly(3,4-ethylenedioxythiophene)polystyrene sulfonate (PEDOT:PSS) and poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (PEG-b-PPG-b-PEG) solution, followed by spray-coating the composites composed of multi-walled carbon nanotube (MWCNT) and pseudocapacitive nanowires as active materials (Fig. 5a).107 The resulting symmetric supercapacitors exhibited specific and areal capacitances of 48.8 F g−1 and 33.8 mF cm−2, respectively (Fig. 5b). In addition, they could maintain their electrochemical properties under various deformations such as folding, twisting, and stretching (Fig. 5c–e). However, the relatively low electrical conductivities of conducting polymer- or MWCNT-based current collectors (102–103 Ω) compared to that of bulk metal-based current collectors increased the total resistance within the electrodes. To further enhance electrical conductivity, conductive metal layers can be applied onto fiber-based substrates using approaches such as electroplating, electroless plating, or polymer-assisted metal deposition.94,103–105 For instance, Chang et al. developed a highly conductive textile by assembling copper sulfide (CuS) NPs with cysteamine (Cys) linkers followed by electroplating with Ni (Fig. 5f). Additionally, they electroplated a NiCo layer with a layered double hydroxide structure onto the textile to create highly conductive and pseudocapacitive textile electrodes.69 The Ni-electroplated textile demonstrated metal-like electrical conductivity (∼0.06 Ω sq−1) and retained 92.5% of its initial state even after 5000 bending cycles, attributed to strong interfacial interactions between the textile substrates and conductive components (Fig. 5g–i). After depositing an electrochemically active NiCo layer onto the Ni textile current collector, the resulting NiCo textile electrodes exhibited a notably high areal capacitance of 12.2 F cm−2, along with low equivalent series resistance (Fig. 5j). However, it is important to note that Ni and NiCo layers should be used in in vitro systems rather than in vivo systems due to their tendency to corrode within the living body.
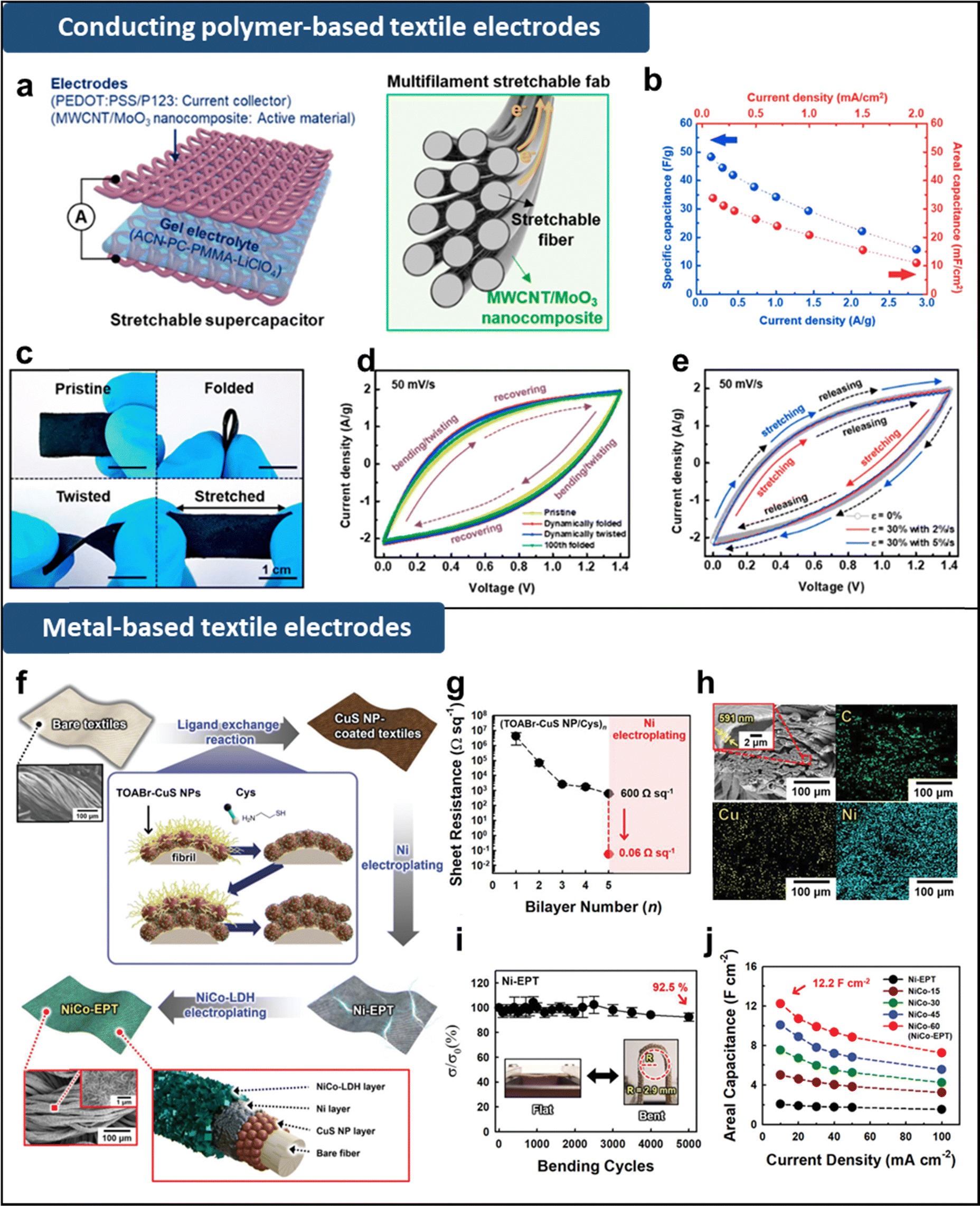 | ||
| Fig. 5 (a) Schematic illustration of stretchable textile supercapacitor electrodes. (b) Specific and area capacitances with variation of current density. (c) Optical images and (d) CV curves of the supercapacitor measured during various dynamic deformation: pristine, folding, twisting, and after 100 folding cycles. (e) CV curves measured during dynamic stretching/releasing cycles at 30% strain with different strain rates of 2 and 5% s−1. Adapted with permission.107 Copyright 2019, American Chemical Society. (f) Schematic illustration of the preparation of NiCo-electroplated textile electrodes. (g) Electrical properties of (CuS NP/Cys)n-coated textiles with increasing bilayer number (n) and after Ni electroplating. (h) FE-SEM and EDX mapping images of Ni-electroplated textile. (i) Relative electrical conductivity of Ni-electroplated textile as a function of bending cycles. (j) Areal capacitance of NiCo-electroplated textile electrodes with various electroplating times of NiCo in comparison with that of Ni-electroplated textile electrodes for various current densities from 10 to 100 mA cm−2. Adapted under the terms of the Creative CommonsAttribution License.69 Copyright 2022, Wiley-VCH. | ||
In addition to natural fiber-based substrates, hydrogels are considered as one of the most promising biocompatible substrates for biocompatible energy devices due to their remarkable advantages, including high biocompatibility, high flexibility, stretchability, and self-healing ability.96,113–124 These properties enable hydrogels to withstand the repeated expansion and contraction experienced by various organs. In addition, hydrogels, acting as efficient reservoirs for electrons and ions, can serve as both electrodes and electrolytes, facilitating the fabrication of all-in-one hydrogel-based supercapacitor.
To prepare these hydrogel-based supercapacitor electrodes, conducting polymers such as polypyrrole (PPy), polyaniline (PANI), and poly(3,4-ethylenedioxythiophene): poly(styrenesulfonate) (PEDOT:PSS) are typically embedded into the hydrogel matrix.111,113,115,121 For example, Zhou et al. fabricated a PANI-hydrogel with interwoven PANI nanofibers, using V2O5·nH2O nanowires as sacrificial 3D templates (Fig. 6a and b).96 Through oxidative polymerization of aniline on V2O5·nH2O nanowires, they constructed an ultrathin PANI nanofiber network, subsequently converted into a pure PANI-hydrogel by removing the templates. The resulting electrode, characterized by its highly porous nature and relatively high electrical conductivity of 0.12 S cm−1, exhibited a high specific capacitance of 636 F g−1 (1540 mF cm−2) and capacitance retention of 83.3% after 10000 charge/discharge cycles (Fig. 6c and d).
 | ||
| Fig. 6 (a) Schematic illustration of fabrication of PANI hydrogel. (b) SEM image of 3D porous network of V2O5 nanowires. (c) CV curves at different scan rate and (d) GCD curves at different scan rate of PANI hydrogel electrodes. Adapted with permission.96 Copyright 2018, American Chemical Society. (e) Schematic illustration of fabrication of PPy@Ag NW-based hydrogel supercapacitor. (f) Macroscopic and FE-SEM images of PPy@Ag NWs foam composite with stretchability and mechanical robustness. (g) Areal and specific capacitance of hydrogel supercapacitor. (h) GCD curves of the hydrogel supercapacitor at a current density of 10 mA cm−2 when stretched to 0–125% strain. Adapted with permission.118 Copyright 2021, Wiley-VCH. | ||
Another approach to providing electrical conductivity is to incorporate metal-based nanomaterials into the hydrogel matrix.115,118,119 As shown in Fig. 6e, open-porous conductive polymer foam was fabricated using a mixture solution containing silver nanowires (Ag NWs), pyrrole monomers, and oxidants.118 Ag NWs provided not only a highly conductive pathway with an electrical conductivity of ∼3.50 S m−1 but also a scaffold for the growth of polypyrrole serving as an active material. Then PVA hydrogel was infiltrated to PPy@Ag NWs foam, followed by freezing and salting out processes. The toughness, flexibility, and stretchability were highly improved by PVA polymer chain coating, while maintaining the open-porous microstructure for fast charge diffusion (Fig. 6f). The all-solid-state supercapacitor fabricated by sandwiching two hydrogel electrodes and one PVA hydrogel separator exhibited high areal capacitance of 5.25 F cm−2 (35.0 F g−1) at 1.0 mA cm−2 and maintained 95.8% of its initial capacitance after 3500 GCD cycles under 20 mA cm−2 (Fig. 6g). Additionally, benefited from the excellent mechanical properties, the device capacitance showed no reduction up to 50% strain and 76% reduction at 125% strain, which was recovered upon releasing the strain (Fig. 6h).
Despite extensive efforts to develop supercapacitors using biocompatible substrates, a recurring problem arises; electrically and electrochemically active components deposited on natural fibers and hydrogels may cause allergic reactions when in contact with human skin. This concern becomes even more pronounced when such supercapacitors are used in the in vivo systems, requiring careful encapsulation of the electrode components to ensure the realization of biologically stable energy storage devices. Therefore, there is an urgent need to apply more stable biocompatible electrode components to these substrates, resulting in energy electrodes with improved stability in both in vivo and in vitro systems.
3.2. Biofuel cells as energy conversion devices
BFCs stand out among diverse energy source devices by utilizing biocatalytic enzymes to convert chemical energy into electrical energy, making them both portable and biocompatible.19,125–132 Furthermore, their potential as implantable energy sources within the human body gives BFCs a significant advantage over alternative power devices. As a result, all components of BFC electrodes are bio-friendly, ensuring no long-term adverse reactions to living organisms, while maintaining high power output and long-term operational stability. Despite these beneficial properties of BFCs, electrically insulating enzymes present a challenge by impeding electron transfer between enzymes and the host electrode (current collector), as well as between adjacent enzymes, thereby limiting the overall energy performance of BFCs.133,134 Moreover, balancing the stringent biological requirements for implantable devices with improved BFC energy output poses a complex and difficult task. However, recent advancements based on interfacial interaction in biocompatible BFC electrodes offer promising solutions for various miniaturized bioelectronics, including in vivo and in vitro systems.In this section, we begin by explaining the electrochemical concept of BFC, and then discuss the recent advancements in BFC electrode design, with a specific focus on the integration of biocompatible components. Additionally, we explore how optimizing the interfacial interactions between enzymes and host electrodes, as well as among adjacent enzymes, can effectively enhance both the power output and operational stability of BFCs.
| Anode: glucose → gluconolactone + 2H+ + 2e− | (1) |
| Cathode: O2 + 4H+ + 4e− → 2H2O | (2) |
| Full electrode system: 2glucose + O2 → 2gluconolactone + 2H2O | (3) |
In most cases, the significantly high power outputs of more than 1 mW cm−2 have been achieved by redox-mediated electron transfer-based BFCs (MET-BFCs). In contrast, conventional direct electron transfer-based BFCs (DET-BFCs) using carbon material-based supports without redox mediators typically exhibit low power output, often only a few to several hundred μW cm−2. Despite this low energy performance of DET-BFCs, recent research trends have focused on developing the high-performance DET-BFCs to address critical issues of MET-BFCs, such as instability, toxicity, and complex synthesis of redox mediators. Specifically, to improve the performance of DET-BFCs, electrochemically inactive and insulating organic compounds within the electrodes should be minimized. Additionally, the biocompatible electrodes should possess higher electrical conductivity and larger active surface areas compared to conventional carbon-based electrodes to enhance the efficiency of electron transfer between the enzymes and the host electrode.
However, despite notable benefits of the carbon fiber or CNT-based host electrodes for the BFCs, their low operational stability, which is due to only physical adsorption between enzymes and host electrode without complementary interfacial interaction, is still controversial. In addition, it should be noted that the electrical conductivity of CNT-based host electrode is much lower than that of the host electrode with bulk metal-like electrical conductivity, which can significantly affect the electron transfer between enzymes and host electrode, and furthermore, the resulting power performance of the BFC electrodes.
We have recently developed more advanced 3D-structured host electrodes, specifically natural cotton textile- and carbon fiber-based host electrodes with robust interfacial interactions and improved electrical conductivity.88,133–135 These electrodes have been successfully utilized in DET-BFCs (Fig. 7). In fabricating the conductive host electrodes from the insulating cotton fibers composed of hydroxyl group (OH)-functionalized cellulose, we initially converted the OH-functionalized cotton surfaces into the outermost NH2-cotton surfaces through a hydrogen-bonding LbL assembly between NH2-functionalized poly(ethylene imine) (PEI) and cotton fibers. It is worth noting that LbL assembly methods offer effective means to fabricate various functional nanocomposite multilayer films with controlled thicknesses, functionalities, and compositions on substrates of different sizes and shapes, utilizing complementary interactions such as electrostatic, hydrogen-bonding, and/or covalent-bonding interactions between adjacent components.
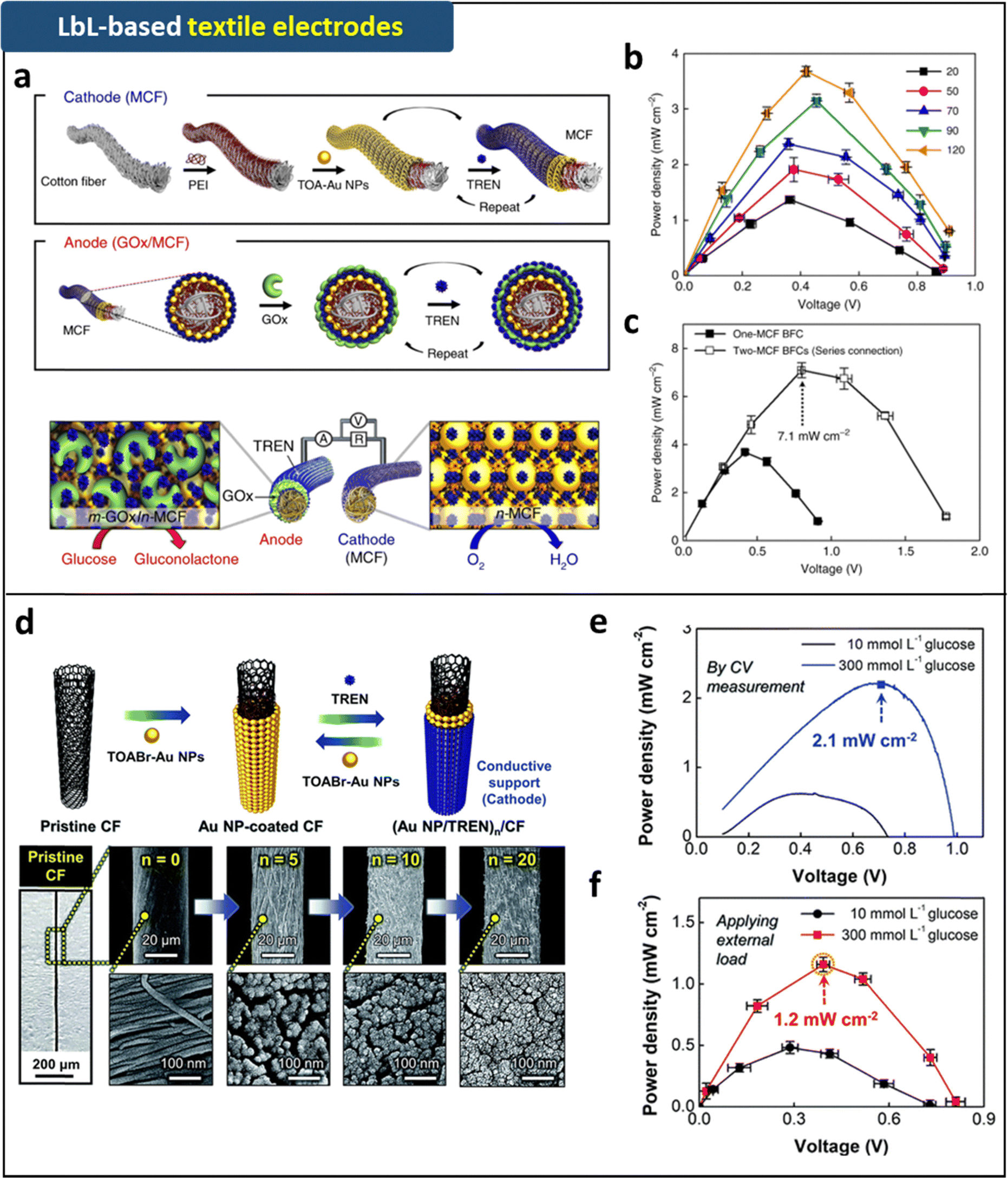 | ||
| Fig. 7 Schematic illustration of (a) fabrication process and (b) operation mechanism of the metallic cotton fiber-based cathode and anode prepared by LRR-LbL assembly. (c) Power output of the metallic cotton fiber-based biofuel cells with external resistors (1 kΩ–10 MΩ) as a function of bilayer number. (d) Power outputs of one and two metallic cotton fiber based-biofuel cells. Adapted under the terms of the CC-BY Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).88 Copyright 2018, Springer Nature. (d) Schematic illustration and FE-SEM images of (TOABr-Au NP/TREN)20/cotton fiber electrodes fabricated by LRR-LbL assembly. (e) Power output of the complete hybrid biofuel cell as a function of potential in 300 mmol L−1 glucose in ambient conditions. (f) Power output of the complete hybrid biofuel cells with a fixed external resistance in the range of 1 kΩ to 10 MΩ. Adapted with permission.135 Copyright 2019, Royal Society of Chemistry. | ||
Following this interfacial LbL-assembly-induced surface modification, the NH2-functionalized cotton fibers were subsequently immersed in TOABr-Au NPs dispersed in toluene. As mentioned earlier, the bulky TOA ligands, which were weakly bound to the surface of Au NPs, were almost completely displaced by the outermost NH2 groups of the cotton fibers due to the high affinity (i.e., covalent-bonding interaction) between the Au NP surface and NH2 groups (Fig. 7). Using this LRR-LbL assembly approach, NH2-functionalized molecular linkers (TREN) in alcohol were sequentially assembled with TOABr-Au NPs in toluene, resulting in the formation of highly conductive cotton fibers serving as host electrodes. In particular, the Au NP multilayers, which were fabricated by small organic linker-based LRR-LbL assembly, could exhibit extremely high electrical conductivity due to the minimized separation distance between adjacent Au NPs (Fig. 7). The electrical conductivities of the formed fibers were estimated to be approximately 3500 S cm−1. Furthermore, the outer surfaces of numerous cellulose fibrils within the fiber host electrode were uniformly coated with Au NP multilayers, suggesting that the conductive cotton electrode could act an as enzyme reservoir as well as a simple host electrode. It should also be noted that the residual toluene solvents were completely removed from the host electrode by a thorough drying process.
Based on this highly conductive host electrode, GOx enzymes containing significant amounts of amino acid groups in pH 7.4 PBS aqueous solution could be further assembled with TOABr-Au NPs in toluene (via LRR-LbL assembly between TOA ligands of Au NPs and neutral NH2 group of GOx) or TREN molecular linkers in the same PBS solution (through electrostatic interaction LbL-assembly between anionic carboxylate ion (COO−) group of GOx and cationic amine (NH3+) group of TREN) on the fiber host electrode. The resulting enzymatic fiber anode exhibited significantly high anodic performance, and no residual organic solvent was detected from the anode due to sufficient washing and drying processes. Notably, when these anodes were paired with cotton fiber-based cathodes (either Au NP multilayer-coated host electrode or Pt-sputtered host electrode), the complete DET-BFC devices exhibited exceptionally high power output (>3 mW cm−2 in 300 mmol L−1 glucose concentration under ambient conditions) and high operational stability (∼62% of initial power output in 10 mmol L−1 glucose buffer after 35 days) (Fig. 7). These results underscore the critical role of interfacial and structural design in enhancing the power output and operational stability of BFC devices.
3.3. Biocompatible soft actuators
There have been significant research efforts to develop soft counterparts to conventional electromagnetic motors/actuators, with the goal of efficiently converting electrical energy into mechanical energy for a wide range human-interfaced robotic application.1,2,4,75,136–138 Although various mechanisms, including electrostatic, electrothermal, and electrochemical, have been proposed for the development of electroactive soft actuator, their progress has been stagnant due to the intrinsic limitations of the materials and their corresponding working mechanisms. Specifically, challenges such as heat management in electrothermal actuators, high operation voltages required for electrostatic actuators, and poor performances of electrochemical actuators present substantial huddles for their integration into human-friendly robotic applications, which require both material and operational compatibility. Development of a novel class of electroactive soft actuator with biocompatible properties, mild operation conditions (operation voltages <3 V, inhibited electrochemical reactions, operation time > 1 h), and high performance (strain >20%, power density >50 kW m−3) is essential for advancing the next-generation soft robotics.The development of hydrogel electrodes with unique features such as electrical conductivity, softness, water/ion inclusion, stimuli-responsiveness, and biocompatibility enabled the creation of new class of soft actuators that cannot be realized by other types of soft actuators. This section introduces and discusses the existing hydrogel actuators based on different mechanisms, including electrostatic, electrochemical, electroosmotic, and electrochemical-osmotic mechanisms, highlighting their features and limitations.
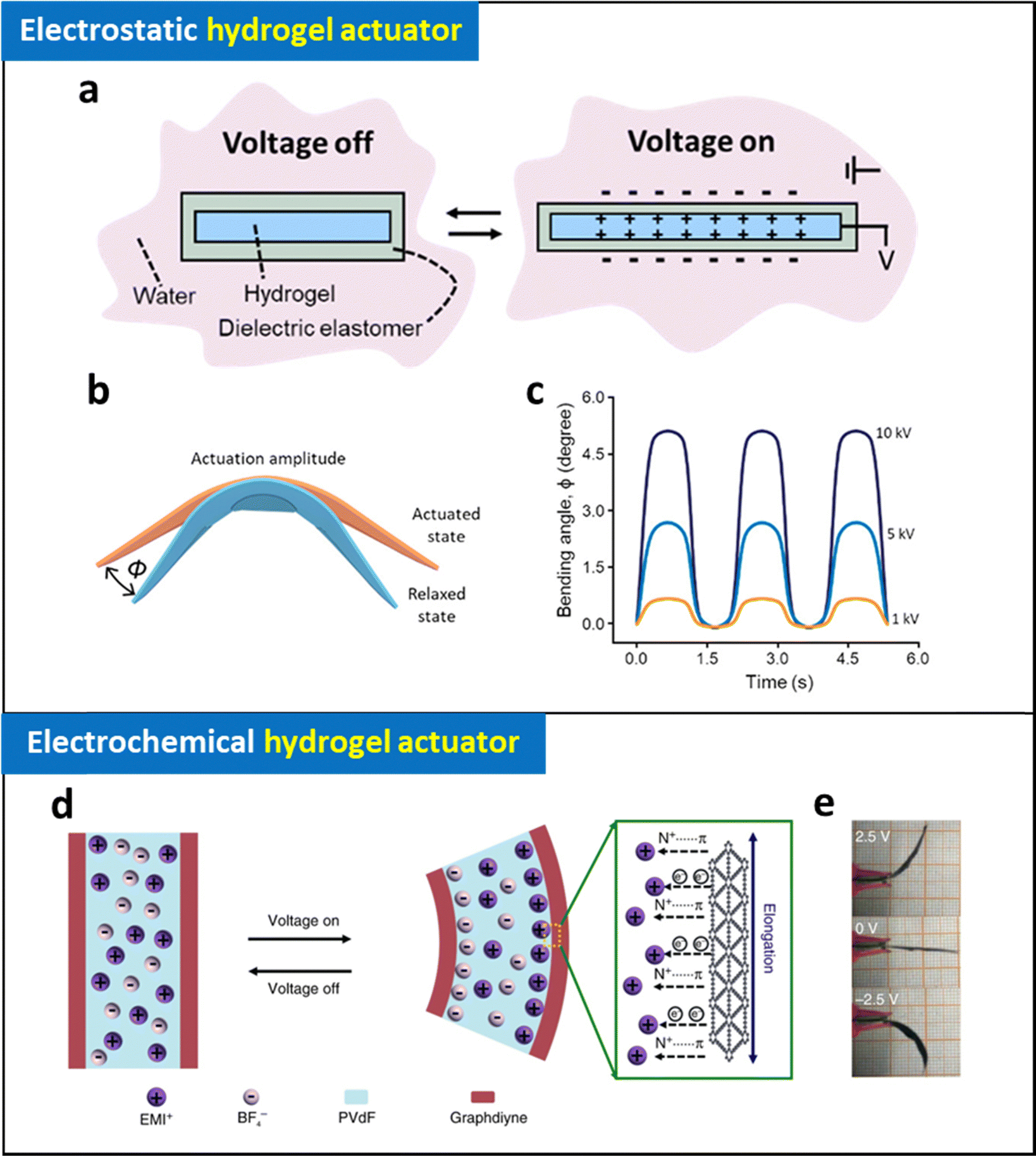 | ||
| Fig. 8 Schematic illustration of (a) operating mechanism and (b) bending of electrostatic hydrogel actuators and (c) their bending angle at different applied voltages. Adapted with permission.143 Copyright 2022, American Chemical Society. (d) Schematic illustration of operating mechanism of electrochemical actuator (graphdiyne-based electrode coated ionic gel) and (e) photographic images of their bending motions at different applied voltages. Adapted under the terms of the CC-BY Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).144 Copyright 2018, Springer Nature. | ||
Extending the capabilities of energy conversion through electrostatic interaction, a fiber-shaped ionic device was developed that combines sensing and actuation in a single device.142 This device consists of fiber-shaped elastomers coated with an organogel, specifically polyacrylamide network dispersed in ethylene glycol with lithium chloride. When an AC voltage of 1500 V is applied to two closely positioned fiber-shaped ionic device, electrostatic interactions generate vibrations in the ionic fibers. Notably, the ionic device also achieves additional functionalities such as adhesion and sensing by adjusting the electric signal, demonstrating multiple energy conversion capabilities within a single device.
Despite their promising capabilities, they face limitations due to their high operating voltages, electric breakdown, and complicated fabrication process. Hydraulically amplified self-healing electrostatic (HASEL) artificial muscles, which utilize hydraulic pressure induced by local electrostatic forces in liquid-state dielectric materials, have been developed to address some of these issues.137,140,141 Additionally, new electrostatic artificial muscles constructed with ionically conductive hydrogel electrodes exhibit outstanding and reliable actuation performance, resulting in transparent, high-speed, and large-strain soft actuators. Despite the promising achievement, electrostatic hydrogel actuators still face significant challenges such as high operating voltages (∼kV) and risk of leakage or damage of dielectric materials, which are detrimental when interfaced with biological tissues. Addressing these issues is crucial for future biomedical applications.
Thin metal layer-coated hydrogel has been widely utilized in developing electrochemical hydrogel actuators. Ionic polymer–metal composites, featuring μm-thick Pt layers prepared by electrodeposition on ionic polymer films such as Nafion or PVDF, have demonstrated bending motions with 1% strain and a response time of a few seconds.71,74,75 Moreover, multi-stacking of the actuators can generate significant force, surpassing that of conventional hydrogel actuators.71 To decrease the thickness of the metallic layer, Yan et al. reported supersonic cluster beam implantation method that the 100-nm thick Au layer can be deposited on PAA-PAN hydrogel. In this case, the formed device exhibited the bending motion with a 2% strain at 2 V.145
Carbon-based electrochemically active materials, such as graphene, CNT, MXene, have been employed to leverage their high capacitance for constructing electrochemical hydrogel actuators.64,70,72,73 Umrao et al., introduced a MXene-based electrode on an ionic gel (Nafion containing [EMIm][BF4] ionic liquid).73 Based on the high capacitance of the device, applying low voltages ranging from 0.1 to 1 V caused larger mobile cations (EMIm+) to be attracted to the negative electrode, leading to expansion and bending motion of the actuator in a controllable and reversible manner. This actuator exhibited a maximum actuation strain of 1.37% and operated at frequencies up to 10 Hz. Notably, it achieved long-term cyclic stability of 97% over 18000 cycles, demonstrating potential for various kinetic art applications, such as flapping butterflies and moving leaves on a tree. Although electrochemical hydrogel actuators can be operated under low voltages (<2 V) with high frequency (∼10 Hz), their performance has generally been limited to less than 2% of strain, restricting their wide range of robotic applications in actual biomedical scenarios.
For example, Na et al. reported a strong and fast hydrogel actuator utilizing the electroosmosis phenomenon that can generate large stress of 0.73 MPa.146 The reported device features polyelectrolyte hydrogels (PSPA polymer with 1 M LiCl aqueous solution) wrapped with stiff semi-permeable membrane. When an electric field (4 V cm−1) is applied to the hydrogels with stiff membrane, it can generate turgor pressure up to 1.44 MPa, demonstrating the ability to break a brick and construct underwater structures. It should be noted that these achievements could not be realized by previously reported actuators. Despite the promising features of electroosmotic actuators, they have suffered from the slow response time (∼several minutes), the requirement of large operation voltages (∼10 V), and the use of rigid and bulky systems. Seamless integration of electrodes on hydrogels is highly desirable to improve response time of the actuation by suppressing undesirable electrochemical reactions.
As described in Section 2.2, electrodes with a unique structure (i.e., interconnected cracked electrodes, wrinkled nanomembrane electrodes) have been successfully introduced on hydrogels by using LRR-LbL assembly methods (Fig. 9a and b).90,91 Based on an exceptional performance of the hydrogel electrodes (electrical conductivity of 4.1 × 104 S cm−1 with 110% stretchability), high degree of electric field can be efficiently applied on hydrogels at mechanically dynamic states. Thanks to these breakthroughs in electrode design, the wrinkled nanomembrane electrode based-hydrogel actuators can be operated at low voltages (less than 3 V) with unprecedentedly high performance based on electroosmotic pumping inside hydrogel matrix. Based on the novel mechanism, small-scale untethered robots can be developed, demonstrating overcoming intrinsic limitations of other types of soft actuators. Although electroosmotic hydrogel actuators demonstrated promise for low-voltage operation with high actuating performance that address the limitation of electrostatic and electrochemical hydrogel actuators, their limited degree of operation stability (∼500 cycles) should be further enhanced for future studies.
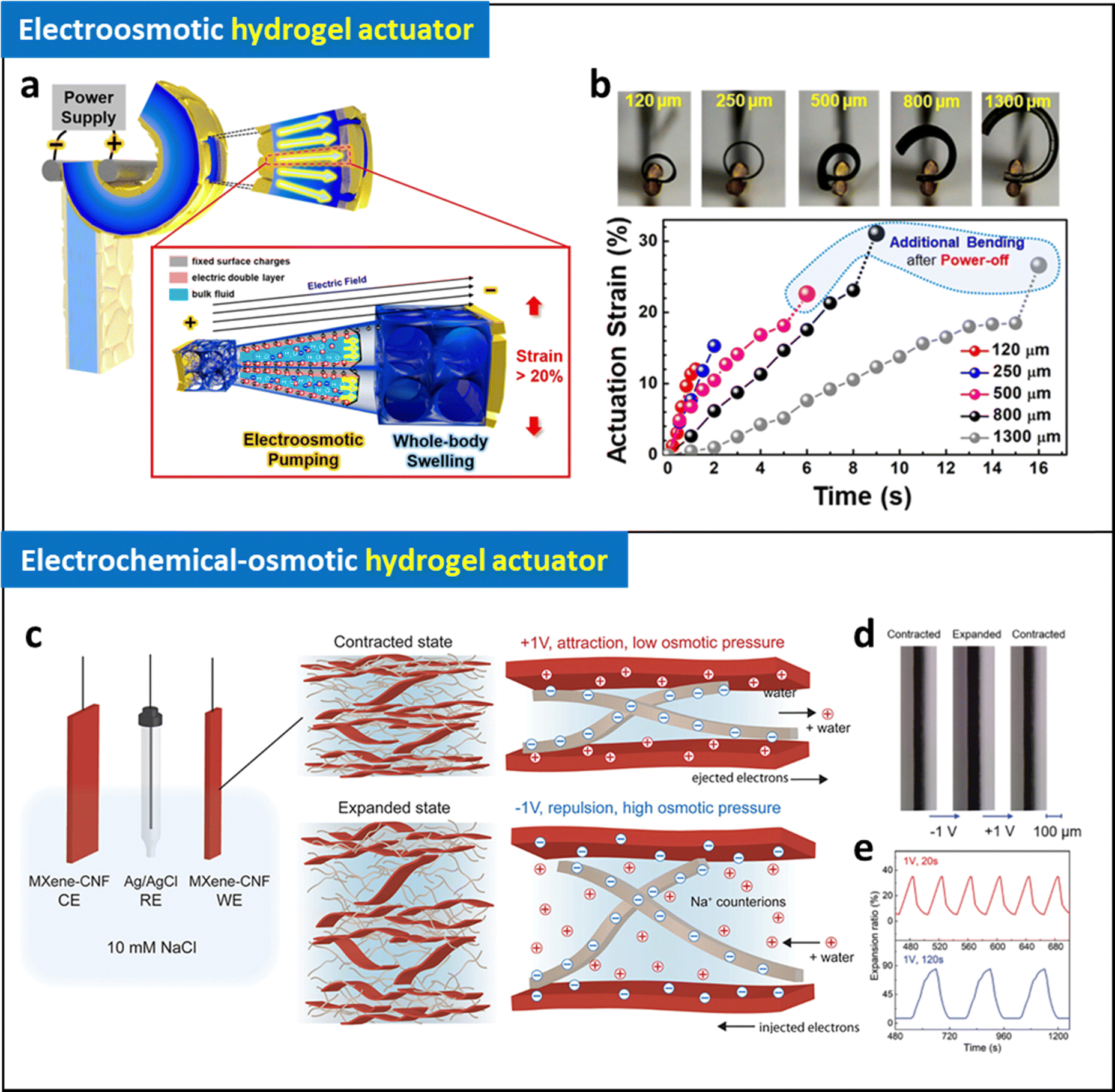 | ||
| Fig. 9 (a) Schematic illustration of operating mechanism of electroosmotic hydrogel actuator. (b) Photographic images and bending strain of the electroosmotic actuators with different thickness as a function of time at 3 V. Adapted with permission.90 Copyright 2020, American Chemical Society. (c) Schematic illustration of operating mechanism of electrochemical-osmotic hydrogel actuator. (d) Photographic images of expanded and contracted actuators with different applied potentials and their expansion ratio as a function of time at 1 V with different frequency. Adapted with permission.95 Copyright 2023, Wiley-VCH. | ||
Benselfelt et al. reported electrochemically controlled anisotropic nanowire composite hydrogels with enhanced conductivity.147 Applying an electrical stimulus to these hydrogels amplifies the electrochemical swelling pressure, enabling the precise electrical manipulation of uniaxial shape deformations. Notably, precise shape deformation and a 300% uniaxial expansion were achieved even at a low voltage of 1 V. Moreover, these ECO hydrogel actuators can exert a volumetric force of approximately 10000 N cm−3, vastly surpassing the capabilities of electroosmotic hydrogel actuators by two orders of magnitude.
As summarized in Table 2, severely different types of biocompatible soft actuators have been developed depending on hydrogel electrodes used, showing crucial impacts of various properties on their performance. It should be noted that not only in soft actuators in general, but also in a variety of other energy conversion devices, their performance can be significantly improved through meticulous interface control of biocompatible electrodes. Additionally, interfacial and structural engineering approach for biocompatible materials can provide a foundational platform for developing high-performance biocompatible soft actuators with enhanced energy conversion and superior operational stability.
| Type | Electrode type | Materials | Performance | Ref. |
|---|---|---|---|---|
| Supercapacitor | Stretchable fabric | MWCNT/MoO3 NW | 33.8 mF cm−2 (at 0.1 mA cm−2) | 107 |
| Textile | NiCo | 12.2 F cm−2 (at 10 mA cm−2) | 69 | |
| Polyester yarn | rGO | 49.4 mF cm−2 (at 1.0 mA cm−2) | 104 | |
| Conducting polymer-based hydrogel | PVA@PANI | 1475 mF cm−2 (at 1.0 mA cm−2) | 124 | |
| Conducting polymer-based hydrogel | V2O5·nH2O NW | 1540 mF cm−2 (at 5.0 mA cm−2) | 96 | |
| Metal nanocomposite-based hydrogel | PPy@Ag NW | 5.25 F cm−2 (at 1.0 mA cm−2) | 118 | |
| Biofuel cell | Microfiber | Carbon fiber, conducting polymer | 137 μW cm−2 at 37 °C | 130 |
| CNT fiber | CNT | ∼0.7 mW cm−2 | 131 | |
| CNT yarn textile | PEDOT, MWCNT | 2.18 mW cm−2 | 132 | |
| Cotton fiber | Au NPs | >3 mW cm−2 | 88 and 135 | |
| Soft actuator | Ionic hydrogel | PAAm (NaCl) | Areal strain 167% at 18 kV | 38 |
| Ionic hydrogel | PAAm (LiCl) | Bending ∼26.9° at 10 kV | 139 | |
| Nanocomposite hydrogel | CNF/MXene CNF/MWCNT | Uniaxial swelling ∼85% at 1 V | 95 | |
| Nanocomposite hydrogel | CNF/MXene CNF/MWCNT | Uniaxial swelling ∼300% at 1 V | 147 | |
| Force ∼10000 N cm−3 |
||||
| Electrode-coated hydrogel | Au (100 nm) | Bending stain ∼2% at 2 V | 145 | |
| Electrode-coated hydrogel | MXene-coated ionic gel | Bending stain ∼1.37% at 1 V | 73 | |
| Electrode-coated hydrogel | Au nanomembrane-coated | Bending stain >50% at 3 V | 91 | |
4. Summary and perspective
The fabrication of high-performance biocompatible and soft energy electrodes with high electrical conductivity, mechanical flexibility, and bio-friendliness is one of the classic issues in realizing the electrodes for a variety of the next-generation wearable, skin-attached, and implantable electronics. In most cases, to fabricate such electrodes, metal and graphene layers have been deposited on the surface of biofriendly soft substrates by vacuum deposition process, chemical reduction process, or chemical vapor deposition method. On the other hand, solution-dispersible conductive components like metal nanocomposites or carbon nanotubes have been applied via spin-coating or simple mixing processes. However, it is very difficult for these conductive components and traditional processes to be stably and uniformly adsorbed onto the soft substrates with various form factors (i.e., porous and curved plastics, textiles, or hydrogel substrates) and sizes. Furthermore, the electrical conductivity of solution-processable conductive components is not suitable for the electrodes that require the bulk metal-like electrical conductivity due to the numerous contact resistances and their intrinsically low electrical conductivities (mainly carbon-based components and conducting polymers). In particular, hydrogel substrates, which are considered as one of the most promising candidates among various bio-friendly and soft substrates, have been unable to achieve stable and dense surface adsorption of various conductive materials. Thus, the interfacial interaction between soft substrate and conductive components as well as the careful control over these organic ligands and surface coverage plays an important role in developing energy electrodes and determining their performance.As described in this review article, the interfacial interaction control over the outermost surfaces of both biocompatible/soft substrates and conductive metal NPs can produce the ultrathin nanocomposite layers that can be used as electrical conductors of energy storage and conversion systems. In particular, converting hydrophobic metal NPs to hydrophilic metal NPs through sequential adsorption assembly based on interfacial interactions can perfectly convert various insulating biocompatible substrates, including even hydrogels, into electrically conductive substrates. Different from conventional coating methodologies (i.e., vacuum deposition process, spin-coating, and slurry casting), this approach ensures a uniform coating of functional NPs over the entire area of porous substrates (paper and cotton textile) and hydrated substrates (hydrogels).
As a result, this unique assembly could significantly enhance the charge transfer kinetics between neighboring NPs as well as between the substrate and NPs while maintaining the mechanical flexibility of biocompatible substrates. These phenomena have been confirmed in applications such as supercapacitors, BFCs, and soft actuators. Despite the advantages of interfacial interaction assembly, such as accurate control over surface chemistry and the surface coverage of the active NPs within the electrodes, the dip-coating process for thin film (nanometer-thick) deposition often causes time-consuming issues. However, these issues can be easily optimized by simply controlling the solution concentration of functional NPs and employing methods such as automatic spray deposition and roll-to-roll processes. In this regard, an effective combination of the interfacial interaction assembly of functional nanomaterials, structural design, and the advanced fabrication technology can be a breakthrough in overcoming the barriers of applying the biocompatible electrodes to practical devices. Therefore, we envision that such an approach can be effectively applied to the energy electrodes for various wearable, portable, and/or implantable devices, including energy storage, conversion devices and biological devices.
Author contributions
J. A. and H. L. contributed equally to this work. J. A., H. L., J. K., and J. C. wrote and revised the manuscript. All authors discussed the results and commented on the manuscript.Data availability
This review is based on previously published studies and does not involve new data collection or analysis. Where previously published data have been used or reproduced the relevant citations are included.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT; Ministry of Science and ICT) (NRF-2021R1A2C3004151 and RS-2024-00352893). This work was also supported from the KIST Institutional Program (Project no.: 2V09840-23-P025) and the KU-KIST Graduate School of Converging Science and Technology Program.References
- J. A. Rogers, T. Someya and Y. Huang, Science, 2010, 327, 1603–1607 CrossRef CAS PubMed.
- S. I. Rich, R. J. Wood and C. Majidi, Nat. Electron., 2018, 1, 102–112 CrossRef.
- J. Ko, R. Berger, H. Lee, H. Yoon, J. Cho and K. Char, Chem. Soc. Rev., 2021, 50, 3585–3628 RSC.
- P. Rothemund, Y. Kim, R. H. Heisser, X. Zhao, R. F. Shepherd and C. Keplinger, Nat. Mater., 2021, 20, 1582–1587 CrossRef CAS PubMed.
- S. Liu, Y. Rao, H. Jang, P. Tan and N. Lu, Matter, 2022, 5, 1104–1136 CrossRef CAS.
- G. D. Cha, D.-H. Kim and D. C. Kim, Korean J. Chem. Eng., 2024, 1–24 CAS.
- D. C. Kim, H. Seung, J. Yoo, J. Kim, H. H. Song, J. S. Kim, Y. Kim, K. Lee, C. Choi and D. Jung, Nat. Electron., 2024, 1–10 Search PubMed.
- H. J. Kim, H. Choi, D.-H. Kim and D. Son, Nano Lett., 2024, 24, 8453–8464 CrossRef CAS PubMed.
- W. Chang, E. Yong, Y. J. Chung, Y. Ko and J. Cho, Small Structures, 2024, 5, 2300330 CrossRef CAS.
- C. Sun, Z. Han, X. Wang, B. Liu, Q. Li, H. Li, J. Xu, J.-M. Cao and X.-L. Wu, Adv. Funct. Mater., 2023, 33, 2305606 CrossRef CAS.
- Z.-Y. Cu, X.-T. Wang, Y.-L. Heng, K.-Y. Zhang, H.-J. Liang, J.-L. Yang, E. H. Ang, P.-F. Wang, Y. You, F. Du and X.-L. Wu, Sci. Bull., 2023, 68, 2302–2306 CrossRef PubMed.
- D. Li, W.-Y. Lai, Y.-Z. Zhang and W. Huang, Adv. Mater., 2018, 10, 1704738 CrossRef PubMed.
- T. Cheng, Y.-Z. Zhang, S. Wang, Y.-L. Chen, S.-Y. Gao, F. Wang, W.-Y. Lai and W. Huang, Adv. Funct. Mater., 2021, 31, 2101303 CrossRef CAS.
- L. Hu, J. W. Choi, Y. Yang, S. Jeong, F. La Mantia, L.-F. Cui and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21490–21494 CrossRef CAS PubMed.
- J. S. Chae, N.-S. Heo, C. H. Kwak, W.-S. Cho, G. H. Seol, W.-S. Yoon, H.-K. Kim, D. J. Fray, A. E. Vilian and Y.-K. Han, Nano Energy, 2017, 34, 86–92 CrossRef CAS.
- X. Chen, L. Yin, J. Lv, A. J. Gross, M. Le, N. G. Gutierrez, Y. Li, I. Jeerapan, F. Giroud and A. Berezovska, Adv. Funct. Mater., 2019, 29, 1905785 CrossRef CAS.
- W. Jeon, J. M. Lee, Y. Kim, Y. Lee, J. Won, S. Lee, W. Son, Y. H. Koo, J. W. Hong and H. Gwac, Adv. Mater., 2024, 2313625 CrossRef CAS PubMed.
- M. Irimia-Vladu, Chem. Soc. Rev., 2014, 43, 588–610 RSC.
- A. Gross, M. Holzinger and S. Cosnier, Energy Environ. Sci., 2018, 11, 1670–1687 RSC.
- Y. Shi, R. Liu, L. He, H. Feng, Y. Li and Z. Li, Smart Mater. Med., 2020, 1, 131–147 CrossRef.
- W. Yang, Y. Gong and W. Li, Front. Bioeng. Biotechnol., 2021, 8, 622923 CrossRef PubMed.
- Y. Zhou, G. H. B. Morris and M. Nair, Cell Rep. Phys. Sci., 2024, 5, 101852 CrossRef.
- M. Tanaka, T. Motomura, M. Kawada, T. Anzai, Y. Kasori, T. Shiroya, K. Shimura, M. Onishi and A. Mochizuki, Biomaterials, 2000, 21, 1471–1481 CrossRef CAS PubMed.
- A. Mochizuki, T. Hatakeyama, Y. Tomono and M. Tanaka, J. Biomater. Sci., Polym. Ed., 2009, 20, 591–603 CrossRef CAS PubMed.
- K. Elouarzaki, M. Bourourou, M. Holzinger, A. Le Goff, R. Marks and S. Cosnier, Energy Environ. Sci., 2015, 8, 2069–2074 RSC.
- E. Delivopoulos, D. J. Chew, I. R. Minev, J. W. Fawcett and S. P. Lacour, Lab Chip, 2012, 12, 2540–2551 RSC.
- K. Kwon, J. U. Kim, S. M. Won, J. Zhao, R. Avila, H. Wang, K. S. Chun, H. Jang, K. H. Lee and J.-H. Kim, Nat. Biomed. Eng., 2023, 7, 1215–1228 CrossRef PubMed.
- J.-H. Kim, A. Vázquez-Guardado, H. Luan, J.-T. Kim, D. S. Yang, H. Zhang, J.-K. Chang, S. Yoo, C. Park and Y. Wei, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2404007121 CrossRef PubMed.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef PubMed.
- X. Xue, Y. Hu, Y. Deng and J. Su, Adv. Funct. Mater., 2021, 31, 2009432 CrossRef CAS.
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS.
- H. Yuk, B. Lu and X. Zhao, Chem. Soc. Rev., 2019, 48, 1642–1667 RSC.
- F. Fu, J. Wang, H. Zeng and J. Yu, ACS Mater. Lett., 2020, 2, 1287–1301 CrossRef CAS.
- L. Hu, P. L. Chee, S. Sugiarto, Y. Yu, C. Shi, R. Yan, Z. Yao, X. Shi, J. Zhi and D. Kai, Adv. Mater., 2023, 35, 2205326 CrossRef CAS PubMed.
- J. Ko, Korean J. Chem. Eng., 2023, 40, 3106–3129 CrossRef CAS.
- Y. Zhang, Y. Tan, J. Lao, H. Gao and J. Yu, ACS Nano, 2023, 17, 9681–9693 CrossRef CAS PubMed.
- T. Zhu, Y. Ni, G. M. Biesold, Y. Cheng, M. Ge, H. Li, J. Huang, Z. Lin and Y. Lai, Chem. Soc. Rev., 2023, 52, 473–509 RSC.
- C. Keplinger, J.-Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed.
- J. Wu, Q. e Zhang, A. a Zhou, Z. Huang, H. Bai and L. Li, Adv. Mater., 2016, 28, 10211–10216 CrossRef CAS PubMed.
- P. Li, Z. Jin, L. Peng, F. Zhao, D. Xiao, Y. Jin and G. Yu, Adv. Mater., 2018, 30, 1800124 CrossRef PubMed.
- K. S. Kumar, L. Zhang, M. S. Kalairaj, H. Banerjee, X. Xiao, C. C. Jiayi, H. Huang, C. M. Lim, J. Ouyang and H. Ren, ACS Appl. Mater. Interfaces, 2021, 13, 37816–37829 CrossRef CAS PubMed.
- S. Noh, H. Y. Gong, H. J. Lee and W.-G. Koh, Materials, 2021, 14, 308 CrossRef CAS PubMed.
- Y. Ohm, C. Pan, M. J. Ford, X. Huang, J. Liao and C. Majidi, Nat. Electron., 2021, 4, 185–192 CrossRef CAS.
- Y. Xu, R. Rothe, D. Voigt, S. Hauser, M. Cui, T. Miyagawa, M. Patino Gaillez, T. Kurth, M. Bornhäuser and J. Pietzsch, Nat. Commun., 2021, 12, 2407 CrossRef CAS PubMed.
- L. Ye, H. Ji, J. Liu, C. H. Tu, M. Kappl, K. Koynov, J. Vogt and H. J. Butt, Adv. Mater., 2021, 33, 2102981 CrossRef CAS PubMed.
- J. Chong, C. Sung, K. S. Nam, T. Kang, H. Kim, H. Lee, H. Park, S. Park and J. Kang, Nat. Commun., 2023, 14, 2206 CrossRef CAS PubMed.
- M. Sun, P. Li, H. Qin, N. Liu, H. Ma, Z. Zhang, J. Li, B. Lu, X. Pan and L. Wu, Chem. Eng. J., 2023, 454, 140459 CrossRef CAS.
- Y. Zhao, Y. Ohm, J. Liao, Y. Luo, H.-Y. Cheng, P. Won, P. Roberts, M. R. Carneiro, M. F. Islam, J. H. Ahn, L. M. Walker and C. Majidi, Nat. Electron., 2023, 6, 206–215 CrossRef CAS.
- H. Chen, L. Yuan, W. Song, Z. Wu and D. Li, Prog. Polym. Sci., 2008, 33, 1059–1087 CrossRef CAS.
- S. Hanson, P. A. Lalor, S. M. Niemi, S. J. Northup, B. D. Ratner, M. Spector, B. H. Vale and J. E. Willson, Biomaterials Science, Elsevier, 1996, pp. 215–242 Search PubMed.
- X. Navarro, T. B. Krueger, N. Lago, S. Micera, T. Stieglitz and P. Dario, J. Peripher. Nerv. Syst., 2005, 10, 229–258 CrossRef PubMed.
- S. Frederick, Wiley Encycl. Chem. Biol., 2007, 1–8 Search PubMed.
- G. Márton, E. Z. Tóth, L. Wittner, R. Fiáth, D. Pinke, G. Orbán, D. Meszéna, I. Pál, E. L. Győri and Z. Bereczki, Mater. Sci. Eng., C, 2020, 112, 110870 CrossRef PubMed.
- S. P. Lacour, S. Benmerah, E. Tarte, J. FitzGerald, J. Serra, S. McMahon, J. Fawcett, O. Graudejus, Z. Yu and B. Morrison, Med. Biol. Eng. Comput., 2010, 48, 945–954 CrossRef PubMed.
- A. Lecomte, E. Descamps and C. Bergaud, J. Neural Eng., 2018, 15, 031001 CrossRef PubMed.
- H. Li, H. Zhao, K. Song, F. Han, Z. Liu and Q. Tian, Nanoscale, 2024, 16, 6402–6428 RSC.
- S. F. Cogan, P. R. Troyk, J. Ehrlich and T. D. Plante, IEEE Trans. Biomed. Eng., 2005, 52, 1612–1614 CrossRef PubMed.
- R. Chen, A. Canales and P. Anikeeva, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- J. W. Seo, K. Kim, K. W. Seo, M. K. Kim, S. Jeong, H. Kim, J. W. Ghim, J. H. Lee, N. Choi and J. Y. Lee, Adv. Funct. Mater., 2020, 30, 2000896 CrossRef CAS.
- T. Stuart, W. J. Jeang, R. A. Slivicki, B. J. Brown, A. Burton, V. E. Brings, L. C. Alarcón-Segovia, P. Agyare, S. Ruiz and A. Tyree, ACS Nano, 2022, 17, 561–574 CrossRef PubMed.
- N. T. Garland, J. W. Song, T. Ma, Y. J. Kim, A. Vázquez-Guardado, A. B. Hashkavayi, S. K. Ganeshan, N. Sharma, H. Ryu and M. K. Lee, Adv. Healthcare Mater., 2023, 12, 2301280 CrossRef CAS PubMed.
- Z. Jiang, N. Chen, Z. Yi, J. Zhong, F. Zhang, S. Ji, R. Liao, Y. Wang, H. Li and Z. Liu, Nat. Electron., 2022, 5, 784–793 CrossRef CAS.
- C. Zhang, L. McKeon, M. P. Kremer, S.-H. Park, O. Ronan, A. Seral-Ascaso, S. Barwich, C. O. Coileáin, N. McEvoy and H. C. Nerl, Nat. Commun., 2019, 10, 1795 CrossRef PubMed.
- G. Wu, X. Wu, Y. Xu, H. Cheng, J. Meng, Q. Yu, X. Shi, K. Zhang, W. Chen and S. Chen, Adv. Mater., 2019, 31, 1806492 CrossRef PubMed.
- Z. Wang, S. Ji, F. Liu, H. Wang, X. Wang, Q. Wang, B. G. Pollet and R. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 29791–29798 CrossRef CAS PubMed.
- Z. H. Guo, M. Liu, Z. Cong, W. Guo, P. Zhang, W. Hu and X. Pu, Adv. Mater. Technol., 2020, 5, 2000544 CrossRef CAS.
- Z. Kang, Y. He, J. Sang, H. Hirahara and D. Chen, Adv. Mater. Interfaces, 2021, 8, 2100651 CrossRef CAS.
- X. Lu, W. Shang, G. Chen, H. Wang, P. Tan, X. Deng, H. Song, Z. Xu, J. Huang and X. Zhou, ACS Appl. Electron. Mater., 2021, 3, 1477–1488 CrossRef CAS.
- W. Chang, D. Nam, S. Lee, Y. Ko, C. H. Kwon, Y. Ko and J. Cho, Adv. Sci., 2022, 9, 2203800 CrossRef CAS PubMed.
- I.-W. P. Chen, M.-C. Yang, C.-H. Yang, D.-X. Zhong, M.-C. Hsu and Y. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 5550–5555 CrossRef CAS PubMed.
- H. S. Wang, J. Cho, D. S. Song, J. H. Jang, J. Y. Jho and J. H. Park, ACS Appl. Mater. Interfaces, 2017, 9, 21998–22005 CrossRef CAS PubMed.
- M. Kotal, J. Kim, R. Tabassian, S. Roy, V. H. Nguyen, N. Koratkar and I. K. Oh, Adv. Funct. Mater., 2018, 28, 1802464 CrossRef.
- S. Umrao, R. Tabassian, J. Kim, V. H. Nguyen, Q. Zhou, S. Nam and I.-K. Oh, Sci. Robot., 2019, 4, eaaw7797 CrossRef PubMed.
- S. Ma, Y. Zhang, Y. Liang, L. Ren, W. Tian and L. Ren, Adv. Funct. Mater., 2020, 30, 1908508 CrossRef CAS.
- H. Zhang, Z. Lin, Y. Hu, S. Ma, Y. Liang, L. Ren and L. Ren, Adv. Sci., 2023, 10, 2206135 CrossRef CAS PubMed.
- G. Decher and J. D. Hong, Makromol. Chem., Macromol. Symp., 1991, 321–327 CrossRef CAS.
- G. Decher, J. D. Hong and J. Schmitt, Thin Solid Films, 1992, 210, 831–835 CrossRef.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS PubMed.
- J. Cho, K. Char, J. D. Hong and K. B. Lee, Adv. Mater., 2001, 13, 1076–1078 CrossRef CAS.
- J. A. Hiller, J. D. Mendelsohn and M. F. Rubner, Nat. Mater., 2002, 1, 59–63 CrossRef CAS PubMed.
- J.-S. Lee, J. Cho, C. Lee, I. Kim, J. Park, Y.-M. Kim, H. Shin, J. Lee and F. Caruso, Nat. Nanotechnol., 2007, 2, 790–795 CrossRef CAS PubMed.
- J. F. Quinn, A. P. R. Johnston, G. K. Such, A. N. Zelikin and F. Caruso, Chem. Soc. Rev., 2007, 36, 707–718 RSC.
- G. K. Such, A. P. R. Johnston and F. Caruso, Chem. Soc. Rev., 2010, 40, 19–29 RSC.
- Y. Ko, Y. Kim, H. Baek and J. Cho, ACS Nano, 2011, 5, 9918–9926 CrossRef CAS PubMed.
- J. J. Richardson, J. Cui, M. Björnmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed.
- Y. Ko, M. Kwon, W. K. Bae, B. Lee, S. W. Lee and J. Cho, Nat. Commun., 2017, 8, 1–11 CrossRef CAS PubMed.
- C. H. Kwon, Y. Ko, D. Shin, M. Kwon, J. Park, W. K. Bae, S. W. Lee and J. Cho, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- Y. Song, D. Kim, S. Kang, Y. Ko, J. Ko, J. Huh, Y. Ko, S. W. Lee and J. Cho, Adv. Funct. Mater., 2019, 29, 1806584 CrossRef.
- J. Ko, D. Kim, Y. Song, S. Lee, M. Kwon, S. Han, D. Kang, Y. Kim, J. Huh, J.-S. Koh and J. Cho, ACS Nano, 2020, 14, 11906–11918 CrossRef CAS PubMed.
- J. Ko, C. Kim, D. Kim, Y. Song, S. Lee, B. Yeom, J. Huh, S. Han, D. Kang and J.-S. Koh, Sci. Robot., 2022, 7, eabo6463 CrossRef PubMed.
- L. Manjakkal, A. Pullanchiyodan, N. Yogeswaran, E. S. Hosseini and R. Dahiya, Adv. Mater., 2020, 32, 1907254 CrossRef CAS PubMed.
- Z. Li, T. Huang, W. Gao, Z. Xu, D. Chang, C. Zhang and C. Gao, ACS Nano, 2017, 11, 11056–11065 CrossRef CAS PubMed.
- M. Liu, Z. Cong, X. Pu, W. Guo, T. Liu, M. Li, Y. Zhang, W. Hu and Z. L. Wang, Adv. Funct. Mater., 2019, 29, 1806298 CrossRef CAS.
- L. Li, W. Tian, A. VahidMohammadi, J. Rostami, B. Chen, K. Matthews, F. Ram, T. Pettersson, L. Wågberg and T. Benselfelt, Adv. Mater., 2023, 35, e2301163 CrossRef PubMed.
- K. Zhou, Y. He, Q. Xu, Q. E. Zhang, A. A. Zhou, Z. Lu, L.-K. Yang, Y. Jiang, D. Ge and X. Y. Liu, ACS Nano, 2018, 12, 5888–5894 CrossRef CAS PubMed.
- H. Sheng, X. Zhang, J. Liang, M. Shao, E. Xie, C. Yu and W. Lan, Adv. Healthcare Mater., 2021, 10, 2100199 CrossRef CAS PubMed.
- G. Li, Z. Zhao, S. Zhang, L. Sun, M. Li, J. A. Yuwono, J. Mao, J. Hao, J. Vongsvivut and L. Xing, Nat. Commun., 2023, 14, 6526 CrossRef CAS PubMed.
- L. Li, D. Li, Y. Wang, T. Ye, E. He, Y. Jiao, L. Wang, F. Li, Y. Li and J. Ding, Adv. Mater., 2023, 35, 2302997 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- A. Noori, M. F. El-Kady, M. S. Rahmanifar, R. B. Kaner and M. F. Mousavi, Chem. Soc. Rev., 2019, 48, 1272–1341 RSC.
- N. T. Garland, R. Kaveti and A. J. Bandodkar, Adv. Mater., 2023, 35, 2303197 CrossRef CAS PubMed.
- L. Liu, Y. Yu, C. Yan, K. Li and Z. Zheng, Nat. Commun., 2015, 6, 7260 CrossRef CAS PubMed.
- X. Pu, L. Li, M. Liu, C. Jiang, C. Du, Z. Zhao, W. Hu and Z. L. Wang, Adv. Mater., 2016, 28, 98–105 CrossRef CAS PubMed.
- Y. Yang, Q. Huang, L. Niu, D. Wang, C. Yan, Y. She and Z. Zheng, Adv. Mater., 2017, 29, 1606679 CrossRef PubMed.
- J. Lv, P. Zhou, L. Zhang, Y. Zhong, X. Sui, B. Wang, Z. Chen, H. Xu and Z. Mao, Chem. Eng. J., 2019, 361, 897–907 CrossRef CAS.
- H. Park, J. W. Kim, S. Y. Hong, G. Lee, H. Lee, C. Song, K. Keum, Y. R. Jeong, S. W. Jin and D. S. Kim, ACS Nano, 2019, 13, 10469–10480 CrossRef CAS PubMed.
- A. Pullanchiyodan, L. Manjakkal, S. Dervin, D. Shakthivel and R. Dahiya, Adv. Mater. Technol., 2020, 5, 1901107 CrossRef CAS.
- X. Li, L. Yuan, R. Liu, H. He, J. Hao, Y. Lu, Y. Wang, G. Liang, G. Yuan and Z. Guo, Adv. Energy Mater., 2021, 11, 2003010 CrossRef CAS.
- S. Woo, D. Nam, W. Chang, Y. Ko, S. Lee, Y. Song, B. Yeom, J. H. Moon, S. W. Lee and J. Cho, Small, 2021, 17, 2007579 CrossRef CAS PubMed.
- S. Selvam and J.-H. Yim, J. Energy Storage, 2023, 58, 106340 CrossRef.
- J. Ahn, W. Chang, Y. Song, Y. Son, Y. Ko and J. Cho, Energy Storage Mater., 2024, 69, 103396 CrossRef.
- Z. Chen, J. W. To, C. Wang, Z. Lu, N. Liu, A. Chortos, L. Pan, F. Wei, Y. Cui and Z. Bao, Adv. Energy Mater., 2014, 4, 1400207 CrossRef.
- W. Li, F. Gao, X. Wang, N. Zhang and M. Ma, Angew. Chem., 2016, 128, 9342–9347 CrossRef.
- L. Mao, C. Guan, X. Huang, Q. Ke, Y. Zhang and J. Wang, Electrochim. Acta, 2016, 196, 653–660 CrossRef CAS.
- J. Han, H. Wang, Y. Yue, C. Mei, J. Chen, C. Huang, Q. Wu and X. Xu, Carbon, 2019, 149, 1–18 CrossRef CAS.
- L. Li, Y. Zhang, H. Lu, Y. Wang, J. Xu, J. Zhu, C. Zhang and T. Liu, Nat. Commun., 2020, 11, 62 CrossRef CAS PubMed.
- M. Hua, S. Wu, Y. Jin, Y. Zhao, B. Yao and X. He, Adv. Mater., 2021, 33, 2100983 CrossRef CAS PubMed.
- H. Huang, F. Yan, G. Wei, H. Shen, L. Yan, S. Xu, X. Liang, W. Zhou and J. Guo, J. Energy Storage, 2021, 44, 103254 CrossRef.
- Y. Liu, H. Zhou, W. Zhou, S. Meng, C. Qi, Z. Liu and T. Kong, Adv. Energy Mater., 2021, 11, 2101329 CrossRef CAS.
- H. Cai, D. Zhang, H. Zhang, M. Tang, Z. Xu, H. Xia, K. Li and J. Wang, Chem. Eng. J., 2023, 472, 144849 CrossRef CAS.
- Q. Lv, X. Li, X. Tian, D. A. Fu, H. Liu, J. Liu, Y. Song, B. Cai, J. Wang and Q. Su, Adv. Energy Mater., 2023, 13, 2203814 CrossRef CAS.
- R. Elashnikov, O. Khrystonko, T. Jilková, S. Rimpelová, J. Prchal, I. Khalakhan, Z. Kolská, V. Švorčík and O. Lyutakov, Adv. Funct. Mater., 2024, 2314420 CrossRef CAS.
- M. Xu, J. Zhu, J. Xie, Y. Mao and W. Hu, Small, 2024, 20, 2305448 CrossRef CAS PubMed.
- S. J. Updike and G. P. Hicks, Nature, 1967, 214, 986–988 CrossRef CAS PubMed.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2002, 124, 12962–12963 CrossRef CAS PubMed.
- X. Xiao, H.-q Xia, R. Wu, L. Bai, L. Yan, E. Magner, S. Cosnier, E. Lojou, Z. Zhu and A. Liu, Chem. Rev., 2019, 119, 9509–9558 CrossRef CAS PubMed.
- I. Jeerapan, J. R. Sempionatto and J. Wang, Adv. Funct. Mater., 2020, 30, 1906243 CrossRef CAS.
- A. Ruff, F. Conzuelo and W. Schuhmann, Nat. Catal., 2020, 3, 214–224 CrossRef CAS.
- T. Chen, S. C. Barton, G. Binyamin, Z. Gao, Y. Zhang, H.-H. Kim and A. Heller, J. Am. Chem. Soc., 2001, 123, 8630–8631 CrossRef CAS PubMed.
- F. Gao, L. Viry, M. Maugey, P. Poulin and N. Mano, Nat. Commun., 2010, 1, 2 CrossRef PubMed.
- C. H. Kwon, S.-H. Lee, Y.-B. Choi, J. A. Lee, S. H. Kim, H.-H. Kim, G. M. Spinks, G. G. Wallace, M. D. Lima and M. E. Kozlov, Nat. Commun., 2014, 5, 3928 CrossRef CAS PubMed.
- C. H. Kwon, M. Kang, M. Kwon, D. Nam, Y. Song, E. Yong, M.-K. Oh, Y. Kim, B. Yeom and J. H. Moon, Appl. Phys. Rev., 2022, 9, 021413 CAS.
- M. Kang, D. Nam, J. Ahn, Y. J. Chung, S. W. Lee, Y. B. Choi, C. H. Kwon and J. Cho, Adv. Mater., 2023, 35, 2304986 CrossRef CAS PubMed.
- C. H. Kwon, Y. Ko, D. Shin, S. W. Lee and J. Cho, J. Mater. Chem. A, 2019, 7, 13495–13505 RSC.
- J. Byun, Y. Lee, J. Yoon, B. Lee, E. Oh, S. Chung, T. Lee, K.-J. Cho, J. Kim and Y. Hong, Sci. Robot., 2018, 3, eaas9020 CrossRef PubMed.
- P. Rothemund, N. Kellaris, S. K. Mitchell, E. Acome and C. Keplinger, Adv. Mater., 2021, 33, 2003375 CrossRef CAS PubMed.
- Q. Deng, H. Jia, C. An, S. Wu, S. Zhao and N. Hu, Composites, Part A, 2023, 165, 107336 CrossRef CAS.
- T. Li, G. Li, Y. Liang, T. Cheng, J. Dai, X. Yang, B. Liu, Z. Zeng, Z. Huang and Y. Luo, Sci. Adv., 2017, 3, e1602045 CrossRef PubMed.
- E. Acome, S. K. Mitchell, T. Morrissey, M. Emmett, C. Benjamin, M. King, M. Radakovitz and C. Keplinger, Science, 2018, 359, 61–65 CrossRef CAS PubMed.
- N. Kellaris, V. Gopaluni Venkata, G. M. Smith, S. K. Mitchell and C. Keplinger, Sci. Robot., 2018, 3, eaar3276 CrossRef PubMed.
- Y. Lee, W. J. Song, Y. Jung, H. Yoo, M.-Y. Kim, H.-Y. Kim and J.-Y. Sun, Sci. Robot., 2020, 5, eaaz5405 CrossRef PubMed.
- C. W. Zhang, W. Zou, H. C. Yu, X. P. Hao, G. Li, T. Li, W. Yang, Z. L. Wu and Q. Zheng, ACS Appl. Mater. Interfaces, 2022, 14, 52430–52439 CrossRef CAS PubMed.
- C. Lu, Y. Yang, J. Wang, R. Fu, X. Zhao, L. Zhao, Y. Ming, Y. Hu, H. Lin and X. Tao, Nat. Commun., 2018, 9, 752 CrossRef PubMed.
- Y. Yan, T. Santaniello, L. G. Bettini, C. Minnai, A. Bellacicca, R. Porotti, I. Denti, G. Faraone, M. Merlini and C. Lenardi, Adv. Mater., 2017, 29, 1606109 CrossRef PubMed.
- H. Na, Y.-W. Kang, C. S. Park, S. Jung, H.-Y. Kim and J.-Y. Sun, Science, 2022, 376, 301–307 CrossRef CAS PubMed.
- T. Benselfelt, J. Shakya, P. Rothemund, S. B. Lindström, A. Piper, T. E. Winkler, A. Hajian, L. Wågberg, C. Keplinger and M. M. Hamedi, Adv. Mater., 2023, 35, 2303255 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |