Elucidating the effects of the sidechain substitution direction on the optoelectronic properties of isomeric diketopyrrolopyrrole-based conjugated polymers for near-infrared organic phototransistors†
Received
13th October 2023
, Accepted 30th November 2023
First published on 1st December 2023
Abstract
Sidechain engineering is a powerful molecular design strategy to tune the solid-state packing and structural ordering of conjugated polymers. While the modification of the type and length of sidechains has become a key strategy in tuning the polymer properties, the effects of the sidechain direction on the optoelectronic properties of polymers have been less investigated. Herein, two new isomeric diketopyrrolopyrrole (DPP)-based conjugated polymers (namely P3BT-in and P3BT-out) with different sidechain substitution directions are designed and synthesized based on the O-alkylated DPP isomer (pyrrolo[3,4-c]pyrrol-1(2H)-one). Through comprehensive theoretical studies and experimental characterization, the effects of the sidechain-substitution direction on the optoelectronic properties of polymers and the corresponding performance of organic transistors and near-infrared organic phototransistors (NIR-OPTs) were systematically investigated. Compared to P3BT-out with sidechains spreading in the outer way, P3BT-in with sidechains in the inner way has better molecular planarity, resulting in increased crystallinity. In addition, P3BT-in has deeper LUMO energy levels and a narrower bandgap of 1.06 eV with a stronger light absorption ability, leading to increased electron mobility and n-type NIR photosensitivity. Overall, tuning the sidechain substitution direction could be a useful strategy for the rational design of functional polymers.
Introduction
Over the years, state-of-the-art donor–acceptor (D–A) conjugated polymers have attracted extensive attention in organic solar cells, organic thin-film transistors (OTFTs), organic thermoelectrics, and organic electrochemical transistors.1–4 Among them, exciting progress has been achieved with OTFTs, which can be implemented into artificial skins, electronic papers, and photonic devices.5,6 Meanwhile, D–A polymers are able to absorb near-infrared (NIR) light owing to their narrow bandgaps. Thus, they can be applied in NIR phototransistors (OPTs), which can convert NIR light into electrical signals to realize the detection of NIR light.2,7–9 Although D–A copolymers have been intensively investigated in photodetector applications,10–14 there are only a handful of instances that can achieve high NIR-OPT performances.
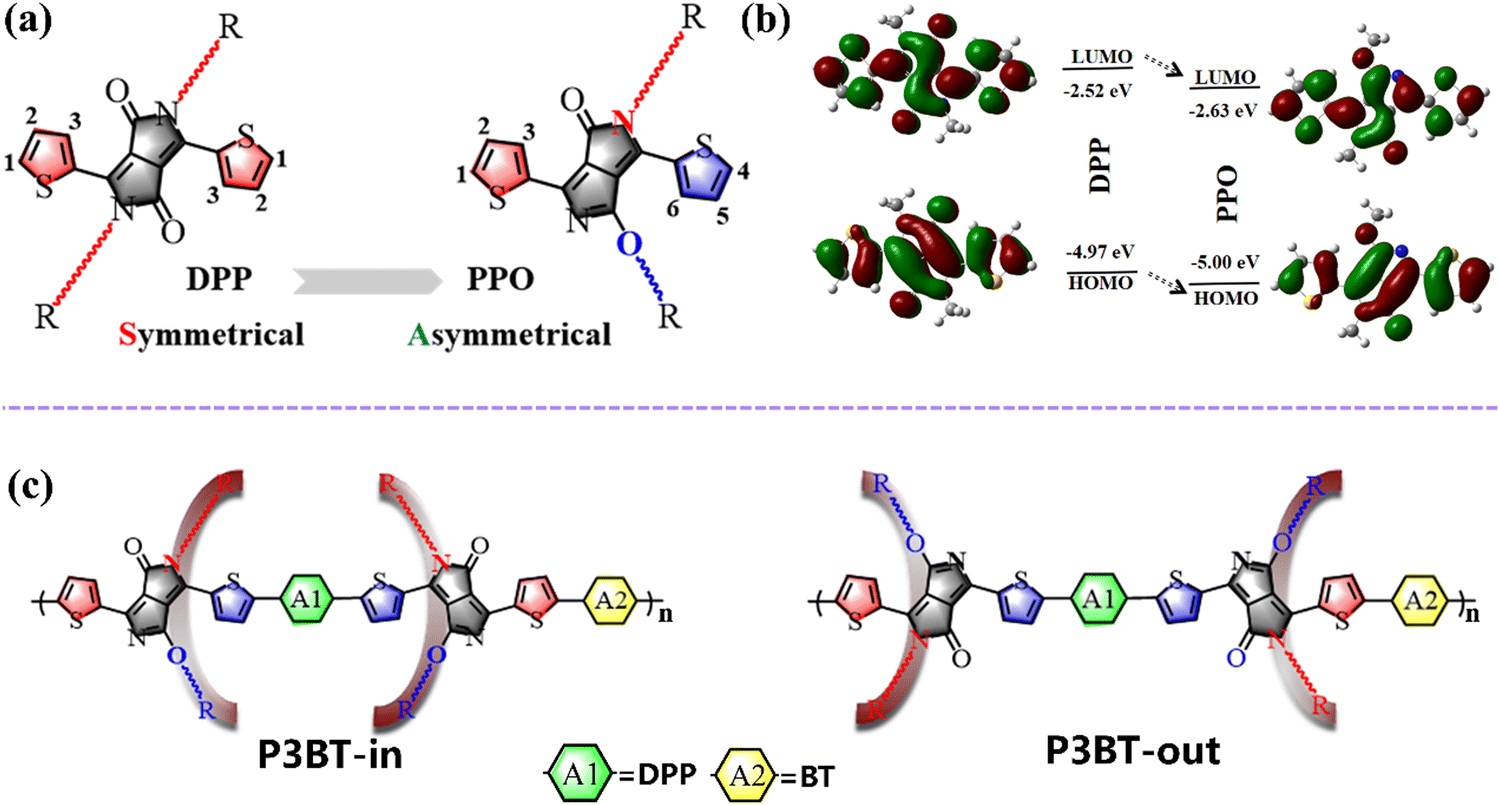
To develop D–A polymers with strong NIR-absorbing ability, various acceptor units, such as diketopyrrolopyrrole (DPP), isoindigo (IID), and naphthalene diimide (NDI), have been used. Among them, DPP has been widely investigated because of its coplanar structure and strong intermolecular interaction.15–17 The basic DPP structural unit consists of three parts: a central bis-lactam core, two flanking aromatic units (usually thiophene), and two side chains at the amide N-position (Fig. 1a left). Over the past decades, the modification of DPP-based units has mainly focused on adjusting the flanking aromatic structures and/or changing the length and type of sidechains.18,19 Alkylation usually occurs on the nitrogen atoms of the DPP unit. However, Frebort et al. reported that the alkyl chains can also be substituted on the oxygen atom under some conditions, leading to an O-alkylated DPP isomer, namely pyrrolo[3,4-c]pyrrol-1(2H)-one (PPO), as shown in Fig. 1a.20 Compared to the DPP unit, the PPO-based molecules have the following advantages. First, the absence of electron donation from the lone pair of electrons on the nitrogen to the π-orbit of the diene moiety may contribute to the lowering of highest occupied molecular orbital (HOMO) or lowest unoccupied molecular orbital (LUMO) energy levels in PPO. Density functional theory (DFT) calculations showed that the HOMO/LUMO levels gradually decreased from DPP to PPO (Fig. 1b). The calculated energy gap of the PPO is 2.37 eV, which is 0.08 eV narrower than that of the DPP. The calculation results indicate that the PPO-based polymer is more electron-deficient and may have a narrower bandgap. Second, two alkyl chains in the PPO are arranged asymmetrically, while they are symmetric in the DPP unit (Fig. 1a). This means that asymmetric N,O-alkylated PPO will provide more possibilities to tune the packing direction of the alkyl chain using the acceptor dimerization (trimerization) process that we proposed previously to reduce polymers’ bandgap and increase the electron mobility.21–25 Currently, there are few reports focusing on the substitution directions of the side groups.26–29 Tuning the sidechain substitution direction of the polymers may lead to intriguing properties of the conjugated polymers with useful structure–property relationships.
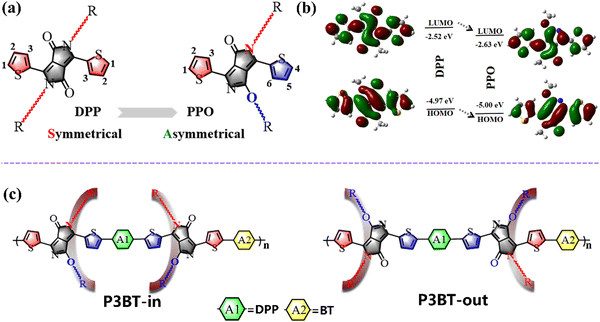 |
| | Fig. 1 (a) Molecular structure of DPP and PPO monomers; (b) the electrostatic surface potential of DPP and PPO units; (c) design strategies and chemical structures of the two isomeric polymers with different N,O-alkylated chain packing directions. | |
In this work, we first designed two isomeric polymers based on PPO (namely P3BT-in and P3BT-out) using an acceptor trimerization strategy (Fig. 1c). Due to PPO's asymmetric substitution direction of sidechains, P3BT-in with the alkyl chains spreading in the inner way and P3BT-out with the alkyl chains spreading in the outer way were designed (Fig. 1c). Specifically, the central DPP unit (A1) in the isomeric polymer P3BT-in is surrounded by two C-type N,O-alkyl chains while the C-type N,O-alkyl chains were facing away from the A1 in P3BT-out. In order to further enhance the electron transport, we employed another electron-deficient unit benzothiadiazole (BT) as the comonomer (A2). Thus, two isomeric polymers, namely P3BT-in and P3BT-out, were synthesized by using the acceptor–acceptor type C–H direct arylation polymerization (A–A DArP) protocol that we developed previously.23 Through characterization using UV-vis-NIR spectrometry, cyclic voltammetry, and DFT calculations, these two isomeric polymers have distinct optoelectronic properties. P3BT-in has a deeper LUMO energy level and broader absorption with the optical bandgap as low as 1.06 eV. Both polymers exhibit ambipolar charge transport behavior in bottom-gate/top-contact OTFTs. Due to better molecular planarity and increased crystallinity, P3BT-in demonstrates higher electron mobilities. With regard to broad absorption extending to the near-infrared region, these two polymers were further applied in NIR-OPTs. Both polymers showed ambipolar NIR-OPT behavior. The P3BT-in-based NIR-OPTs showed the highest n-type photosensitivity P value of 1.70 under the NIR intensity of 4.6 mW cm−2, which is much higher than that of P3BT-out. The P3BT-in also demonstrates a high photoresponsivity of 12.31 A W−1 under the NIR intensity of 0.6 mW cm−2, which is one order of magnitude higher than that of one DPP-acepleiadylene derivative reported recently (1.30 A W−1).2 Overall, this work not only presents two novel isomeric DPP-based conjugated polymers with interesting optoelectronic properties but also offers highly valuable structure–property correlations for rational design of functional polymer semiconductors.
Results and discussion
Materials synthesis and characterization
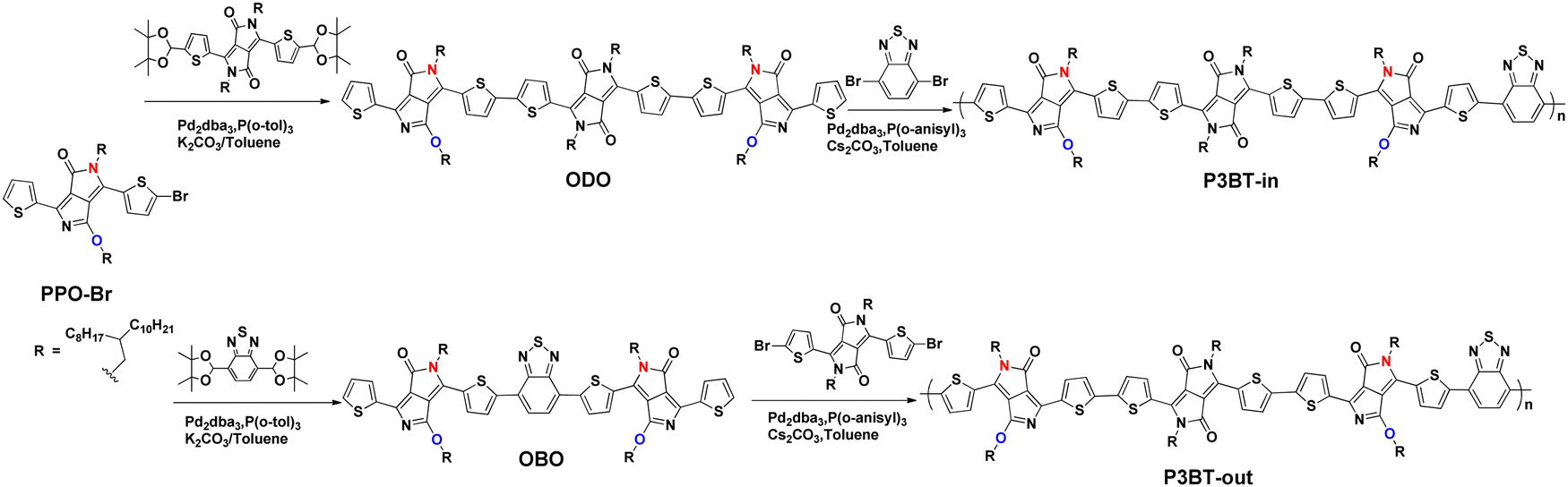
The synthetic routes to the monomers (ODO and OBO) and the isomeric copolymers (P3BT-in and P3BT-out) are outlined in Scheme 1, and the details are described in the ESI.† The initial PPO-Br was first prepared by a monobromine substitution protocol according to the literature.30 Note that this protocol only produces one asymmetric monobromine product PPO-Br, with a high yield of over 80%. We speculate that this is because the electron cloud densities are significantly different on the 1 and 4 position of the thiophene units of PPO (Fig. 1b). Due to the electron-withdrawing properties of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN, the 4-position of thiophene is more electron-deficient. As a result, the electrophilic bromide-substitution reaction is more difficult to occur. Then, ODO and OBO monomers were obtained by Suzuki coupling reactions of PPO-Br with DPP or BT-based diboronic acid pinacol ester derivative (Scheme 1; synthetic details are provided in ESI†). The structures of ODO and OBO were unambiguously verified by 1H NMR, 13C NMR, and matrix-assisted laser desorption/ionization coupled to time-of-flight (MALDI-TOF) mass spectra (Fig. S1–S6, ESI†). Finally, these monomers were subjected to the DArP, yielding the isomeric copolymers P3BT-in and P3BT-out (Scheme 1; synthetic details in ESI†). In fact, we also tried to synthesize these polymers by Stille or Suzuki polymerization. However, the bromination of ODO and OBO under neutral conditions failed. The reason for the failure is the same as that of PPO-Br (see above). To avoid the tedious bromination process, we utilized a more efficient DArP protocol to synthesize the isomeric polymers. The combination of the Pd2dba3 catalyst, tris(o-methoxyphenyl) phosphine, Cs2CO3, and PivOH provides the optimal DArP condition. The crude polymers were purified by Soxhlet extraction with methanol for 12 h, acetone for 12 h, and hexane for 12 h to remove the low-molecular-weight fractions. Finally, the chloroform-extracted fractions were reprecipitated into chilled methanol to yield the target polymers. The resulting polymers have sufficient solubility in the common organic solvents, such as chloroform, chlorobenzene, and dichlorobenzene. High-temperature gel permeation chromatography (HT-GPC) (using polystyrene as the standard samples and 1,2,4-trichlorobenzene as the eluent at 150 °C) was carried out to show the number-average molecular weight (Mn) of the polymers as follows: P3BT-in (Mn = 27.1 kg mol−1, PDI = 2.1), P3BT-out (Mn = 20.8 kg mol−1, PDI = 2.3), (Table 1 and Fig. S7, ESI†). A thermogravimetric analysis (TGA) was carried out under flowing nitrogen at the heating rate of 10 °C min−1 to understand the thermal stability of the polymers. All polymers demonstrated excellent thermal stability with decomposition temperatures (the temperature of 5% weight loss, Td) values exceeding 300 °C (Fig. S8, ESI†), indicating good thermal stability.
O and CN, the 4-position of thiophene is more electron-deficient. As a result, the electrophilic bromide-substitution reaction is more difficult to occur. Then, ODO and OBO monomers were obtained by Suzuki coupling reactions of PPO-Br with DPP or BT-based diboronic acid pinacol ester derivative (Scheme 1; synthetic details are provided in ESI†). The structures of ODO and OBO were unambiguously verified by 1H NMR, 13C NMR, and matrix-assisted laser desorption/ionization coupled to time-of-flight (MALDI-TOF) mass spectra (Fig. S1–S6, ESI†). Finally, these monomers were subjected to the DArP, yielding the isomeric copolymers P3BT-in and P3BT-out (Scheme 1; synthetic details in ESI†). In fact, we also tried to synthesize these polymers by Stille or Suzuki polymerization. However, the bromination of ODO and OBO under neutral conditions failed. The reason for the failure is the same as that of PPO-Br (see above). To avoid the tedious bromination process, we utilized a more efficient DArP protocol to synthesize the isomeric polymers. The combination of the Pd2dba3 catalyst, tris(o-methoxyphenyl) phosphine, Cs2CO3, and PivOH provides the optimal DArP condition. The crude polymers were purified by Soxhlet extraction with methanol for 12 h, acetone for 12 h, and hexane for 12 h to remove the low-molecular-weight fractions. Finally, the chloroform-extracted fractions were reprecipitated into chilled methanol to yield the target polymers. The resulting polymers have sufficient solubility in the common organic solvents, such as chloroform, chlorobenzene, and dichlorobenzene. High-temperature gel permeation chromatography (HT-GPC) (using polystyrene as the standard samples and 1,2,4-trichlorobenzene as the eluent at 150 °C) was carried out to show the number-average molecular weight (Mn) of the polymers as follows: P3BT-in (Mn = 27.1 kg mol−1, PDI = 2.1), P3BT-out (Mn = 20.8 kg mol−1, PDI = 2.3), (Table 1 and Fig. S7, ESI†). A thermogravimetric analysis (TGA) was carried out under flowing nitrogen at the heating rate of 10 °C min−1 to understand the thermal stability of the polymers. All polymers demonstrated excellent thermal stability with decomposition temperatures (the temperature of 5% weight loss, Td) values exceeding 300 °C (Fig. S8, ESI†), indicating good thermal stability.
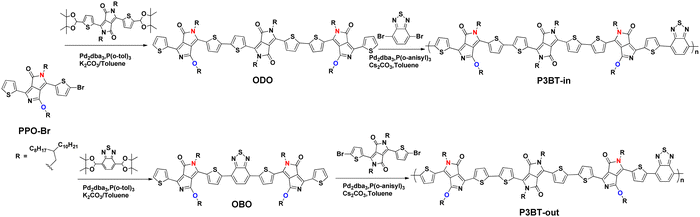 |
| | Scheme 1 Synthetic route of monomers and isomeric polymers. | |
Table 1 Summary of molecular weights, thermal decomposition temperatures, and optical and electrochemical properties of the polymers
| Polymer |
M
n
[kDa] |
PDI |
T
d
[°C] |
λ
maxsol
[nm] |
λ
maxfilm
[nm] |
λ
onset [nm] |
E
optg [eV] |
E
LUMO
[eV] |
E
HOMO
[eV] |
|
Measured from HT-GPC using 1,2,4-trichlorobenzene (TCB) as eluent at 150 °C.
Temperature at 5% weight loss.
Absorption maximum in the dilute chloroform solution.
Absorption maximum in the pristine film cast from chloroform solution.
Estimated from cyclic voltammetry measurements.
|
| P3BT-in |
27.1 |
2.1 |
302 |
903 |
904 |
1175 |
1.06 |
−3.64 |
−5.45 |
| P3BT-out |
20.8 |
2.3 |
311 |
838, 913 |
858 |
1102 |
1.13 |
−3.61 |
−5.54 |
Optoelectronic properties
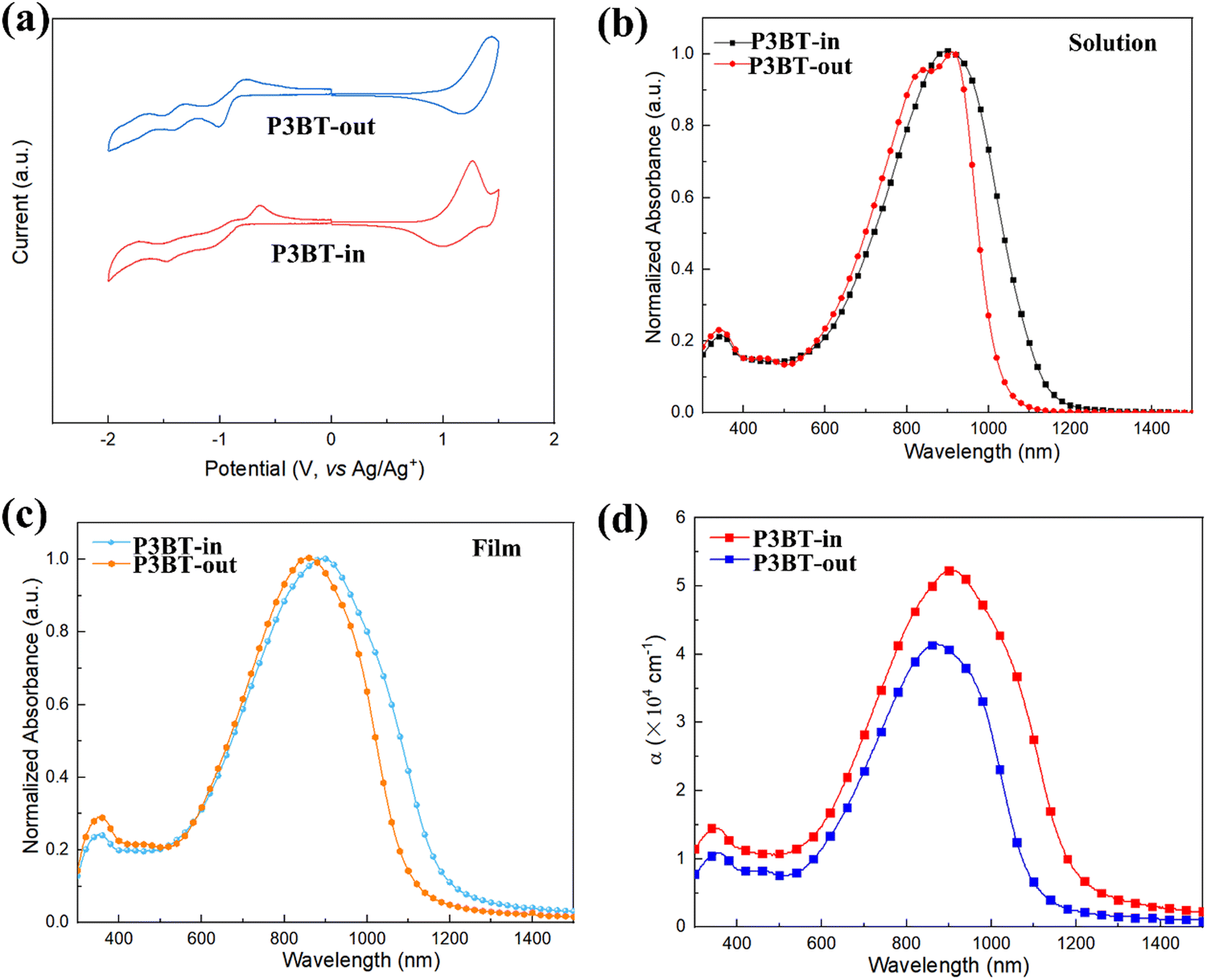
The cyclic voltammetry (CV) measurements were conducted to estimate the experimental frontier molecular orbitals (FMO) energies. As shown in Fig. 2a, both polymers exhibited pronounced oxidation and reduction processes. The HOMO/LUMO energy levels (EHOMO's and ELUMO's) calculated from the onset potentials are summarized in Table 1. The ELUMO of P3BT-in (ELUMO = −3.64 eV) was deeper than that of P3BT-out by 0.03 eV. On the other hand, the EHOMO of P3BT-in (EHOMO = −5.45 eV) was 0.09 eV higher than that of P3BT-out (EHOMO = −5.54 eV). This difference leads to the narrower band gap of P3BT-in, which is consistent with the subsequent UV-vis-NIR absorption spectrum.
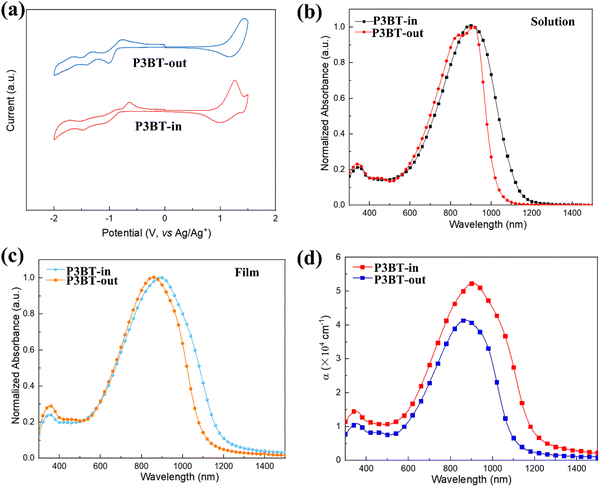 |
| | Fig. 2 (a) Cyclic voltammetry (CV) of polymer films; (b) and (c) UV-vis-NIR absorption spectra of P3BT-in and P3BT-out (b) in solution and (c) as films; (d) absorption coefficients (α) of the polymer thin films. | |
UV-vis-NIR absorption spectra of these isomeric polymers in CHCl3 and thin films are shown in Fig. 2b and c, with the corresponding data summarized in Table 1. Two main absorption bands for the polymers can be observed in the solution, where the band at 300–400 nm is assigned to the π–π* transition of the conjugated backbone, and the lower-energy band (600–1200 nm) is tentatively attributed to the intramolecular charge transfer. It is worth pointing out that the absorption profile of the N,N-alkylated DPP monomer showed two absorption peaks while there was only one single peak in the absorption profile of N,O-alkylated PPO. This is because the intramolecular hydrogen bonds within the molecules are broken in the case of PPO, weakening its aggregation as previously reported in the literature.23 Interestingly, there is a significant difference in the absorption of two isomeric polymers in the solution. As shown in Fig. 2b, two absorption peaks centered at 838 nm and 912 nm were observed for P3BT-out, similar to many previously reported DPP-based materials.31–33 For P3BT-in, the lower-energy absorption band red shifts with only one peak centered at 903 nm. Furthermore, there is a more pronounced red shift in P3BT-in compared to that in P3BT-out, as thin films (Fig. 2c), leading to a narrower band gap of 1.06 eV, which is consistent with the CV results. Fig. 2d shows the absorption coefficients (α) of the isomeric polymers P3BT-in and P3BT-out thin films. The corresponding maximum α values are 5.4 and 4.1 × 105 cm−1 for P3BT-in and P3BT-out, respectively. Compared with P3BT-out, P3BT-in has a stronger intensity of near-infrared absorption. Therefore, P3BT-in is able to harvest more incident NIR photons for efficient charge carrier generation and thus would be beneficial for NIR-OPT applications.
Theoretical calculations
Recently, DFT methods using hybrid functions have emerged as very powerful theoretical methods describing optoelectronic and geometric properties of conjugated organic systems due to the inclusion of the electron correlation.34 The hybrid functional B3LYP/6-31g(d,p) basic set has become a standard method used to study organic chemistry due to the good compromise between the computational cost, coverage, and accuracy of results.35 To gain insights into the effect of sidechain substitution direction on the backbone geometry and electronic structures, DFT calculations of the two polymers were conducted at the B3LYP/6-31g(d,p) level. The long N,O-alkylated chains were truncated to a methoxy group for simplicity of calculation. Fig. 3a–d shows the top view and side view of the optimized ground-state molecular configurations. One can observe large torsion angles (15–20°) near O-alkylated chains (Fig. 3a and b). For example, P3BT-in has dihedral angles of 15.0° and 15.2° between PPO and its flanking thiophene unit. P3BT-out has even larger dihedral angles of 18.2° and 19.0°. We suggest the presence of O-alkyl chains can distort the molecular structure, leading to lower planarity. However, the distortion extent is different. For P3BT-in, the O-alkyl chains spread in the inner way. Because of the intramolecular hydrogen bonding of the DPP-thiophene moiety, O-alkyl chains only slightly distort the molecular structures of the center region, leading to relatively better backbone planarity. For P3BT-out, the O-alkyl chains spread in the outer way. Because of the lack of hydrogen bonding and other noncovalent interactions in the outer region, the steric hindrance is even larger. For the calculated FMOs levels, the HOMO/LUMO values of the two isomeric polymers are −3.27/−4.83 eV and −3.06/−4.53 eV for P3BT-in and P3BT-out, respectively (Fig. 3e–h), correlating well with the CV values.
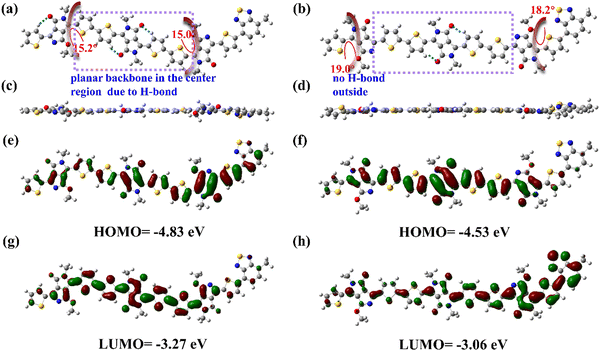 |
| | Fig. 3 DFT-calculated backbone geometry and electronic structures of polymers P3BT-in (a), (c), (e), and (g) and P3BT-out (b), (d), (f), and (h). (a) and (b) Top view and (c) and (d) side view of the optimized ground-state molecular configurations. (e) and (f) HOMO and (g) and (h) LUMO profiles for the two polymers. | |
Charge transport characteristics
To investigate the effect of sidechain substitution direction on the charge transport properties of both polymers, OTFTs were fabricated and measured. The devices based on these two isomeric polymers were fabricated based on the bottom-gate/top-contact (BG/TC) architecture. The process details are described in the ESI.† Typical transfer and output I–V curves and parameters of the OTFTs are shown in Fig. S9 (ESI†) and Table 2. Both isomeric polymers show the n-type dominant ambipolar charge transport properties. Electron mobility is one order of magnitude higher than hole mobility. For example, P3BT-in demonstrates an electron mobility of 4 × 10−3 cm2 V−1 s−1 which is ten times higher than the hole mobility (4 × 10−4 cm2 V−1 s−1). This is because these two polymers are composed of four acceptors, which is beneficial for electron injection and transport. Compared to P3BT-out, P3BT-in exhibits a higher electron mobility, which might be due to its deeper LUMO level and higher backbone planarity.
Table 2 The parameters for the NIR OPTs under an NIR intensity of 4.6 mW cm−2
| Polymer |
μ
h
/μ
e (cm2 V−1 s−1) |
V
th (V) |
I
on/Ioff ratio |
R (A W−1) |
P
|
| P3BT-in |
4 × 10−4/4 × 10−3 |
−6/47 |
102/102 |
3.40 (p)/3.30 (n) |
0.44 (p)/1.70 (n) |
| P3BT-out |
4 × 10−4/3 × 10−3 |
−7/48 |
102/102 |
1.47 (p)/0.13 (n) |
1.22 (p)/0.71 (n) |
NIR-sensing OPTs
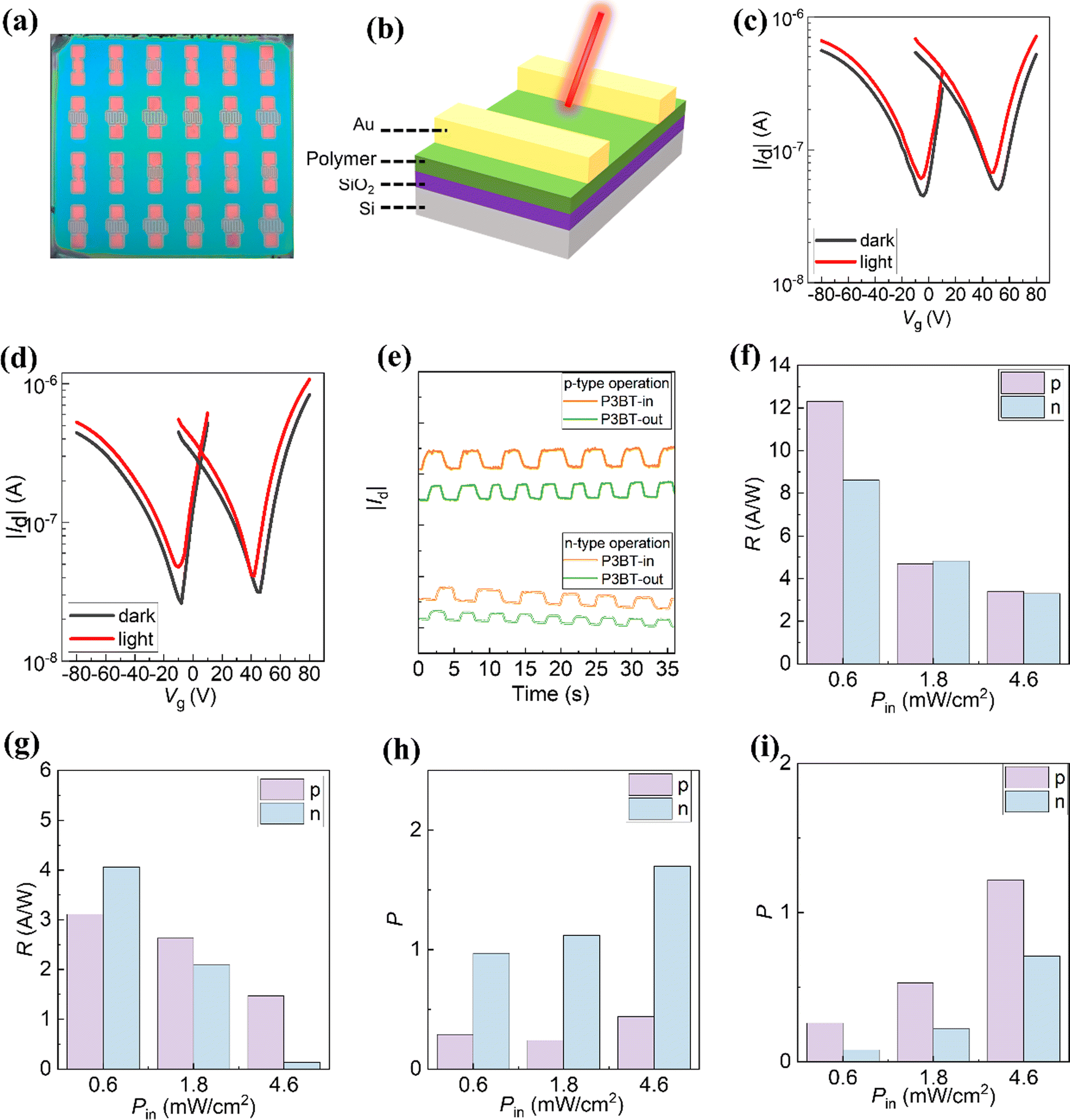
Driven by their simultaneous high NIR absorption, we investigated the photoresponse performance of NIR OPTs on the basis of these two isomeric polymers. The OPT devices were fabricated in the BG/TC configuration, which is the same as OTFTs. Fig. 4a and b show the photograph and device structure (BG/TC) of the OPT devices, respectively. First, we evaluated the optoelectronic properties of the OPT devices in the dark and under monochromic NIR light illumination (λ = 810 nm) with an intensity of 4.6 mW cm−2. As expected, all the OPT devices showed ambipolar transfer curves (Fig. 4c and d). Excitingly, the drain currents increased under the exposure of the NIR light, indicating a high NIR photosensitivity (Fig. 4c and d). From the transfer curves in the dark and under illumination, two important parameters that quantitatively describe the light detection capability of OPTs, namely, photosensitivity (P) and responsivity (R), were calculated using the following fundamental equations.| | | P = (Ilight − Idark)/Idark | (1) |
| | | R = (Ilight − Idark)/Pinc | (2) |
where Ilight and Idark are the IDS values in the dark and under illumination, Pinc is the incident illumination power.
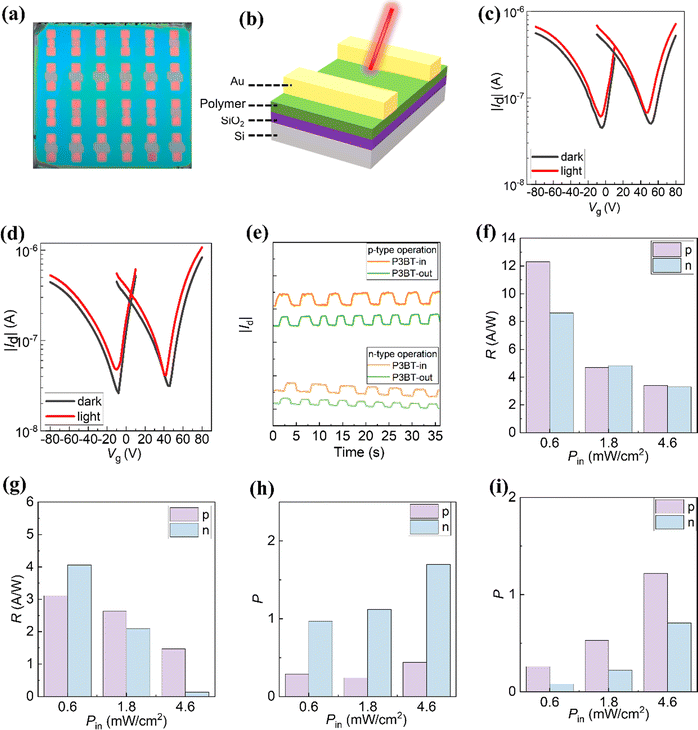 |
| | Fig. 4 (a) Photograph of NIR OPTs. (b) The BG/TC device structure of NIR OPTs. (c) and (d) Transfer curves of the (c) P3BT-in and (d) P3BT-out NIR OPTs in the dark and under NIR light illumination (810 nm, 4.6 mW cm−2). (e) Photocurrent response upon the repeated optical on/off modulations of the NIR light. The responsivity (R) and photo/dark current ratio (P) under different light illumination (f) and (h) for P3BT-in (g) and (i) and P3BT-out. | |
The derived R and P values were plotted against different gate voltages, as shown in Fig. S10 (ESI†). These values generally increase with the increasing gate voltage because a strong electric field leads to efficient charge separation and transfer. Under the light intensity of 4.6 mW cm−2, the maximum (R, P) values for p-type P3BT-in and P3BT-out are (3.40 A W−1, 0.44) and (1.47 A W−1, 1.22), respectively. The maximum (R, P) values for n-type P3BT-in and P3BT-out are (3.30 A W−1, 1.70) and (0.13 A W−1, 0.71), respectively (Table 2). In particular, the P3BT-in-based OPTs have higher values of (R, P) in the n-channel operation compared to the P3BT-out-based OPTs under 810 nm light illumination. This is mainly because the P3BT-in has a higher NIR absorption ability and electron mobility. This result suggests that the photoresponse performance of the NIR OPTs can be optimized by adjusting the sidechain substitution direction. As shown in Fig. 4e, the optical on/off modulations at λ = 810 nm reveal that the drain currents of NIR-OPTs are stable for both the p-channel and n-channel mode sensing operations. From the pulse current changing, P3BT-in's photosensitivity is indeed stronger than that of P3BT-out.
To further investigate the sidechain substitution direction on the photoresponse of NIR-OPTs, the transfer curves of the devices based on P3BT-in and P3BT-out were characterized under different light illumination. The detailed trend of photo-sensing performances is summarized in Fig. 3f–i. As shown in Fig. S11 (ESI†), in the case of both p-channel and n-channel modes, the drain currents in the transfer curves gradually increase with the increase of NIR light intensity. In addition, the transfer curves are slightly shifted by increasing the NIR light intensity, which can be attributed to the change of the threshold voltage level owing to the photogenerated charges induced by the NIR light in the channel layers. Fig. 3f and g show the photoresponsivity under different light illumination. As the NIR light intensity increases, the photoresponsivity shows a gradual reduction. This is because of the increased charge recombination of the photogenerated charge carriers in the polymer channel layers at high NIR light intensity, which is consistent with the previous reports.13,14,36,37 P3BT-in demonstrates the highest photoresponsivity of 12.31 A W−1 under the NIR intensity of 0.6 mW cm−2, which is one order of magnitude higher than that of the high-performance DPP-based NIR material reported recently.2 In addition, as the NIR light intensity increases, the photosensitivity (P) of the NIR-OPTs gradually increases. For the P3BT-in-based OPTs, the P value is relatively higher for the n-channel operation than for the p-channel operation (Fig. 3h). In contrast, the P value for n-channel operation is lower in the P3BT-out-based OPTs (Fig. 3i). This result indicates that the electron carriers are more susceptible under light in P3BT-in-based OPTs while hole carriers in P3BT-out-based OPTs are more active. Overall, P3BT-in has better photoelectric detection performance than P3BT-out. We believe that the strengthened NIR absorption and enhanced electron mobility are the main reasons for the performance improvement of the P3BT-in-based devices.
Film microstructures and morphologies
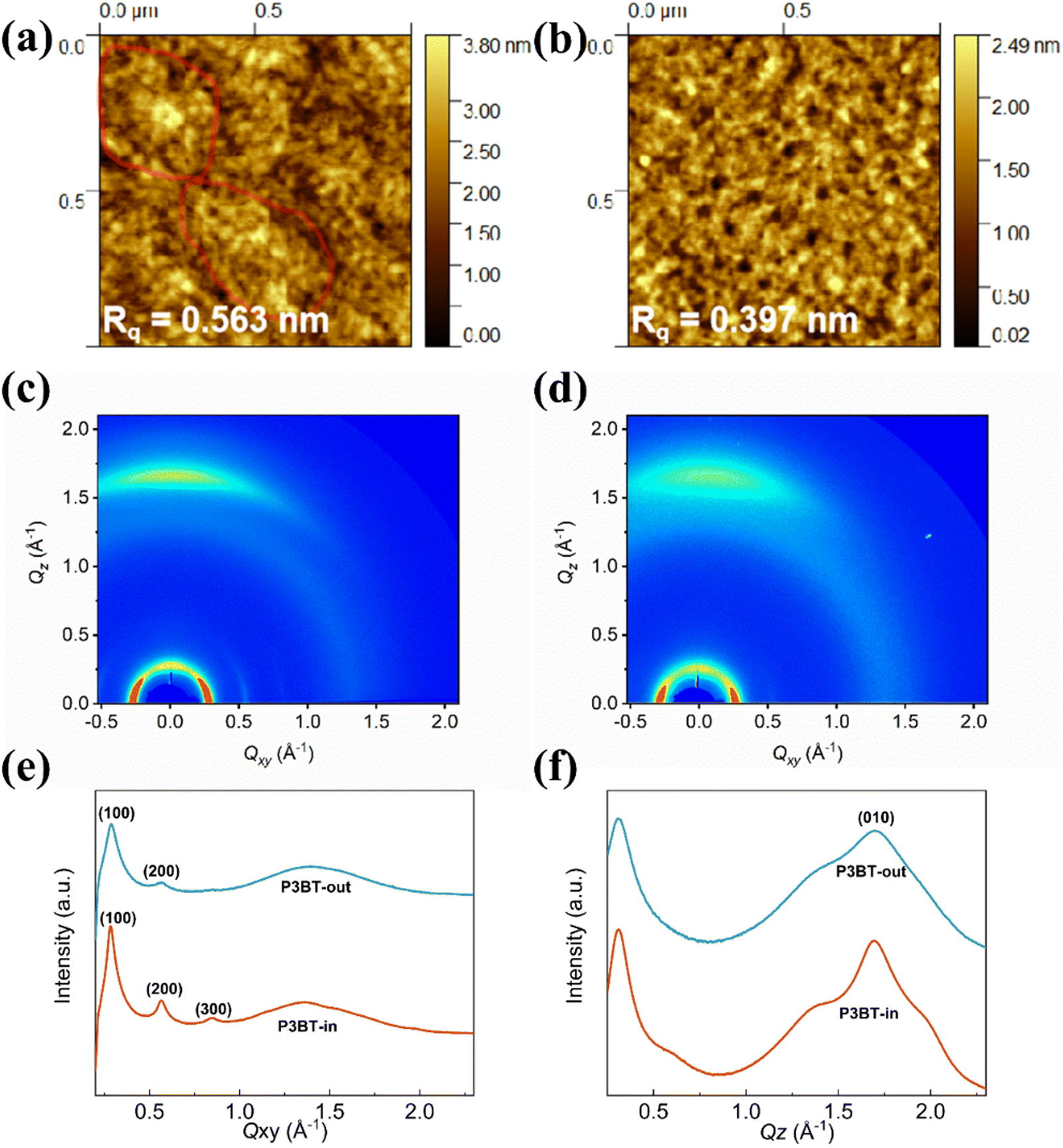
To address the structure–property relationship, both polymer thin films were characterized using atomic force microscopy (AFM) and grazing-incidence incidence wide-angle X-ray scattering (GIWAXS).38–40 The thin films were fabricated via spin coating at 3000 rpm for 60 s onto the n-octadecyltrimethoxysilane (OTS)-modified SiO2/Si substrates and subsequently annealed at an optimal temperature of 150 °C (same conditions of fabricating OTFTs). As depicted in Fig. 5a and b, the surface topography image of the P3BT-in film showed relatively large crystalline domain networks, while the P3BT-out film showed mesh-like morphologies. Thus, P3BT-in has a slightly rougher surface with the rms roughness of 0.563 nm than that of P3BT-out.
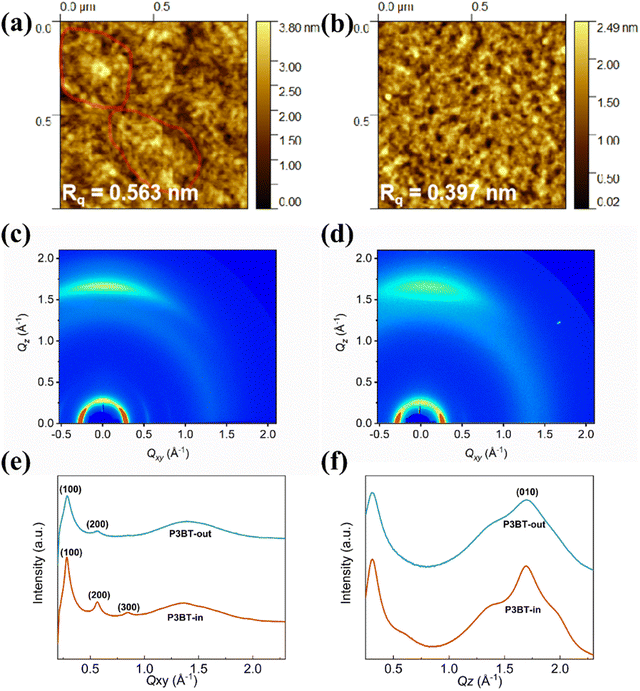 |
| | Fig. 5 AFM height images of the polymer thin films (a) P3BT-in and (b) P3BT-out. (c) and (d) Grazing incidence wide-angle X-ray scattering (GIWAXS) pattern of (c) P3BT-in and (d) P3BT-out. The extracted line profiles of the GIWAXS patterns in the (e) in-plane and (f) out-of-plane direction. | |
To gain insights into the microstructural ordering features of both polymers, GIWAXS measurements were carried out (Fig. 5c and d). The detailed π–π stacking and lamellar packing distances in the GIWAXS patterns of the two isomeric polymer films are summarized in Table S1 (ESI†). As shown in Fig. 5e, f and Table S1 (ESI†), both the isomeric polymer films exhibited predominant face-on orientation, which can be proved by their lamellar scattering in the in-plane (IP) direction and the obvious (010) π–π stacking peak in the out-of-plane (OOP) direction. The lamellar packing distances of P3BT-in and P3BT-out were calculated to be 21.7 and 22.0 Å, and the corresponding crystalline coherence lengths (CCL) were 193.9 and 111.4 Å, respectively. Although both polymers have similar π–π stacking distances of 3.7 Å, the CCL of P3BT-in was significantly larger than that of P3BT-out. Also, the lamellar scattering in the IP direction is up to the third order (300) for P3BT-in. These results suggest that the polymer P3BT-in has higher interchain ordering (crystallinity) and more intensified intermolecular interactions when compared to P3BT-out. Owing to the intramolecular hydrogen bonding of the DPP-thiophene moiety of the center region, O-alkyl chains spreading in the inner way only slightly distort the molecular structures, leading to relatively better backbone planarity, as evidenced by the DFT results. The higher interchain ordering (crystallinity) is beneficial for charge carrier transport, thus leading to better performance of OTFTs and OPTs.
Conclusion
In summary, two new isomeric polymers based on PPO units were synthesized. The two polymers shared an identical conjugated core but with different sidechain substitution directions. Through comprehensive theoretical studies and experimental characterizations, the effects of the N,O-alkylated chain substitution direction on the optoelectronic properties, polymer backbone conformation, and corresponding performance of OTFTs and NIR-OPTs were systematically investigated. Compared to the P3BT-out with sidechains spreading in the outer way, P3BT-in with sidechains in the inner way has better molecular planarity, deeper LUMO energy levels, narrower bandgap, and stronger light absorption ability, resulting in higher crystallinity, increased electron mobility in OTFTs, and higher NIR photosensitivity/photoresponsivity in OPTs. Overall, tuning the sidechain substitution direction could be a useful molecular strategy for advanced functional polymers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the National Key R&D Program of China (No. 2018YFA0703200), National Natural Science Foundation of China (Grant No. 61890940, 51903051, 22375051, and 52203216), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (SSH2021010), and the Natural Science Foundation of Shanghai (21ZR1406900). We thank beamline BL14B1/15U (Shanghai Synchrotron Radiation Facility) for providing the beam time and help.
References
- H. Guo, C.-Y. Yang, X. Zhang, A. Motta, K. Feng, Y. Xia, Y. Shi, Z. Wu, K. Yang, J. Chen, Q. Liao, Y. Tang, H. Sun, H. Y. Woo, S. Fabiano, A. Facchetti and X. Guo, Nature, 2021, 599, 67–73 CrossRef CAS PubMed.
- P. Liu, L. Fu, X.-Y. Tang, R. Xue, L. Zhang, J. Cao and X.-Y. Wang, J. Mater. Chem. C, 2023, 11, 10149–10153 RSC.
- H. Tang, Y. Liang, C. Liu, Z. Hu, Y. Deng, H. Guo, Z. Yu, A. Song, H. Zhao, D. Zhao, Y. Zhang, X. Guo, J. Pei, Y. Ma, Y. Cao and F. Huang, Nature, 2022, 611, 271–277 CrossRef CAS PubMed.
- J. Chen, Z. Yan, L. Tang, M. A. Uddin, J. Yu, X. Zhou, K. Yang, Y. Tang, T. J. Shin, H. Y. Woo and X. Guo, Macromolecules, 2018, 51, 5352–5363 CrossRef CAS.
- H. Jinno, K. Fukuda, X. Xu, S. Park, Y. Suzuki, M. Koizumi, T. Yokota, I. Osaka, K. Takimiya and T. Someya, Nat. Energy, 2017, 2, 780–785 CrossRef CAS.
- S. Wang, J. Xu, W. Wang, G.-J. N. Wang, R. Rastak, F. Molina-Lopez, J. W. Chung, S. Niu, V. R. Feig, J. Lopez, T. Lei, S.-K. Kwon, Y. Kim, A. M. Foudeh, A. Ehrlich, A. Gasperini, Y. Yun, B. Murmann, J. B. H. Tok and Z. Bao, Nature, 2018, 555, 83–88 CrossRef CAS PubMed.
- D. Zhu, D. Ji, L. Li and W. Hu, J. Mater. Chem. C, 2022, 10, 13312–13323 RSC.
- T. Han, Z. Wang, N. Shen, Z. Zhou, X. Hou, S. Ding, C. Jiang, X. Huang, X. Zhang and L. Liu, Nat. Commun., 2022, 13, 1332 CrossRef CAS PubMed.
- J. Huang, J. Lee, J. Vollbrecht, V. V. Brus, A. L. Dixon, D. X. Cao, Z. Zhu, Z. Du, H. Wang, K. Cho, G. C. Bazan and T.-Q. Nguyen, Adv. Mater., 2020, 32, 1906027 CrossRef CAS PubMed.
- H. Son, T. Kim, C. Lee, H. Kim and Y. Kim, J. Mater. Chem. C, 2022, 10, 3951–3958 RSC.
- Y. Cho, T. Kim, W. Lee, H. Kim and Y. Kim, J. Mater. Chem. C, 2023, 11, 2970–2976 RSC.
- S. Kim, D. Lee, J. Lee, Y. Cho, S.-H. Kang, W. Choi, J. H. Oh and C. Yang, Chem. Mater., 2021, 33, 7499–7508 CrossRef CAS.
- Y. Tang, W. Ge, P. Deng, Q. Zhang, Y. Liao, Z. Xiong, W. Lan, B. Wei and Y. Lei, J. Mater. Chem. C, 2020, 8, 16915–16922 RSC.
- X. Wang, D. Pan, M. Sun, F. Lyu, J. Zhao and Q. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 26187–26195 CrossRef CAS PubMed.
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647–663 CrossRef CAS.
- Y. Wang, T. Hasegawa, H. Matsumoto and T. Michinobu, J. Am. Chem. Soc., 2019, 141, 3566–3575 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, W. S. C. Roelofs, S. C. J. Meskers, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 1565–1570 CrossRef CAS PubMed.
- Z. Yuan, B. Fu, S. Thomas, S. Zhang, G. DeLuca, R. Chang, L. Lopez, C. Fares, G. Zhang, J.-L. Bredas and E. Reichmanis, Chem. Mater., 2016, 28, 6045–6049 CrossRef CAS.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636–2642 CrossRef CAS PubMed.
- Š. Frebort, Z. Eliáš, A. Lyčk, S. Luňák Jr, J. Vyňuchal, L. Kubáč, R. Hrdina and L. Burgert, Tetrahedron Lett., 2011, 52, 5769–5773 CrossRef.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- T. Shen, W. Li, Y. Zhao, Y. Liu and Y. Wang, Adv. Mater., 2023, 35, 2210093 CrossRef CAS PubMed.
- T. Shen, W. Li, Y. Zhao, Y. Liu and Y. Wang, Matter, 2022, 5, 1953–1968 CrossRef CAS.
- T. Izawa, E. Miyazaki and K. Takimiya, Adv. Mater., 2008, 20, 3388–3392 CrossRef CAS.
- C. Zhu, Z. Zhao, H. Chen, L. Zheng, X. Li, J. Chen, Y. Sun, F. Liu, Y. Guo and Y. Liu, J. Am. Chem. Soc., 2017, 139, 17735–17738 CrossRef CAS PubMed.
- J. Li, K. Yang, D. Wang, B. Liu, Y. Wang, S. Y. Jeong, Z. Chen, H. Y. Woo and X. Guo, Macromolecules, 2023, 56, 2339–2347 CrossRef CAS.
- X. Luo, D. T. Tran, N. M. Kadlubowski, C. H. Y. Ho, P. Riley, F. So and J. Mei, Macromolecules, 2018, 51, 8486–8492 CrossRef CAS.
- A. Yoshitake, H. Kudo, H. Matsumoto and M. Tokita, Macromol. Rapid Commun., 2021, 42, 2100311 CrossRef CAS PubMed.
- Y. Chen, Z. Wu, L. Ding, S. Zhang, Z. Chen, W. Li, Y. Zhao, Y. Wang and Y. Liu, Adv. Funct. Mater., 2023, 33, 2304316 CrossRef CAS.
- G. Qian, J. Qi, J. A. Davey, J. S. Wright and Z. Y. Wang, Chem. Mater., 2012, 24, 2364–2372 CrossRef CAS.
- B. Zhao, K. Sun, F. Xue and J. Ouyang, Org. Electron., 2012, 13, 2516–2524 CrossRef CAS.
- B. Fu, L. Sun, L. Liu, D. Ji, X. Zhang, F. Yang and W. Hu, Sci. China Mater., 2022, 65, 2779–2785 CrossRef CAS.
- Q. Liu, Y. Wang, A. Kohara, H. Matsumoto, S. Manzhos, K. Feron, S. E. Bottle, J. Bell, T. Michinobu and P. Sonar, Adv. Funct. Mater., 2020, 30, 1907452 CrossRef CAS.
- S. M. Bouzzine, S. Bouzakraoui, M. Bouachrine and M. Hamidi, THEOCHEM, 2005, 726, 271–276 CrossRef CAS.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef CAS PubMed.
- C. Wang, X. Zhang and W. Hu, Chem. Soc. Rev., 2020, 49, 653–670 RSC.
- H. Dong, H. Zhu, Q. Meng, X. Gong and W. Hu, Chem. Soc. Rev., 2012, 41, 1754–1808 RSC.
- S. Otep, Y. Wang, A. Kohara, H. Matsumoto, T. Mori and T. Michinobu, ACS Appl. Polym. Mater., 2019, 1, 2302–2312 CrossRef CAS.
- B. Fan, L. Ying, Z. Wang, B. He, X. F. Jiang, F. Huang and Y. Cao, Energy Environ. Sci., 2017, 10, 1243–1251 RSC.
- S. Chen, L. Feng, T. Jia, J. Jing, Z. Hu, K. Zhang and F. Huang, Sci. China Chem., 2021, 64, 1192–1199 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 * and
Yunqi
Liu
* and
Yunqi
Liu