
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational bottom-up synthesis of sulphur-rich porous carbons for single-atomic platinum catalyst supports†
Koki
Chida
 a,
Takeharu
Yoshii
*a,
Ryo
Kawaguchi
a,
Masataka
Inoue
b,
Fumito
Tani
b,
Tatsuki
Sobue
c,
Shunsuke
Ohtani
c,
Kenichi
Kato
c,
Tomoki
Ogoshi
*cd,
Shoko
Nakahata
e,
Kazuhide
Kamiya
*ef and
Hirotomo
Nishihara
ag
a,
Takeharu
Yoshii
*a,
Ryo
Kawaguchi
a,
Masataka
Inoue
b,
Fumito
Tani
b,
Tatsuki
Sobue
c,
Shunsuke
Ohtani
c,
Kenichi
Kato
c,
Tomoki
Ogoshi
*cd,
Shoko
Nakahata
e,
Kazuhide
Kamiya
*ef and
Hirotomo
Nishihara
ag
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan. E-mail: takeharu.yoshii.b3@tohoku.ac.jp
bInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
cDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: ogoshi@sbchem.kyoto-u.ac.jp
dWPI Nano Life Science Institute (WPI-NanoLSI), Kanazawa University, Kakuma-machi, Kanazawa, Ishikawa 920-1192, Japan
eResearch Center for Solar Energy Chemistry, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: kamiya.kazuhide.es@osaka-u.ac.jp
fInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University Suita, Osaka, 565-0871, Japan
gAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
First published on 9th July 2024
Abstract
Single-atomic metal catalysts are attractive for green chemistry in terms of their outstanding catalytic performance and savings in precious metal usage owing to maximized metal utilization, including anode catalysts in polymer electrolyte fuel cells (PEFCs). Heteroatom-doped porous carbons are extensively used as supports, where the heteroatoms contribute to the immobilization of single-atomic metals. However, high-content doping of heteroatoms, especially sulphur (S), into carbon supports is still challenging because S species can be readily desorbed during heat treatment. Herein, we present a bottom-up fabrication approach for S-rich porous carbons from molecular precursors via a thermal polymerization process. A simple carbonization of molecules with thermally stable S-containing building blocks and polymerizable ethynyl moieties at 900 °C yields microporous carbon materials with record-high S content (over 15 wt%). The abundant S species function as an effective anchoring site for single-atomic platinum (Pt) species. Toward anode catalysts in PEFCs, the prepared single-atomic Pt catalysts efficiently promote the electrochemical hydrogen oxidation reaction, whose activity is comparable to that of commercial Pt/C, despite the significantly low Pt loading amount.
 Takeharu Yoshii | Takeharu Yoshii (b. 1992) received his Ph.D. degree in Engineering from Osaka University under the supervision of Professor Hiromi Yamashita in 2020. Since 2020, he has been an Assistant Professor at the Institute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, working with Professor Hirotomo Nishihara. He was conferred the title of Prominent Research Fellow at Tohoku University in 2023. From 2023 to the present, he has been participating in the JST PREST “Future Materials” program. His work focusses on the design and synthesis of nanostructured heterogeneous catalysts, especially carbon-based single-atomic metal catalysts for green chemistry. |
1. Introduction
A variety of catalytic processes have brought significant improvements to the chemical industry.1,2 Platinum (Pt)-based catalysts are indispensable to industry for their superior catalytic performance in a variety of reactions.3–5 With the growing demand for rechargeable batteries toward achieving a sustainable society, Pt has recently been extensively used as a catalyst for both the anode and cathode in polymer electrolyte fuel cells (PEFCs).6,7 However, Pt is one of the precious metals, and there are practical issues with its limited availability and high cost. Therefore, there is an urgent need to optimize catalyst design for green chemistry.8 From this point of view, the ultimate reduction of metal sizes down to atomic dispersion, i.e., single-atomic catalysts (SACs), is one of the effective strategies to significantly improve the atomic utilization efficiency for saving precious metal usage.9 At the same time, SACs can ensure the advantages of traditional heterogeneous catalysts, such as stability and recyclability. To date, many researchers have reported single-atomic metal catalysts on a variety of support materials, including metal oxides,10–12 zeolites,13,14 layered materials,15,16 and carbon materials,17–20 for a wide range of catalytic reaction systems. Metal oxides, zeolites and layered materials are generally unsuitable for electrocatalysis due to their low electron conductivity. Porous carbon materials, on the other hand, possess outstanding advantages especially for electrocatalysis, owing to their high porosity, electrical conductivity, and chemical/thermal stability.21In order to immobilize single-atomic metal species onto carbon supports, it is necessary to create “anchoring sites” like defects on the carbon surface due to the poor interaction between the metal species and the hexagonal lattice structure of graphene sheets. Heteroatom dopants, including nitrogen (N), boron (B), sulphur (S), and phosphorus (P), can coordinate single-atomic metal species, and thus, doped carbon materials are widely used as supports for SACs.22–24 In particular, N-doped carbon materials have been intensively studied as supports for transition metal-based SACs. On the other hand, S doping is known to be more effective than N doping for single-atomic noble metal species like Pt, due to the strong bonding between Pt and S.25 Various methods have been reported to prepare S-doped carbon materials, such as direct conversion of small organic compounds or natural products26–28 and post-processing of carbon materials via chemical reactions.29,30 There are simultaneous requirements for supports for SACs: high heteroatom content, controlled chemical structure, and developed pores. In this regard, S-doping techniques for SACs have not yet been well established.31 Even when a large number of S species were introduced into porous carbon frameworks, the chemical state of S species could not be uniformly controlled and contained oxidized species (–SOx), which would not act as anchoring sites for metal species and would be unstable during electrocatalysis. For example, transition metal-assisted carbonization of small organic compounds has been developed, allowing the fabrication of S-doped porous carbon with a high S content of 12 wt% and a controlled pore structure. However, this methodology was not able to precisely and homogeneously control the chemical structure around the S species and required a complex synthetic process.32 Direct carbonization of precursor molecules with well-defined S species could be a promising approach, but in practice, low-temperature decomposition or evaporation of precursors leads to the production of low-S-containing carbons in low yield.33
We have recently revealed that the carbonization of organic molecules via solid-state polymerization is an effective strategy for the formation of graphene-based porous frameworks with high carbonization yields (∼90 wt%).34 Simple carbonization of metalloporphyrin crystals with polymerizable moieties yields three-dimensional (3D) carbon materials with ordered structures.35–39 Furthermore, carbon materials with a precisely controlled pore size at the angstrom level have been successfully produced by the carbonization of 3D aromatic molecules (tetraphenylmethane and tetrabiphenylmethane) with ethynyl groups.37 Thus, well-designed molecules possessing thermally stable building blocks and polymerizable moieties can be used as precursors to fabricate 3D microporous carbons with molecular-level design in high yields. These studies inspired the application of this bottom-up methodology to the synthesis of carbon materials doped with high concentrations of heteroatoms.
Herein, we report the synthesis of porous carbon materials with a high S content by a simple carbonization approach as a platform for single-atomic Pt species for electrocatalysis. First, design guidelines for two-dimensional (2D) precursor molecules are established towards porous carbons. We further extended our study to the synthesis of S-doped carbons from a precursor molecule possessing benzotrithiophene units with ethynyl moieties. The ethynyl moieties are cross-linked with each other to produce rigid frameworks during heat treatment, offering a high S content (>15 wt%) and a specific surface area (>700 m2 g−1) with high carbonization yields (>59 wt%). Furthermore, single-atomic Pt species can be coordinated to S-dopants in carbon. Interestingly, the prepared single-atomic Pt catalysts show high catalytic activity equivalent to that of commercial Pt/C for the electrochemical hydrogen oxidation reaction (HOR), in spite of a significantly lower Pt content, with high durability and low side reaction activity.
2. Experimental
Chemicals
Reagents and solvents of the best grade available were purchased from commercial suppliers and were used without further purification unless otherwise noted.Synthesis
Pyrene (1) was obtained commercially (Wako Pure Chemical Industries, Ltd). 1-Ethynylpyrene (2) was synthesized according to the previous work (see Scheme S1a†).40,411H NMR (400 MHz, CDCl3): 8.60 (d, J = 9.2 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.19 (d, J = 9.2 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.12 (d, J = 8.0 Hz, 2H), 8.06 (d, J = 8.8 Hz, 1H), 8.04 (t, J = 7.6 Hz, 1H), 3.63 (s, 1H).
A pyrene-based molecule with four ethynyl moieties (1,3,6,8-tetraethynylpyrene, 3) was synthesized according to the previous work (see Scheme S1b†).42
1H NMR (400 MHz, CDCl3): δ 3.67 (s, 4H), 8.49 (s, 2H), 8.69 (s, 4H).
A molecule with a thiophene skeleton and ethynyl moieties (2,5,8-tri(triethynyl)benzo[1,2-b:3,4-b:5,6-b′′]-trithiophene, 1S) was obtained according to previous work (see Scheme S2†).43,44
1H NMR (400 MHz, CDCl3): δ 3.52 (s, 3H), 7.73 (s, 3H).
Benzotrithiophene (2S) was obtained commercially (Wako Pure Chemical Industries, Ltd).
Heat treatment of the samples was conducted under a N2 flow in a tubular furnace by using a temperature heating program as shown in Scheme S3.† The heat-treated samples are denoted as X_T, where X indicates the type of precursor (2, 3 and 1S) and T is the heat treatment temperature (700 or 900 °C).
Characterization
1H NMR spectra were recorded at 400 MHz using a JNM-ECS400 spectrometer (Jeol Resonance Inc.). 1H chemical shifts were expressed as values relative to tetramethylsilane (TMS). The thermal decomposition behaviours of the precursors were assessed by simultaneous analysis systems including thermogravimetry, differential scanning calorimetry, and mass spectroscopy (TG-DSC-MS). TG-DSC was conducted on a STA 449 Jupiter (Netzsch) from 60 °C to 900 °C at a heating rate of 10 °C min−1 under a He flow (150 mL min−1). The emission gas from TG-DSC was analysed by using a quadrupole mass spectrometer (JMS-Q1500GC, JEOL). Scanning electron microscopy (SEM) images were obtained on a FE-SEM using a Hitachi-S4800. Powder XRD (PXRD) patterns for all the samples were collected using a MiniFlexII (Rigaku Co., Tokyo, Japan) between 2 and 40° (2θ) with a 0.01° step (conditions: Cu-Kα radiation, 45 kV, 15 mA, 1.5418 Å). Transmission electron microscopy (TEM) images of the carbonized samples were obtained using a JEOL JEM-2010 operated at 100 kV. Raman spectra were obtained on a LabRAM HR resolution spectrometer (HORIBA) with a laser wavelength of 532 nm, and the intensity was normalized by the G-band. X-ray photoelectron spectroscopy (XPS) analysis was performed using a JEOL JPS-9200 (Al-Kα radiation) for S 2p spectra and a Kratos Analytical Co. Axis Ultra (Al-Kα radiation) for Pt 4f spectra. For analysis, the binding energy of a peak corresponding to sp2 carbon was calibrated to 284.5 eV.45 Elemental analysis was conducted using a MICRO CORDER JM10 and JM S (J-Science Lab. Inc.). N2 adsorption and desorption isotherms were obtained using a BELSORP-max system (MicrotracBEL Corp.) and QUADRASORB EVO4 (Quantachrome Instruments) at −196 °C. The Brunauer–Emmett–Teller (BET) surface area (SBET) was calculated using the multipoint BET method from the adsorption data in the range of P/P0 = 0.01–0.05.46 The pore size distribution was calculated with the non-local density functional theory (NLDFT) using a slit-pore model.47 Adsorption with various molecular probes was measured using a BELSORP-max system (MicrotracBEL Corp.) at 25 °C. Molecular probes of different kinetic diameters, including CO2 (0.33 nm at 25 °C), ethane (0.40 nm at 25 °C), n-butane (0.43 nm at 25 °C), n-hexane (0.49 nm at 25 °C), and tetrachloromethane (0.60 nm at 25 °C), were used for adsorption to evaluate the pore size of the adsorbents.37,48,49 Pt L3-edge X-ray absorption fine structure (XAFS) spectra were recorded in transmission mode by synchrotron radiation at the Beamline BL01B1 in SPring-8, JASRI, Harima, Japan, using an Si (111) monochromator. Fourier transform was applied to the k3-weighted normalized extended X-ray absorption fine structure (EXAFS) data over the range of 3.0 < k (Å−1) < 12 to obtain radial structure functions using Athena and Artemis.50 Curve fitting analysis for EXAFS spectra was performed using a quick first shell offered by Artemis. The coordination number of Pt-S and Pt-Cl bonding for cis-[Pt(DMSO)2Cl2] was fixed to 2 to determine S02: backscattering amplitude factor. Then, the calculated S02 was also employed for the curve fitting analysis of Pt-1S_900. Wavelet transform analysis of EXAFS was conducted using the Morlet function as a mother wavelet.51Electrochemical hydrogen oxidation reaction performance
For the preparation of working electrodes, 1.0 mg of 3_900 or 1S_900 was mixed with 5 wt% Nafion™ solution (20 μL) and ethanol (300 μL), and then, the suspension was sonicated. For 1S_900, 80 μL of catalyst ink was dropped onto a 0.125 cm2 polished rotating disk electrode (RDE) with a catalyst loading of 2.0 mg cm−2. For 3_900, 16 μL of catalyst ink was dropped onto a 0.125 cm2 polished RDE with a catalyst loading of 0.4 mg cm−2. For an impregnation process of Pt species, the as-prepared working electrodes of 1S_900 or 3_900 were immersed in an aqueous solution of 10 mM K2[PtCl4] and rotated for 5 h, and then washed with deionized water to prepare Pt-modified catalysts (denoted as Pt-1S_900 and Pt-3_900, respectively). As a reference catalyst, commercial 20 wt% Pt/C (HiSPEC3000, Johnson Matthey) was used. The catalyst ink of Pt/C was prepared by the same method described above. Then, the as-prepared catalyst ink of 20 wt% Pt/C was dropped onto a 0.125 cm2 polished RDE with a catalyst loading of 2.0 mg cm−2. The electrocatalytic hydrogen oxidation reaction (HOR) performance was evaluated at room temperature by the RDE method at a rotation rate of 2500 rpm in 0.1 M HClO4 aq. (pH: 1.0) under a H2 flow (10 sccm). A saturated Ag/AgCl electrode and a glassy carbon electrode were used as the reference and counter electrodes, respectively. The potential [V] was converted from Ag/AgCl to the reversible hydrogen electrode (RHE) scale using the following equation (eqn (1)).| Potential (RHE) = potential (Ag/AgCl) + 0.059 × pH + 0.1967 | (1) |
Linear sweep voltammetry (LSV) was evaluated between 0 and 0.4 V vs. RHE with a scan rate of 2 mV sec−1 in Ar or H2 saturated 0.1 M HClO4 aq. (pH: 1.0). The oxygen reduction reaction (ORR) activity was also measured by LSV between 0.2 and 1.2 V vs. RHE with a scan rate of 2 mV sec−1 at a rotation rate of 2500 rpm in 0.1 M HClO4 aq. (pH: 1.0) under an O2 flow (10 sccm).
3. Results and discussion
Synthesis of 3D porous carbon from 2D precursor molecules
To prepare design guidelines for precursors towards 3D porous carbon materials, three types of pyrene-based molecules with or without ethynyl moieties (1–3) were investigated (see Fig. 1). The thermal behaviours of these precursors were first examined using TG-DSC-MS (Fig. 1a–c and Fig. S1†). 1 evaporates when the temperature reaches 300 °C, with endothermic DSC peaks and the corresponding MS spectrum of 1 itself. In contrast, 2 and 3 molecules exhibit exothermic DSC peaks at around 160 °C, which correspond to the thermal polymerization of ethynyl moieties. The thermal decomposition of 2 and 3 molecules is significantly prevented, thanks to the polymerization, and the resulting yields at 900 °C are 45 wt% and 78 wt% in the case of 2 and 3, respectively. Based on these thermal analyses, 2 and 3 were employed as carbon precursors, carbonized at 900 °C under an N2 atmosphere, and the resulting materials are denoted as 2_900 and 3_900, respectively. As shown in the photos in Fig. S2a and S2b,† molecule 2 has largely changed its powder shape during the carbonization process, while molecule 3 is carbonized while maintaining the shape. Moreover, SEM observation revealed that the morphology of 3 at the μm level is retained well even after the carbonization at 900 °C (Fig. S2c and S2d†), unlike the case of 2 (Fig. S2e and S2f†). Here, the TG-DSC profile for precursor 2 (see Fig. 1b) shows a sharp endothermic peak without weight loss at around 120 °C, which is attributed to the melting of the molecule before polymerization. On the other hand, no melting-derived peak can be observed for precursor 3 (see Fig. 1c). Thus, precursor 2 changed its morphology due to carbonization via melting, while precursor 3 underwent solid-state polymerization and subsequent carbonization. These results show that introducing multiple polymerizable moieties into aromatic units can avoid their melting and evaporation process, allowing morphology-preserved carbonization via polymerization. From PXRD patterns (Fig. S3†), both crystalline structures of precursors are converted to amorphous structures after the carbonization process. A diffraction peak at around 25° of 2_900 can be assigned to carbon (002), i.e., graphene stacking structures. In the case of 3_900, the (002) peak is broader and weaker than that of 2_900, suggesting that the graphene stacking is not well developed, probably due to the formation of 3D graphene frameworks by carbonization via solid-state polymerization. Raman spectra of 2_900 and 3_900 (Fig. 1d) exhibit D- and G-bands at 1355 and 1590 cm−1, respectively, indicating the formation of defective carbon structures.52 The ID/IG ratios of 2_900 and 3_900 are calculated to be 0.92 and 1.0, respectively (Table S1†), suggesting no significant difference in the quality of the obtained carbon materials. However, porous texture properties are quite different from each other. N2 adsorption/desorption isotherms of a series of 2 and 3 are summarized in Fig. 1e and f, respectively. Both precursors 2 and 3 are poorly porous (SBET: 1 and 24 m2 g−1, respectively). 2_900 also shows a non-porous structure even after carbonization (SBET: 0 m2 g−1). On the other hand, the microporous structure is well developed through the carbonization process in the case of 3, and 3_900 exhibits an SBET of 630 m2 g−1. Taking these characterization results into consideration, introducing multiple ethynyl moieties into 2D aromatic compounds enables the prevention of the liquid-phase transition during heat treatment, but also allows carbonization via solid-state polymerization, resulting in the formation of microporous carbon materials. | ||
| Fig. 1 TG-DSC profiles of (a) 1, (b) 2 and (c) 3. (d) Raman spectra of 2_900 and 3_900. N2 adsorption/desorption isotherms of (e) 2 and 2_900, and (f) 3 and 3_900. | ||
Synthesis of sulphur-doped 3D porous carbon
We then aimed to synthesize highly S-doped porous carbon materials on the basis of the above-suggested design guidelines. Fig. 2a schematically illustrates the preparation of 3D S-doped carbon in this study. Firstly, the precursor molecule (1S), including the thiophene skeleton and multiple ethynyl moieties, was synthesized according to the previous work (see also Scheme S2†).43,44 Then, we investigated the thermal behaviour of 1S using TG-DSC-MS as shown in Fig. 2b and c. As a control sample, commercial benzotrithiophene (denoted as 2S) with no polymerizable moieties was employed. While 2S completely disappears at around 300 °C, 1S demonstrates a high carbonization yield (59 wt%) even after carbonization at 900 °C, exceeding that of previous works (typically less than 40 wt% over 700 °C).53,54 Additionally, an intense exothermic peak can be observed at around 120 °C for 1S, suggesting that ethynyl moieties in 1S are thermally polymerized with each other. Alternatively, 2S exhibits endothermic peaks at around 150 °C and 300 °C, which correspond to its melting and gasification processes, respectively. The MS spectra for 1S show a small amount of desorbed gas species (CO2 or CS, m/z = 44) up to 300 °C (Fig. 2c), while 2S exhibits only m/z = 246, derived from the precursor itself (Fig. S4†). These thermal analyses clearly indicate that ethynyl moieties avoid the gasification of the precursor via the formation of cross-linked frameworks, subsequently yielding a carbon material with high yield. When the temperature reaches 600 °C, sulphur-containing gas species are gradually desorbed including CS2 and SO2 from 1S. It should be noted that SO2 and CO2 were probably generated by reactions with the contaminated air during the TG-DSC-MS measurements, which are unavoidable in this system. Based on these results, S-doped carbon materials were prepared by the carbonization of 1S at 700 or 900 °C under a N2 atmosphere, according to Scheme S3,† and the as-prepared samples are denoted as 1S_700 and 1S_900, respectively. The PXRD pattern of 1S exhibits some peaks corresponding to its crystalline structure (Fig. S5†). These sharp peaks disappear after the carbonization process, indicating its conversion to an amorphous structure. Raman spectra of 1S_700 and 1S_900 (Fig. 2d) demonstrate D- and G-bands at 1355 and 1590 cm−1, respectively, indicating the formation of defective carbons, similar to 2_900 and 3_900 (see Fig. 2d and Table S1†).52 TEM images of carbonized 1S samples (Fig. 2e and S6†) show no graphene stacking structures but numerous micropores as a bright contrast.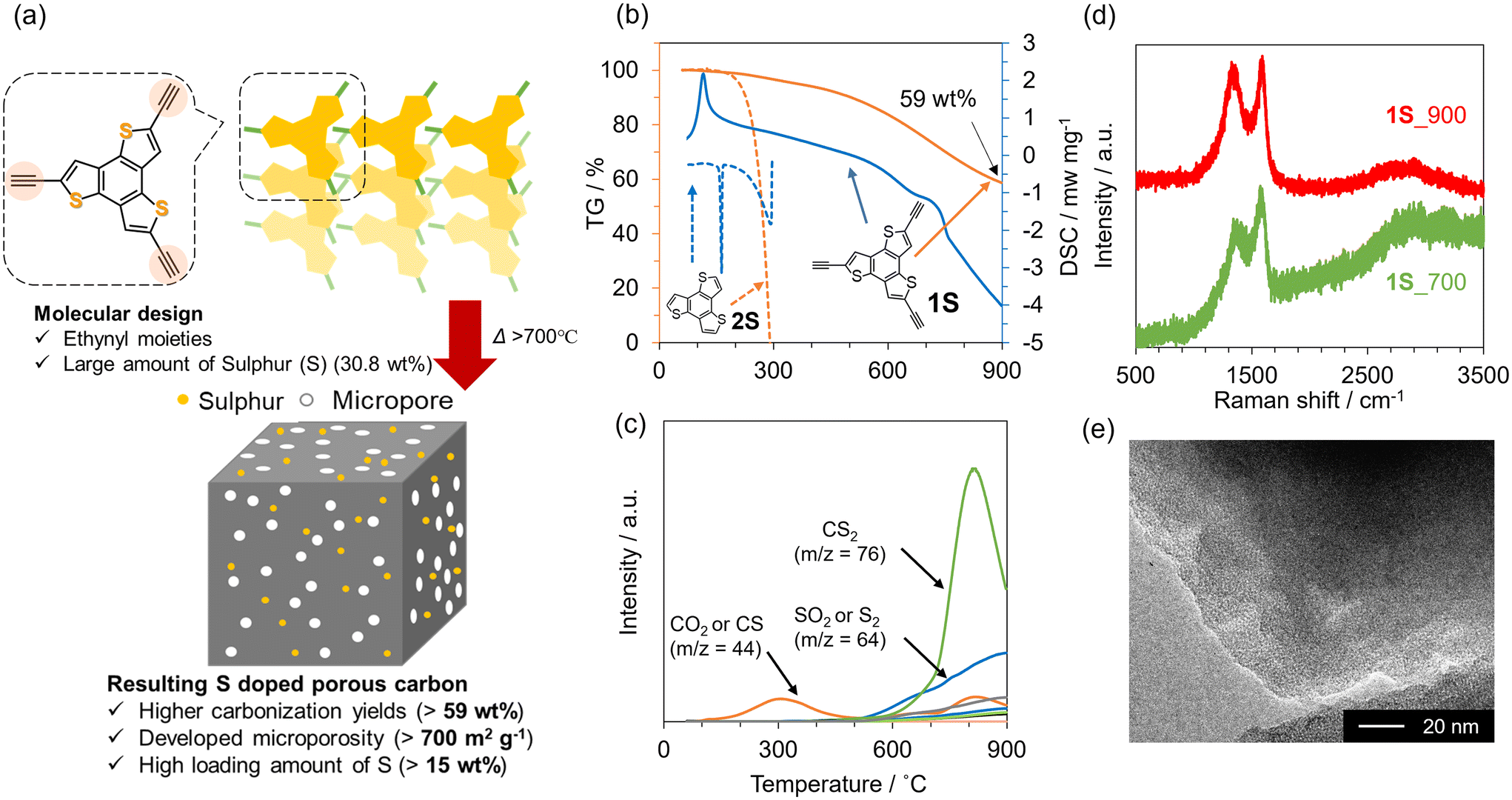 | ||
| Fig. 2 (a) Molecular design of the precursor and the schematic synthetic strategy for highly S-doped porous carbons. (b) TG-DSC profiles for 1S (solid lines) and 2S (dashed lines) and (c) the corresponding MS spectra for 1S. (d) Raman spectra of 1S_700 and 1S_900. (e) TEM image of 1S_900. | ||
To investigate the chemical nature of 1S and carbonized samples, XPS analysis was performed. In the S 2p XPS spectra of 1S (Fig. 3a), the S 2p1/2 and S 2p3/2 orbitals are observed at 164.5 eV and 163.5 eV, respectively. These peaks can be assigned to thiophene-type S.55,56 In the case of 1S_700 and 1S_900 samples, S 2p1/2 and S 2p3/2 peaks are also observed at similar binding energies for 1S (Fig. 3b and c), with the absence of the oxidized S species at around 168 eV.57 Previous works have reported that the carbonization of organic molecules or polymers including thiophene-type S species yielded S-doped carbon materials possessing oxidized S and thiophene-type S species.28,58 Thus, the heat treatment of precursor 1Svia solid-state polymerization would contribute to the retention of thiophene-type S species even after carbonization up to 900 °C. Furthermore, the S content of 1S_700 and 1S_900 is as high as 19.9 and 15.6 wt%, respectively, as determined from the elemental analysis (Table S2†). The sulphur content of the precursor molecule (1S) is calculated to be 30.8 wt% based on the molecular formula. Thus, the yield of sulphur is more than 50% in the whole process even after the carbonization process at 900 °C.
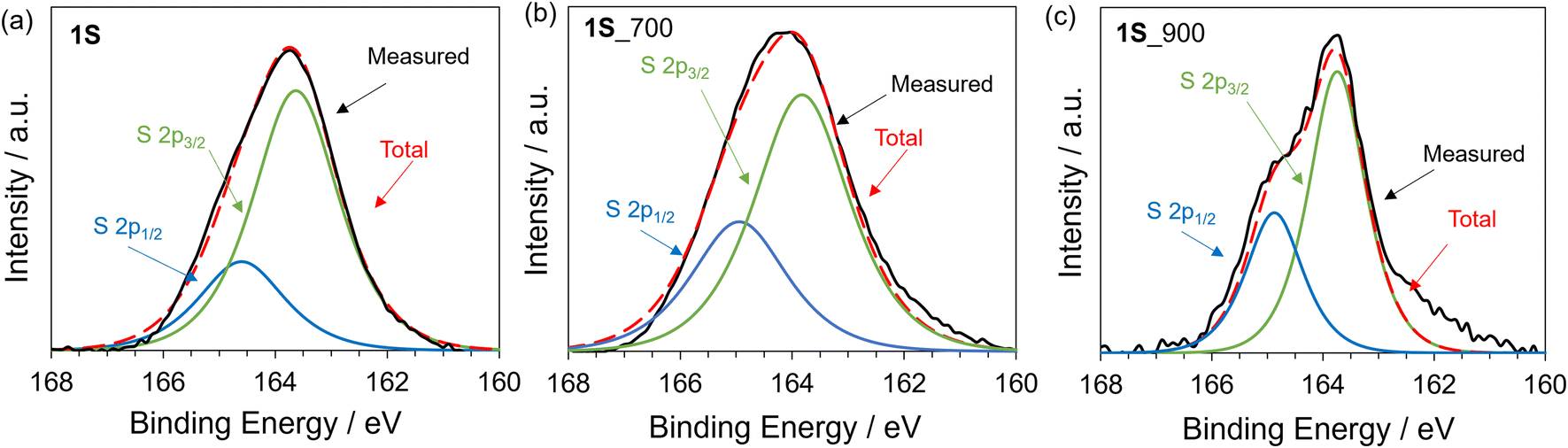 | ||
| Fig. 3 S 2p XPS spectra of (a) 1S, (b) 1S_700, and (c) 1S_900. | ||
N2 adsorption/desorption isotherms of a series of carbonized 1S samples are summarized in Fig. 4a. The precursor 1S is almost non-porous, but the carbonized samples exhibit a clear type-I isotherm, indicating the development of a microporous structure. Indeed, 1S_700 and 1S_900 exhibit high SBET of 714 and 795 m2 g−1, respectively (Table S3†). Pore-size distributions from the NLDFT analysis suggest the formation of a micropore less than 1 nm (Fig. S7†). Thus, the molecular probe method was applied to estimate the pore size more accurately, revealing that a pore of 4.8 Å is formed in 1S_900 (Fig. 4b and S8†). In Fig. 4c and d, we compare the sulphur contents and SBET values of 1S_700 and 1S_900 with the previously reported S-doped carbon materials prepared at 700 or 900 °C (see also Table S4†).27,28,59–67 Interestingly, S-doped carbons synthesized in this work achieve a well-balanced porosity and sulphur content; especially, the highest S content is attained after the heat treatment at 900 °C while maintaining a high specific surface area. Therefore, the precursor design of 1S, with thiophene-type S species and ethynyl moieties, provides multiple benefits, including high carbonization yields, high S contents and high porosity in the resulting carbons.
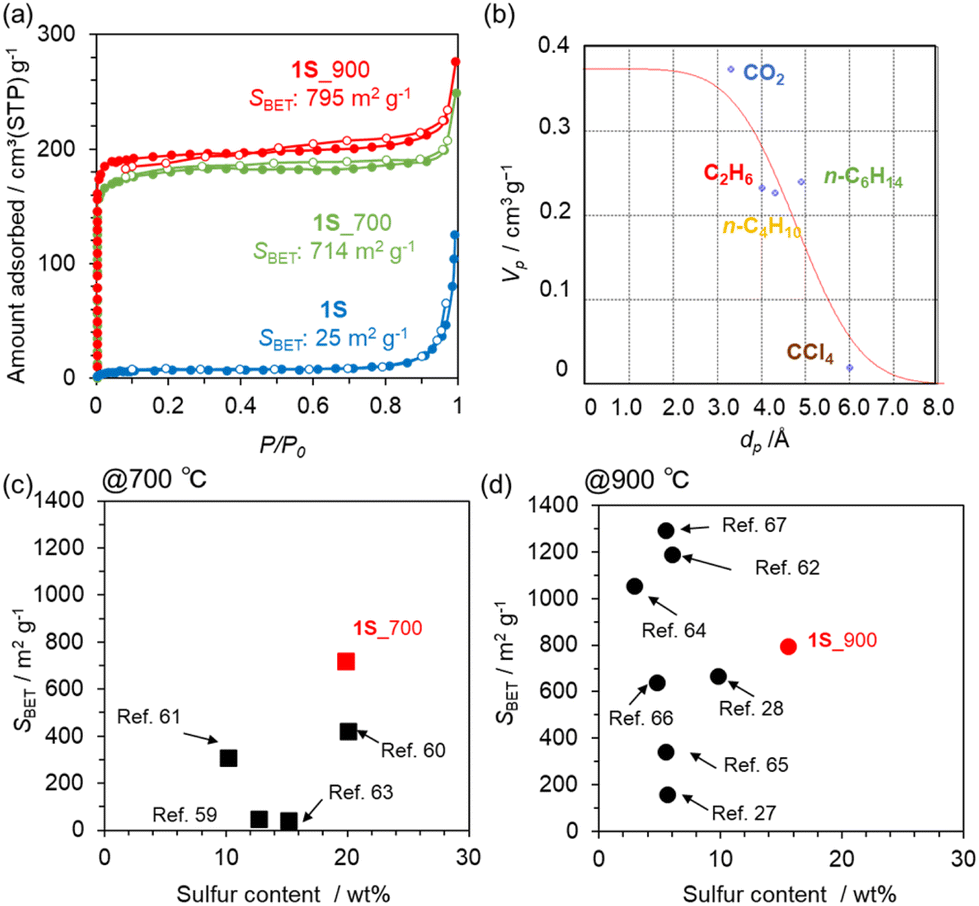 | ||
| Fig. 4 (a) N2 adsorption/desorption isotherms of 1S, 1S_700, and 1S_900 samples. (b) Micropore volume (Vp) of 1S_900 estimated by the Dubinin–Astakhov method, plotted against the kinetic diameter (dp) of molecular probes. Comparison plots of sulphur content and the SBET of S-doped carbons prepared at (c) 700 °C and (d) 900 °C in previous reports and in this work. | ||
Application for single-atomic platinum catalyst supports
We applied the S-doped porous carbons for an electrochemical catalyst support, as sulphur species has lone electron pairs which could be coordinated with metal species. 1S_900 was employed as the catalyst support here in terms of electrical conductivity, and Pt loading was carried out according to a previous work.68 The as-prepared sample is denoted as Pt-1S_900. Pt modification was also executed using 3_900, microporous carbon without S species (denoted as Pt-3_900). Fig. 5a shows the Pt 4f XPS spectra of both Pt-modified samples. The loading amounts of Pt species on Pt-1S_900 and Pt-3_900 are determined to be 0.7 and 0.1 wt%, respectively, from XPS analysis (Table S5†). Since the specific surface areas of both samples are not much different (see Table S3†), the large difference in Pt loading strongly suggests that the S species in 1S_900 contribute to the immobilization of Pt metal species. In addition, Pt-1S_900 has a higher binding energy than the Pt metal in the Pt 4f spectra, indicating the oxidized state of Pt species. Previous works have revealed that the oxidation state of atomically dispersed Pt species generally fluctuates from 2+ to 4+, due to interactions between Pt atoms and catalyst supports.69,70 In order to examine the local structure of Pt species, Pt L3-edge XAFS analysis was carried out. In the XANES spectra (Fig. 5b), the intensity of the white line edge of Pt-1S_900 is identical to that of the cis-[Pt(DMSO)2Cl2] reference, indicating that the oxidation state of Pt is divalent in Pt-1S_900.69 Fourier transforms of the EXAFS spectra of Pt foil and PtO2 show distinct peaks corresponding to Pt–Pt and Pt–O bonding at 2.55 and 1.60 Å, respectively (Fig. 5c). In clear contrast, Pt-1S_900 exhibits a peak at 1.96 Å, which is consistent with the cis-[Pt(DMSO)2Cl2] reference, being ascribed to Pt–S or Pt–Cl bonding. In addition, Pt–Pt bonding is not detected in Pt-1S_900, demonstrating that Pt species dominantly present as atomically dispersed Pt atoms coordinated with S species. Further quantitative analysis of the coordination structure was carried out by fitting the R-space of Pt-1S_900 with Pt–S and Pt–Cl bonding (Table 1). The coordination numbers of Pt–S and Pt–Cl bonding in the first coordination shell of Pt-1S_900 are estimated to be 2.6 and 2.4, respectively, which are close to those for cis-[Pt(DMSO)2Cl2]. This fitting result implies that the coordination structure of Pt species in Pt-1S_900 is similar to that of the Pt(II) complex. To further unveil the local environment of Pt species, we performed wavelet transform (WT) analysis (Fig. 5d), which allows a resolution of the backscattering atoms in both k- and R-spaces.71 The WT of EXAFS signals for Pt foil indicate a single intensity maximum at around 9.3 Å−1 due to the Pt–Pt contribution. In contrast, the WT of EXAFS shows an intensity maximum at around 6 Å−1 for Pt-1S_900, cis-[Pt(DMSO)2Cl2], and PtO2, which can be assigned to the Pt–S/O contribution of the first nearest-neighbour coordination shell. The similarity between the Pt coordination of Pt-1S_900 and cis-[Pt(DMSO)2Cl2] can also be clearly recognized from WT. Combining the FT and WT of EXAFS analysis, it can be concluded that the Pt species in Pt-1S_900 are atomically dispersed in the divalent state coordinated to the S species, without aggregation into particles.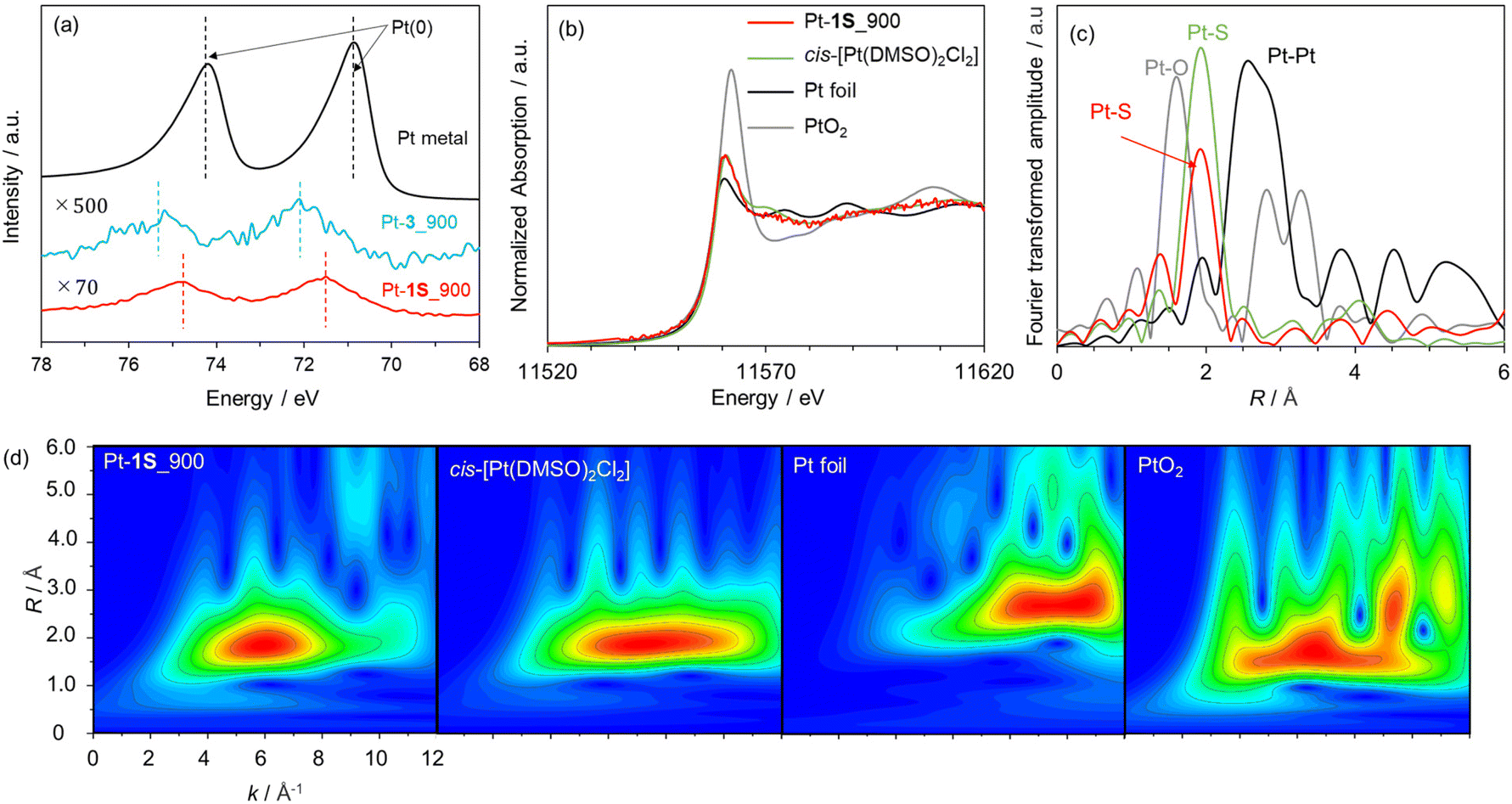 | ||
| Fig. 5 (a) Pt 4f XPS spectra of Pt-1S_900, Pt-3_900, and Pt metal. (b) Pt L3-edge XANES spectra and (c) Fourier transforms of k3-weighted EXAFS oscillations for Pt-1S_900, cis-[Pt(DMSO)2Cl2], Pt foil, and PtO2. (d) Wavelet transforms of Pt-1S_900, cis-[Pt(DMSO)2Cl2], Pt foil, and PtO2. | ||
The electrocatalytic performance of Pt-1S_900 for the hydrogen oxidation reaction (HOR) was evaluated by linear sweep voltammetry (LSV) in H2 saturated 0.1 M HClO4 aq. Commercial Pt/C and Pt-3_900 were employed as control samples. Under Ar-saturated conditions, none of the catalysts (Pt-1S_900, Pt-3_900, and Pt/C) exhibit faradaic current (Fig. S9†). In contrast, 1S_900 displays a clear increase in current from 0 V vs. RHE (namely, without overpotential) under the H2-saturated conditions, similar to Pt/C (Fig. 6a). No faradaic current can be observed in Pt-3_900, probably due to a much smaller loading amount or an overoxidized state of Pt species compared to 1S_900 (see Fig. 5a and Table S5†). These results reveal that single-atomic Pt species supported on S-doped carbon function as an efficient electrocatalyst for the HOR. Notably, Pt-1S_900 shows catalytic activity equivalent to Pt/C, in spite of a much lower Pt loading amount (less than one-twelfth, see Table S5†). Such high catalytic performance could be attributed to the higher atom utilization efficiency of atomically dispersed Pt species. Furthermore, there was no significant change in the Pt content of Pt-1S_900 before and after the electrochemical test, demonstrating no leaching of Pt species even under acidic conditions (Table S5†). The stability test was carried out by LSV for 100 cycles (Fig. 6b). The high catalytic activity of Pt-1S_900 was well retained even after 100 cycles, further indicating the durability of S-coordinated Pt species for the electrochemical HOR. When considering its practical application in PEFCs, air contamination inevitably happens in the anode chamber during the start-up of PEFCs, and the cathodic oxygen reduction reaction (ORR) occurs as a side reaction on the Pt anode catalysts. This phenomenon induces a decrease in power generation and the deterioration of carbon supports. Thus, we further investigated the electrocatalytic ORR activity. Fig. 6c shows the polarization curves of Pt-1S_900 and Pt/C for the ORR. The current density of Pt-1S_900 is significantly smaller compared to Pt/C, indicating its extremely low catalytic activity for the ORR. Therefore, single-atomic Pt coordinated to S-doped carbons can be regarded as one of the ideal anode catalysts for PEFCs in terms of activity per Pt amount, durability, and low ORR reactivity.
 | ||
| Fig. 6 (a) Polarization curves for the HOR of Pt-1S_900, Pt-3_900 and Pt/C under a H2 flow. (b) Polarization curves for the HOR of Pt-1S_900 for the 1st and 100th cycle. (c) Polarization curves for the ORR of Pt-1S_900 and Pt/C. | ||
4. Conclusions
In summary, we have initially established the guidelines for an efficient bottom-up preparation of 3D porous carbon materials; molecular precursors with multiple polymerizable ethynyl groups are converted to microporous carbons in high yields via solid-state polymerization. According to the guidelines, we have also succeeded in the fabrication of S-rich porous carbons from the simple carbonization of the precursor molecule including the thiophene skeleton and ethynyl moieties. Notably, this work achieved a high content (over 15 wt%) of sulphur species with a homogeneous chemical structure in well-developed microporous carbons, even after carbonization at as high as 900 °C. This can be attributed to the formation of rigid 3D cross-linked frameworks by ethynyl moieties, suppressing the decomposition during the carbonization process. We further applied the prepared S-rich porous carbon materials as supports for single-atomic Pt species. High catalytic activity for the electrochemical HOR is demonstrated, equivalent to that of commercial Pt/C, in spite of a much smaller Pt loading amount. Moreover, Pt coordinated S-doped carbons show high long-term stability and low catalytic activity for a side reaction of the ORR, suggesting suitable catalytic performance for their practical application in PEFC anodes.From the perspective of green chemistry, the HOR, the target of this study, is a green catalytic process. However, the use of organic solvents, including toluene, CHCl3, TEA, THF, and NMP, is unavoidable in the organic synthesis of precursor molecules for catalyst supports, although only for temporary use during catalyst synthesis. Future developments in green organic chemistry, such as solid-state cross-coupling reactions, are expected to reduce the use of organic solvents. With regard to recyclability, the prepared catalysts consist of C, H, S, and Pt species. C, H, and S are abundant resources, but the recovery of Pt species from the catalyst is crucial. Here, C, H and S are combustible, so Pt species can be recovered by burning the catalyst after use, as is the case with conventional Pt/C catalysts. In terms of saving in Pt usage, reducing the Pt amount in cathode catalysts is also an urgent issue in PEFCs, since cathode catalysts require more Pt than anode catalysts. However, the prepared single-atomic Pt catalysts are active for the HOR but inactive for the ORR. Therefore, unfortunately, this methodology cannot contribute to the reduction of Pt usage in cathode sides at this stage. The synthetic approach for highly S-doped carbon materials presented in this study is expected to be extended to diverse heteroatom systems such as N, B and P. For example, highly N-doped carbons can be used in single-atomic transition metal catalyst supports, opening up to a variety of reaction systems such as the ORR and CO2 conversion. Therefore, the present rational design for highly hetero-doped porous carbons provides potential applications toward various catalytic systems that are important for green chemistry.
Data availability
All data supporting the findings of this study are available within the article and its ESI† file.Author contributions
Investigation: K. C., R. K., T. F., T. S. and S. N.; formal analysis: K. C., R. K., T. S. and S. N.; supervision: T. Y., S. O., K. Kato, T. O., K. Kamiya and H. N.; project administration: T. Y., T. O., K. Kamiya and H. N.; writing – original draft: K. C.; writing – review & editing: T. Y. and H. N. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST CREST Grant no. JPMJCR18R3; JSPS KAKENHI Grant no. 23H02063, 21K14490 and 22KJ0294; the “Five-star Alliance” in “NJRC Mater. & Dev.”; and JST SICORP Grant no. JPMJSC2112. The authors thank Prof. R. Sakamoto and Prof. Y. Nishina for precious discussions and Dr N. Hashimoto for the support toward XAFS analysis. The XAFS experiments using synchrotron radiation were performed at the Beamline BL01B1 in SPring-8, Harima, Japan (proposal no. 2023A1690).References
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef PubMed.
- R. Liu, H. Wu, J. H. Shi, X. M. Xu, D. Zhao, Y. H. Ng, M. L. Zhang, S. J. Liu and H. Ding, Catal. Sci. Technol., 2022, 12, 6945–6991 RSC.
- M. Paulis, H. Peyrard and M. Montes, J. Catal., 2001, 199, 30–40 CrossRef CAS.
- R. Buitrago-Sierra, M. J. García-Fernández, M. M. Pastor-Blas and A. Sepúlveda-Escribano, Green Chem., 2013, 15, 1981 RSC.
- D. Banham and S. Y. Ye, ACS Energy Lett., 2017, 2, 629–638 CrossRef CAS.
- M. N. Islam, A. B. M. Basha, V. O. Kollath, A. P. Soleymani, J. Jankovic and K. Karan, Nat. Commun., 2022, 13, 6157 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- X. F. Yang, A. Q. Wang, B. T. Qiao, J. Li, J. Y. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- R. Lang, X. R. Du, Y. K. Huang, X. Z. Jiang, Q. Zhang, Y. L. Guo, K. P. Liu, B. T. Qiao, A. Q. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- S. De, A. S. Burange and R. Luque, Green Chem., 2022, 24, 2267–2286 RSC.
- Y. W. Liu, Z. Li, Q. Y. Yu, Y. F. Chen, Z. W. Chai, G. F. Zhao, S. J. Liu, W. C. Cheong, Y. Pan, Q. H. Zhang, L. Gu, L. R. Zheng, Y. Wang, Y. Lu, D. S. Wang, C. Chen, Q. Peng, Y. Q. Liu, L. M. Liu, J. S. Chen and Y. D. Li, J. Am. Chem. Soc., 2019, 141, 9305–9311 CrossRef CAS PubMed.
- Q. Zhang, S. Q. Gao and J. H. Yu, Chem. Rev., 2023, 123, 6039–6106 CrossRef CAS PubMed.
- T. Yoshii, K. Tamaki, Y. Kuwahara, K. Mori and H. Yamashita, J. CO2 Util., 2021, 52, 101691 CrossRef CAS.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- B. Marinho, M. Ghislandi, E. Tkalya, C. E. Koning and G. de With, Powder Technol., 2012, 221, 351–358 CrossRef CAS.
- K. Nakatsuka, T. Yoshii, Y. Kuwahara, K. Mori and H. Yamashita, Phys. Chem. Chem. Phys., 2017, 19, 4967–4974 RSC.
- T. Yoshii, K. Nakatsuka, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Lett., 2017, 46, 789–791 CrossRef CAS.
- S. J. Liu, L. C. Bai, A. P. van Muyden, Z. J. Huang, X. J. Cui, Z. F. Fei, X. H. Li, X. L. Hu and P. J. Dyson, Green Chem., 2019, 21, 1974–1981 RSC.
- J. Maruyama, H. Sato, Y. Takao, S. Maruyama, S. Kato, K. Kamiya, K. Chida, T. Yoshii, H. Nishihara and F. Tani, Nanoscale, 2023, 15, 9954–9963 RSC.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- J. T. Zhang and L. M. Dai, ACS Catal., 2015, 5, 7244–7253 CrossRef CAS.
- T. Asefa and X. X. Huang, Chem. – Eur. J., 2017, 23, 10703–10713 CrossRef CAS PubMed.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- S. S. Desa, T. Ishii and K. Nueangnoraj, ACS Omega, 2021, 6, 24902–24909 CrossRef CAS PubMed.
- G. G. Zhao, Y. Zhang, G. Q. Zou, Y. Zhang, W. W. Hong, Y. L. Jiang, W. Xu, H. L. Shuai, H. S. Hou and X. B. Ji, J. Mater. Chem. A, 2018, 6, 11488–11495 RSC.
- J. P. Paraknowitsch, A. Thomas and J. Schmidt, Chem. Commun., 2011, 47, 8283–8285 RSC.
- N. Parveen, M. O. Ansari, S. A. Ansari and M. H. Cho, J. Mater. Chem. A, 2016, 4, 233–240 RSC.
- W. Q. Han, J. Cumings, X. S. Huang, K. Bradley and A. Zettl, Chem. Phys. Lett., 2001, 346, 368–372 CrossRef CAS.
- W. Kicinski, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1–32 CrossRef CAS.
- Z. Y. Wu, S. L. Xu, Q. Q. Yan, Z. Q. Chen, Y. W. Ding, C. Li, H. W. Liang and S. H. Yu, Sci. Adv., 2018, 4, eaat0788 CrossRef CAS PubMed.
- Z. S. Wu, Y. Z. Tan, S. Zheng, S. Wang, K. Parvez, J. Qin, X. Shi, C. Sun, X. Bao, X. Feng and K. Mullen, J. Am. Chem. Soc., 2017, 139, 4506–4512 CrossRef CAS PubMed.
- T. Yoshii, K. Chida, H. Nishihara and F. Tani, Chem. Commun., 2022, 58, 3578–3590 RSC.
- H. Nishihara, T. Hirota, K. Matsuura, M. Ohwada, N. Hoshino, T. Akutagawa, T. Higuchi, H. Jinnai, Y. Koseki, H. Kasai, Y. Matsuo, J. Maruyama, Y. Hayasaka, H. Konaka, Y. Yamada, S. Yamaguchi, K. Kamiya, T. Kamimura, H. Nobukuni and F. Tani, Nat. Commun., 2017, 8, 109 CrossRef PubMed.
- K. Chida, T. Yoshii, K. Takahashi, M. Yamamoto, K. Kanamaru, M. Ohwada, V. Deerattrakul, J. Maruyama, K. Kamiya, Y. Hayasaka, M. Inoue, F. Tani and H. Nishihara, Chem. Commun., 2021, 57, 6007–6010 RSC.
- T. Ogoshi, Y. Sakatsume, K. Onishi, R. Tang, K. Takahashi, H. Nishihara, Y. Nishina, B. D. L. Campeon, T. Kakuta and T. A. Yamagishi, Commun. Chem., 2021, 4, 75 CrossRef CAS PubMed.
- K. Chida, T. Yoshii, N. Hiyoshi, T. Itoh, J. Maruyama, K. Kamiya, M. Inoue, F. Tani and H. Nishihara, Carbon, 2023, 201, 338–346 CrossRef CAS.
- K. Chida, T. Yoshii, M. Ohwada, Y. Hayasaka, J. Komeda, R. Sakamoto, J. Maruyama, K. Kamiya, M. Inoue, F. Tani and H. Nishihara, Catal. Today, 2023, 411, 113830 CrossRef.
- R. S. Kathayat and N. S. Finney, J. Am. Chem. Soc., 2013, 135, 12612–12614 CrossRef CAS PubMed.
- L. Rocard, A. Berezin, F. De Leo and D. Bonifazi, Angew. Chem., Int. Ed., 2015, 54, 15739–15743 CrossRef CAS PubMed.
- S. Bernhardt, M. Kastler, V. Enkelmann, M. Baumgarten and K. Müllen, Chem. – Eur. J., 2006, 12, 6117–6128 CrossRef CAS PubMed.
- C. J. Sun, P. F. Wang, H. Wang and B. H. Han, Polym. Chem., 2016, 7, 5031–5038 RSC.
- Y. R. Shi, M. A. Gerkman, Q. F. Qiu, S. R. Zhang and G. G. D. Han, J. Mater. Chem. A, 2021, 9, 9798–9808 RSC.
- S. T. Jackson and R. G. Nuzzo, Appl. Surf. Sci., 1995, 90, 195–203 CrossRef CAS.
- K. Kaneko and C. Ishii, Colloids Surf., 1992, 67, 203–212 CrossRef CAS.
- Z. Y. Ryu, J. T. Zheng, M. Z. Wang and B. J. Zhang, Carbon, 1999, 37, 1257–1264 CrossRef CAS.
- Z. H. Hu, N. Maes and E. F. Vansant, J. Porous Mater., 1995, 2, 19–23 CrossRef CAS.
- M. Sadakane, K. Kodato, T. Kuranishi, Y. Nodasaka, K. Sugawara, N. Sakaguchi, T. Nagai, Y. Matsui and W. Ueda, Angew. Chem., Int. Ed., 2008, 47, 2493–2496 CrossRef CAS PubMed.
- K. Ma, L. Zhao, Z. Jiang, Y. Huang and X. Sun, Polym. Compos., 2018, 39, 1223–1233 CrossRef CAS.
- N. Hashimoto, K. Mori and H. Yamashita, J. Phys. Chem. C, 2023, 127, 20786–20793 CrossRef CAS.
- M. J. Matthews, M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus and M. Endo, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, R6585–R6588 CrossRef CAS.
- Y. Guo, Z. Zeng, Y. Liu, Z. Huang, Y. Cui and J. Yang, J. Mater. Chem. A, 2018, 6, 4055–4067 RSC.
- G. Y. Zhao, D. F. Yu, H. Zhang, F. F. Sun, J. W. Li, L. Zhu, L. Sun, M. Yu, F. Besenbacher and Y. Sun, Nano Energy, 2020, 67, 104219 CrossRef CAS.
- Y. Z. Su, Z. Q. Yao, F. Zhang, H. Wang, Z. Mics, E. Cánovas, M. Bonn, X. D. Zhuang and X. L. Feng, Adv. Funct. Mater., 2016, 26, 5893–5902 CrossRef CAS.
- H. J. Peng, V. Montes-Garcia, J. Raya, H. L. Wang, H. P. Guo, F. Richard, P. Samori and A. Ciesielski, J. Mater. Chem. A, 2023, 11, 2718–2725 RSC.
- A. Ivanovskaya, N. Singh, R. F. Liu, H. Kreutzer, J. Baltrusaitis, T. V. Nguyen, H. Metiu and E. McFarland, Langmuir, 2013, 29, 480–492 CrossRef CAS PubMed.
- Z. G. Wang, P. J. Li, Y. F. Chen, J. R. He, W. L. Zhang, O. G. Schmidt and Y. R. Li, Nanoscale, 2014, 6, 7281–7287 RSC.
- L. Qie, W. M. Chen, X. Q. Xiong, C. C. Hu, F. Zou, P. Hu and Y. H. Huang, Adv. Sci., 2015, 2, 1500195 Search PubMed.
- W. Zhong, X. S. Lv, Q. W. Chen, M. M. Ren, W. L. Liu, G. D. Li, J. X. Yu, M. Li, Y. Dai and L. Z. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 37850–37858 CrossRef CAS PubMed.
- Q. Q. Wang, X. F. Ge, J. Y. Xu, Y. C. Du, X. Zhao, L. Si and X. S. Zhou, ACS Appl. Energy Mater., 2018, 1, 6638–6645 CrossRef CAS.
- H. M. Cui, J. G. Xu, J. S. Shi and C. Zhang, J. CO2 Util., 2021, 50, 101582 CrossRef CAS.
- S. Louisia, R. C. Contreras, M. Heitzmann, M. R. Axet, P. A. Jacques and P. Serp, Catal. Commun., 2018, 109, 65–70 CrossRef CAS.
- J. Y. Tian, Z. M. Liu, Z. H. Li, W. G. Wang and H. Y. Zhang, RSC Adv., 2017, 7, 12089–12097 RSC.
- L. P. Wang, W. S. Jia, X. F. Liu, J. Z. Li and M. M. Titirici, J. Energy Chem., 2016, 25, 566–570 CrossRef.
- S. Sfaelou, X. D. Zhuang, X. L. Feng and P. Lianos, RSC Adv., 2015, 5, 27953–27963 RSC.
- Y. P. Guo, Z. Q. Zeng, Y. J. Liu, Z. G. Huang, Y. Cui and J. Y. Yang, J. Mater. Chem. A, 2018, 6, 4055–4067 RSC.
- R. Kamai, K. Kamiya, K. Hashimoto and S. Nakanishi, Angew. Chem., 2016, 128, 13378–13382 CrossRef.
- Q. Q. Yan, D. X. Wu, S. Q. Chu, Z. Q. Chen, Y. Lin, M. X. Chen, J. Zhang, X. J. Wu and H. W. Liang, Nat. Commun., 2019, 10, 4977 CrossRef CAS PubMed.
- J. Liu, M. G. Jiao, L. L. Lu, H. M. Barkholtz, Y. P. Li, Y. Wang, L. H. Jiang, Z. J. Wu, D. J. Liu, L. Zhuang, C. Ma, J. Zeng, B. S. Zhang, D. S. Su, P. Song, W. Xing, W. L. Xu, Y. Wang, Z. Jiang and G. Q. Sun, Nat. Commun., 2017, 8, 15938 CrossRef CAS PubMed.
- H. Funke, A. C. Scheinost and M. Chukalina, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 094110 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental synthetic schemes, TG-DSC-MS spectra, SEM images, TEM images, PXRD patterns, pore-size distributions, molecular probe results, HOR test results, and supplemental tables. See DOI: https://doi.org/10.1039/d4gc02055c |
| This journal is © The Royal Society of Chemistry 2024 |