
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of phenanthrylboroles and formal nitrene insertion to access azaborapyrenes†
Harie
Zacharias
a,
Ayesha
Begum
a,
Jianhua
Han
b,
Tyler A.
Bartholome
a,
Todd B.
Marder
 b and
Caleb D.
Martin
*a
b and
Caleb D.
Martin
*a
aDepartment of Chemistry and Biochemistry, Baylor University, One Bear Place #97348, Waco, Texas 76798, USA. E-mail: caleb_d_martin@baylor.edu
bInstitut für Anorganische Chemie, and Institute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 16th July 2024
Abstract
Extended conjugation has been introduced into boroles but only on the 2,3- or 4,5-positions of the central BC4 core. In this work we synthesize phenanthrylboroles that also have conjugation on the 3,4-positions and demonstrate their insertion reactivity with azides to furnish B,N-analogues of pyrene.
The borole core was first synthesized in 1963 by Köster and Benedikt with two fused phenyl groups as a 9-borafluorene,1 with the monocyclic variant being prepared by Eisch and coworkers in 1969 (Fig. 1).2 Both syntheses followed unsuccessful attempts by other teams attributed to the sensitivity of these compounds because of their four π-electron anti-aromatic state.3 The fused phenyl groups in 9-borafluorenes diminish the anti-aromaticity as well as perturb the photophysical properties by extending conjugation.4 There has been effort devoted to the synthesis of borole-containing polycyclic aromatic hydrocarbons (PAHs) beyond 9-borafluorene by extending conjugation with thiophene, pyrrole, furan, and pyridine heteroaromatics on the core.5 Purely organic groups have garnered less attention with the monoaryl boraindene6 and the binaphthyl-fused analogue 12-boradibenzofluorene being prepared.7 In these systems, the photophysical properties of the borole are greatly influenced by the fusion of arene rings. In all borole-containing PAHs, groups have been fused to the 2,3-positions and 4,5-positions, with no examples having an arene fused at the 3,4-positions.
Boroles are effective reagents to access six-membered heteroaromatic species by the insertion of a lone pair-bearing heteroatom into the five-membered boracycle, which is thermodynamically driven by the conversion of an anti-aromatic ring to an aromatic ring.8 This approach has been very effective to prepare 1,2-azaborine species by reaction with an azide that releases N2 as a benign byproduct resulting in formal nitrene insertion of the α-nitrogen or, in rare cases, inserting the γ-nitrogen without N2 extrusion.9 The monocyclic species generally react at ambient temperature while the polycyclic variants, 9-borafluorene10 and 12-boradibenzofluorene,7 require heating due to their lesser degree of anti-aromaticity. Introducing a B,N isostere in place of a CC unit in PAHs also serves as a means to alter the electronic properties.11 Thus, the preparation of new polycyclic borole frameworks presents synthetic opportunities to access B,N-containing PAHs. In this work, we disclose a borole that features a phenanthryl backbone to introduce conjugation extending along the three C–C linkages in boroles and investigate its nitrene insertion reactivity to target a B,N-containing pyrene.
Boron/tin transmetalation is an effective method to prepare borole species.12 A stannole featuring methyl groups on tin and a phenanthryl backbone (1) was prepared by treating 4,5-diiodophenanthrene with 2.1 equivalents of nBuLi and reaction with Me2SnCl2 (Fig. 2a). The 1H NMR spectrum features a methyl resonance at 0.64 ppm with diagnostic satellites from coupling to the NMR active isotopes of tin (2JSn–H = 32 Hz). Reactions of 1 with BCl3 and PhBCl2 in CDCl3 were monitored by in situ11B and 1H NMR spectroscopy with no reaction occurring at 23 °C after 3 days. Heating to 75 °C resulted in new 11B signals emerging at 63.3 and 63.1 ppm for the reactions with BCl3 and PhBCl2, respectively, with complete consumption of both the borane reagent and 1 after 48 hours. The 11B resonances are in the region of those reported for 9-borafluorenes.4a,b Upon workup, the products were isolated and, from the reaction with BCl3, we were able to grow single crystals suitable for X-ray diffraction confirming the structure of the phenanthryl borole 2Cl (Fig. 2b). Comparing the metrical parameters of 2Cl to 9-Cl-9-borafluorene,13 a notable difference is a shorter C2–C3 bond in 2Cl [1.431(8) Å] versus that in 9-Cl-9-borafluorene [1.481(3) Å], attributed to the C2–C3 bond being within an aromatic ring in 2Cl (see ESI,† Table S2). In the 1H NMR spectrum of 2Cl, four resonances are observed, each integrating to two, consistent with the symmetric structure of a phenanthrylborole. The corresponding 1H NMR spectrum of 2Ph displays the same pattern of four aryl signals, but also shows signals for the five phenyl hydrogen atoms.
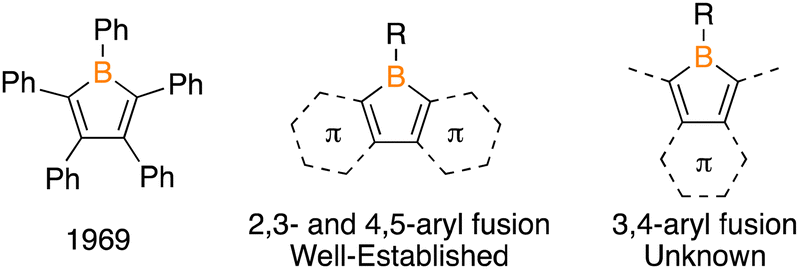 | ||
| Fig. 1 Monocyclic borole, general structure of known aryl-fused variants, and the unknown 3,4-aryl fused variant. | ||
To assess the Lewis acidity and propensity for insertion reactivity, Gutmann–Beckett acceptor numbers (AN) for 2Cl and 2Ph in C6D6 were determined to be 80.3 and 77.6, respectively. Comparing the values to the related 9-borafluorene compounds with the same substitution reveals an enhancement of Lewis acidity in the phenanthryl over the biphenyl compounds with the same trend of the chloro species being more Lewis acidic than the phenyl analogue (9-Cl-9-borafluorene = 78.7, 9-Ph-9-borafluorene = 73.4).10a The value for 2Ph is less than that of pentaphenylborole (79.2), but the value of a monocyclic chloroborole has not been reported. Given that 9-Cl-9-borafluorene and 9-Ph-9-borafluorene undergo insertion,14 this suggests that phenanthrylboroles 2Cl and 2Ph should be poised for such reactivity.
With 2Cl and 2Ph in hand, we targeted the insertion of an azide to access B,N-containing analogues of pyrene. In examining the literature, one article on 1,2-species was published by Dewar and coworkers in 1964.15 Their synthetic strategy was a dehydrohalogenative coupling reaction of an amino phenanthrene with a chloroborane that underwent an intramolecular cyclization to furnish the B,N-pyrene. Other groups have prepared different isomers,16 but Dewar's work is the only report of a B,N pyrene with this connectivity.15 We felt our method could furnish the species akin to Dewar's compounds.
Treatment of 2Cl and 2Ph with benzyl azide (PhCH2N3) at 23 °C resulted in the immediate formation of bubbles consistent with N2 evolution and a loss of the yellow colour to give a colourless solution. Analyzing the reactions by in situ11B NMR spectroscopy revealed upfield shifts from 2Cl and 2Ph within 15 min (2Cl: 63.3 to 37.1 ppm; 2Ph: 63.1 to 29.5 ppm). A break in symmetry of the aryl resonances in the 1H NMR on the phenanthrene backbone for both species is consistent with a formal nitrene insertion. The methylene hydrogens derived from the benzyl azide were observed at 5.52 and 5.43 ppm, for the reactions with 2Ph and 2Cl, respectively. The structure of the chloro species was confirmed to be the desired BN-pyrene by a single crystal X-ray diffraction study (Fig. 2). Compound 3Ph is sufficiently stable for purification by flash column chromatography on silica gel, while 3Cl is moisture sensitive.17
 | ||
| Fig. 2 (a) Synthesis of phenanthrylboroles 2Cl and 2Ph. (b) Solid-state structure of 2Cl. (c) Solid-state structure of 3Cl. Hydrogen atoms are omitted for clarity, and thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for 2Cl: B(1)–Cl(1) 1.746(8), B(1)–C(1) 1.580(1), C(1)–C(2) 1.417(9), C(2)–C(3) 1.431(8), C(3)–C(4) 1.444(10), B(1)–C(4) 1.541(10); for 3Cl: B(1)–Cl(1) 1.789(3), B(1)–N(1) 1.406(3), N(1)–C(1) 1.418(3), C(1)–C(2) 1.424(3), C(2)–C(3) 1.434(3), C(3)–C(4) 1.422(3), C(4)–B(1) 1.540(4). | ||
Examining the metrical parameters of 3Cl and comparing them to the structures of 9,10-B,N-phenanthrenes10,18 revealed analogous parameters within the tricyclic framework (see ESI,† Table S3). The C2–C3 bond length is similar [1.473(3) Å vs. 1.469(3) Å] and lengthened from the borole precursor [2Cl = 1.431(8) Å]. The B–N bond distance [1.406(3) Å] is expectedly longer than the C–C bond length of pyrene [1.344(2) Å]19 and shorter than the B–N bond of the internal BN-pyrene reported by Piers and coworkers [1.456(4) Å].16a
The photophysical properties of 2Ph, 3Cl, and 3Ph were examined by UV/Vis and fluorescence spectroscopy (Table 1, Fig. 3); however, compound 2Cl was too sensitive to handle at the requisite low concentrations for these studies. The absorption spectra feature strong bands in the UV region (253–373 nm); excitation in CH2Cl2 resulted in emission at λem = 365–393 nm, with modest quantum yields (φf = 0.21–0.27) and short lifetimes (τ = 4.5–13.1 ns; Table 1) compared to that of pyrene.20 Compound 2Ph exhibits absorption maxima at 254 nm and 297 nm and emission maxima at 365 nm and 375 nm with a quantum yield of 0.22. It is possible that there is an extremely weak and broad absorption around 350 nm, but this remains difficult to discern even at higher concentrations. The absorption maxima of 3Cl and 3Ph are identical to one another, with peaks at 254 nm and 370 nm, significantly blue-shifted compared to those of the internal BN-pyrene reported by Piers and coworkers,16a but consistent with the absorption data (e.g., λabs = 365 nm) reported by Dewar and coworkers for the parent ‘5,4-borazaropyrene’ as they referred to the BHNH-containing analogue of our compounds of type 3, and the extremely weak S1 ← S0 transition (λabs = 362 nm in toluene, and 372 nm in cyclohexane) for pyrene itself. The emission spectra of 3Cl and 3Ph exhibit very small Stokes shifts (360 cm−1) consistent with those of classic pyrene derivatives, indicating small changes in geometry between the ground and excited states,20 with emission maxima showing clear vibrational bands at 375 nm and 393 nm, and shoulders at lower energy (cf. λem of pyrene = 372 nm in toluene); moderate quantum yields of 0.21 and 0.27, respectively, are observed.
| Compound | λ abs [nm] (ε [104 M−1 cm−1]) | λ em [nm] | Stokes shift [cm−1] | φ f | τ [ns] | k r [107 s−1] | k nr [107 s−1] |
|---|---|---|---|---|---|---|---|
| a Excited at 254 nm. b The non-radiative rate constants (knr) were calculated from knr = (1 − φf)/τ. The radiative rate constants (kr) were calculated from kr = φf/τ. c As there may well be a very weak and broad absorption around 350 nm, it is likely not appropriate to use the 254 nm absorption to calculate a Stokes shift. | |||||||
| 2Ph | 254 (2.0) | 365, 375 | 22% | 13.1 | 1.7 | 6.0 | |
| 3Cl | 254 (5.2), 370 (0.6) | 375, 393 | 360 | 21% | 4.5 | 4.7 | 17.6 |
| 3Ph | 253 (6.9), 370 (1.3) | 375, 393 | 360 | 27% | 4.5 | 6.0 | 16.2 |
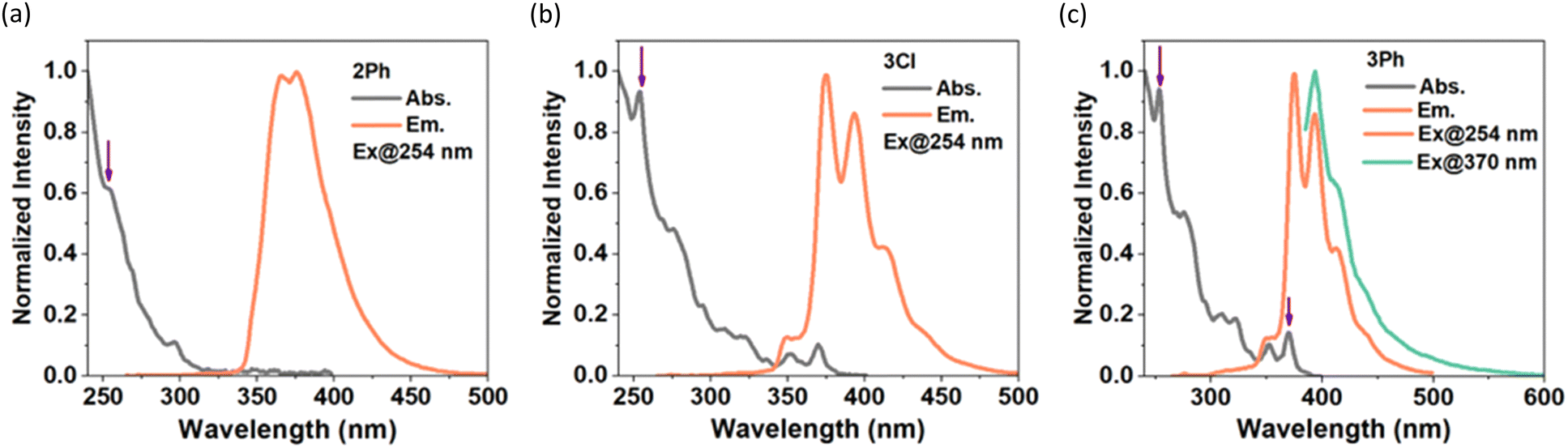 | ||
| Fig. 3 Absorption and emission spectra of (a) 2Ph, (b) 3Cl, and (c) 3Ph in CH2Cl2 (< 10−5 M). The linear dependence of absorbance on concentration is presented in Fig. S23 (ESI†), and excitation spectra are provided in Fig. S24 (ESI†). | ||
In summary, we have established synthetic routes for two types of tetracyclic boron heterocycles, the first being boroles featuring a phenanthrene framework that represent the first of the borole family featuring aryl fusion on the 3,4-positions of the central BC4 core. Reaction of the fused borole with benzyl azide furnishes a pyrene analogue with a BN unit in place of one of the CC bonds. The reactivity disclosed diversifies the chemicals space in the fields of boroles, pyrenes, and boron-containing polycyclic aromatic hydrocarbons as a whole.
C. D. M. is grateful to the Welch Foundation (Grant No. AA-1846) and the National Science Foundation (Award No. 1753025 and 2349851) for their generous support of this work. T. B. M. thanks the Julius-Maximilians-Universität Würzburg for support. C. D. M. and J. H. thank the Alexander von Humboldt Foundation for an Experienced Researcher Fellowship and a Postdoctoral Fellowship, respectively. Josina Bohlen and Nele Wieprecht are thanked for their comments on the project. Dr John Tidwell is thanked for assistance with X-ray crystallography.
Data availability
The data underlying this study are available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. Köster, G. Benedikt, W. Larbig, K. Reinert and G. Rotermund, Angew. Chem., 1963, 75, 1079–1090 CrossRef; (b) R. Koster and G. Benedikt, Angew. Chem. Int. Ed., 1963, 2, 323–324 CrossRef.
- J. J. Eisch, N. K. Hota and S. Kozima, J. Am. Chem. Soc., 1969, 91, 4575–4577 CrossRef CAS.
- (a) E. H. Braye, W. Hübel and I. Caplier, J. Am. Chem. Soc., 1961, 83, 4406–4413 CrossRef CAS; (b) J. M. Davidson and C. M. French, J. Chem. Soc., 1960, 191–195 RSC; (c) W. Zhang, B. Zhang, D. Yu and G. He, Sci. Bull., 2017, 62, 899–900 CrossRef CAS PubMed.
- (a) X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed; (b) J. He, F. Rauch, M. Finze and T. B. Marder, Chem. Sci., 2021, 12, 128–147 RSC; (c) W. Yang, K. E. Krantz, L. A. Freeman, D. A. Dickie, A. Molino, A. Kaur, D. J. D. Wilson and R. J. Gilliard Jr., Chem. – Eur. J., 2019, 25, 12512–12516 CrossRef CAS PubMed; (d) K. E. Krantz, S. L. Weisflog, N. C. Frey, W. Yang, D. A. Dickie, C. E. Webster and R. J. Gilliard Jr., Chem. – Eur. J., 2020, 26, 10072–10082 CrossRef CAS PubMed.
- (a) A. Iida and S. Yamaguchi, J. Am. Chem. Soc., 2011, 133, 6952–6955 CrossRef CAS PubMed; (b) W. Zhang, D. Yu, Z. Wang, B. Zhang, L. Xu, G. Li, N. Yan, E. Rivard and G. He, Org. Lett., 2019, 21, 109–113 CrossRef CAS PubMed; (c) A. Iida, A. Sekioka and S. Yamaguchi, Chem. Sci., 2012, 3, 1461–1466 RSC; (d) J. He, F. Rauch, A. Friedrich, J. Krebs, I. Krummenacher, R. Bertermann, J. Nitsch, H. Braunschweig, M. Finze and T. B. Marder, Angew. Chem., Int. Ed., 2021, 60, 4833–4840 CrossRef CAS PubMed; (e) J. He, F. Rauch, I. Krummenacher, H. Braunschweig, M. Finze and T. B. Marder, Dalton Trans., 2021, 50, 355–361 RSC; (f) A. F. Alahmadi, J. Zuo and F. Jäkle, Polym. J., 2023, 55, 433–442 CrossRef CAS; (g) S. M. Barbon, V. N. Staroverov and J. B. Gilroy, Angew. Chem., Int. Ed., 2017, 56, 8173–8177 CrossRef CAS PubMed.
- (a) W. Schacht and D. Kaufmann, Angew. Chem., Int. Ed. Engl., 1987, 26, 665–666 CrossRef; (b) A. Y. Houghton, V. A. Karttunen, W. E. Piers and H. M. Tuononen, Chem. Commun., 2014, 50, 1295–1298 RSC.
- M. O. Akram, J. R. Tidwell, J. L. Dutton, D. J. D. Wilson, A. Molino and C. D. Martin, Inorg. Chem., 2022, 61, 9595–9604 CrossRef CAS PubMed.
- (a) X. Su, J. J. Baker and C. D. Martin, Chem. Sci., 2020, 11, 126–131 RSC; (b) S. Yruegas, D. C. Patterson and C. D. Martin, Chem. Commun., 2016, 52, 6658–6661 RSC; (c) H. Braunschweig, M. A. Celik, F. Hupp, I. Krummenacher and L. Mailänder, Angew. Chem., Int. Ed., 2015, 54, 6347–6351 CrossRef CAS PubMed; (d) H. Braunschweig, M. A. Celik, T. Dellermann, G. Frenking, K. Hammond, F. Hupp, H. Kelch, I. Krummenacher, F. Lindl, L. Mailänder, J. H. Müssig and A. Ruppert, Chem. – Eur. J., 2017, 23, 8006–8013 CrossRef CAS PubMed; (e) J. H. Barnard, S. Yruegas, K. Huang and C. D. Martin, Chem. Commun., 2016, 52, 9985–9991 RSC; (f) T. Bischof, N. Wieprecht, S. Fuchs, L. Endres, I. Krummenacher, M. Michel, C. Mihm, H. Braunschweig and M. Finze, Inorg. Chem., 2023, 62, 21329–21335 CrossRef CAS PubMed; (g) J. H. Barnard, P. A. Brown, K. L. Shuford and C. D. Martin, Angew. Chem., Int. Ed., 2015, 54, 12083–12086 CrossRef CAS PubMed; (h) P. A. Brown, C. D. Martin and K. L. Shuford, Phys. Chem. Chem. Phys., 2019, 21, 18458–18466 RSC; (i) S. Kirschner, S.-S. Bao, M. K. Fengel, M. Bolte, H.-W. Lerner and M. Wagner, Org. Biomol. Chem., 2019, 17, 5060–5065 RSC.
- (a) S. A. Couchman, T. K. Thompson, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Chem. Commun., 2014, 50, 11724–11726 RSC; (b) H. Braunschweig, C. Hörl, L. Mailänder, K. Radacki and J. Wahler, Chem. – Eur. J., 2014, 20, 9858–9861 CrossRef CAS PubMed.
- (a) S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Commun., 2018, 54, 6808–6811 RSC; (b) W. Zhang, G. Li, L. Xu, Y. Zhuo, W. Wan, N. Yan and G. He, Chem. Sci., 2018, 9, 4444–4450 RSC.
- (a) Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed; (b) C. R. McConnell and S.-Y. Liu, Chem. Soc. Rev., 2019, 48, 3436–3453 RSC; (c) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef; (d) Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef CAS PubMed.
- (a) P. E. Romero, W. E. Piers, S. A. Decker, D. Chau, T. K. Woo and M. Parvez, Organometallics, 2003, 22, 1266–1274 CrossRef CAS; (b) P. A. Chase, W. E. Piers and B. O. Patrick, J. Am. Chem. Soc., 2000, 122, 12911–12912 CrossRef CAS; (c) J. L. Bohlen, L. Endres, R. Drescher, K. Radacki, M. Dietz, I. Krummenacher and H. Braunschweig, Chem. Sci., 2023, 14, 9010–9015 RSC; (d) C. Fan, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2009, 48, 2955–2958 CrossRef CAS PubMed.
- S. Biswas, I. M. Oppel and H. F. Bettinger, Inorg. Chem., 2010, 49, 4499–4506 CrossRef CAS PubMed.
- (a) J. Kashida, Y. Shoji and T. Fukushima, Chem. – Asian J., 2019, 14, 1879–1885 CrossRef CAS PubMed; (b) K. R. Bluer, L. E. Laperriere, A. Pujol, S. Yruegas, V. A. K. Adiraju and C. D. Martin, Organometallics, 2018, 37, 2917–2927 CrossRef CAS.
- M. J. S. Dewar and W. H. Poesche, J. Org. Chem., 1964, 29, 1757–1762 CrossRef CAS.
- (a) M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, Angew. Chem., Int. Ed., 2007, 46, 4940–4943 CrossRef CAS PubMed; (b) M. Müller, H. Neitz, C. Höbartner and H. Helten, Org. Lett., 2024, 26, 1051–1055 CrossRef PubMed; (c) Y. Appiarius, P. J. Gliese, S. A. W. Segler, P. Rusch, J. Zhang, P. J. Gates, R. Pal, L. A. Malaspina, K. Sugimoto, T. Neudecker, N. C. Bigall, S. Grabowsky, A. A. Bakulin and A. Staubitz, J. Phys. Chem. C, 2022, 126, 4563–4576 CrossRef CAS PubMed; (d) S. Wang, D.-T. Yang, J. Lu, H. Shimogawa, S. Gong, X. Wang, S. K. Mellerup, A. Wakamiya, Y.-L. Chang, C. Yang and Z.-H. Lu, Angew. Chem., Int. Ed., 2015, 54, 15074–15078 CrossRef CAS PubMed.
- While only benzyl azide insertion is reported in this communication, this reaction should be compatible with other organic azides.
- I. Valencia, P. García-García, D. Sucunza, F. Mendicuti and J. J. Vaquero, J. Org. Chem., 2021, 86, 16259–16267 CrossRef CAS PubMed.
- C. Frampton, K. Knight, N. Shankland and K. Shankland, J. Mol. Struct., 2000, 520, 29–32 CrossRef CAS.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, multinuclear NMR spectra, X-ray crystallographic data, and photophysical studies (PDF). CCDC 2357740 and 2357741. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02863e |
| This journal is © The Royal Society of Chemistry 2024 |